
Abstract
Mesenchymal stem cells (MSCs) have been used as therapeutic tools not only for their ability to differentiate toward different cells, but also for their unique immunomodulatory properties. However, it is still unknown how MSCs may affect immunity during hepatitis B virus (HBV) infection. This study was designed to explore the effect of bone marrow-derived MSCs (BM-MSCs) on hepatic natural killer (NK) cells in a mouse model of acute HBV infection. Mice were injected with 1 × 106 BM-MSCs, which stained with chloromethyl derivatives of fluorescein diacetate fluorescent probe, 24 h before hydrodynamic injection of viral DNA (pHBV1.3) through the tail vein. In vivo imaging system revealed that BM-MSCs were accumulated in the injured liver, and they attenuated immune-mediated liver injury during HBV infection, as shown by lower alanine aminotransferase levels, reduced proinflammatory cytokine production, and decreased inflammatory cell infiltration in the liver. Importantly, administration of BM-MSCs restrained the increased expression of natural-killer group 2, member D (NKG2D), an important receptor required for NK cell activation in the liver from HBV-infected mice. BM-MSCs also reduced NKG2D expression on NK cells and suppressed the cytotoxicity of NK cells in vitro. Furthermore, BM-MSC-derived transforming growth factor-β1 suppressed NKG2D expression on NK cells. As a consequence, BM-MSC treatment enhanced HBV gene expression and replication in vivo. These results demonstrate that adoptive transfer of BM-MSCs influences innate immunity and limits immune-mediated liver injury during acute HBV infection by suppressing NK cell activity. Meanwhile, the effect of BM-MSCs on prolonging virus clearance needs to be considered in the future.
Introduction
H
Recent studies have demonstrated that non-virus-specific innate immune cells are present at high frequencies in the liver and participate in HBV control and/or pathogenesis [4]. As a key component of innate immunity, natural killer (NK) cells are greatly enriched in the liver and play crucial roles in defense against HBV infection. Once activated, NK cells can secrete high levels of proinflammatory and anti-inflammatory cytokines, which exert key roles in the resistance of viral infection and regulation of innate and adaptive immune responses. However, overactivation of NK cells may lead to liver injury [5].
NK cells may be dramatically important for chronic hepatitis B (CHB) patients because they are shown to be partially functionally tolerant. Recent reports have shown that the functions of NK cells are hampered in persistent HBV carrier, which may result from transforming growth factor-β1 (TGF-β1)-induced downregulation of natural-killer group 2 member D (NKG2D)/DAP10 and 2B4/SAP expression on NK cells [6]. Accumulating evidence suggests that NK cells not only play an important role in controlling viral hepatitis, liver fibrosis, and liver tumorigenesis, but also contribute to the pathogenesis of liver injury and inflammation [7].
Mesenchymal stem cells (MSCs) are multipotent progenitor cells representing an attractive therapeutic tool for regenerative medicine. During the last decade, there is a growing realization that MSCs possess unique immunomodulatory properties, being capable of suppressing the proliferation and functions of T, B, and NK cells [8 –11], and modifying dendritic cell differentiation and maturation [11,12]. However, it is unknown how MSCs may affect NK cell functions in HBV-induced liver injury.
In this study, we used a mouse model of acute HBV infection caused by hydrodynamic injection with plasmid containing a 1.3-fold overlength HBV genome (pHBV1.3). We attempted to evaluate the immunomodulatory capacity of bone marrow-derived MSCs (BM-MSCs) on hepatic NK cells during the early stage of viral infection and explore the underlying mechanism. Our results indicated that adoptive transfer of BM-MSCs in mice ameliorates immune-mediated liver injury, but enhances the virus expression and replication. More importantly, the mechanism may be, at least in part, a consequence of the suppression of NK cell cytotoxicity in the liver by BM-MSC administration. These data suggest that BM-MSCs have an important influence on immunopathology during acute HBV infection at the cost of prolonging virus clearance.
Materials and Methods
Materials
RPMI 1640 medium, Dulbecco's modified Eagle's medium (DMEM), α-Minimum Essential Medium (α-MEM), 0.25% trypsin, and phosphate-buffered solution (PBS) were purchased from Hyclone (Logan, UT). Fetal bovine serum (FBS) was from Gibco (Grand Island, NY). Percoll medium used to separate liver mononuclear cells (MNCs) was from GE Healthcare (Waukesha, WI). Alanine aminotransferase (ALT) was measured using commercial assay kits (Jiancheng Bioengineering, Nanjing, China). Polymerase chain reaction (PCR) reagents were from Applied Biosystems (Foster City, CA). CellTracker™ green fluorescent chloromethyl derivatives of fluorescein diacetate (CMFDA) dye used as a marker to label cell movement was from Life Technology (Carlsbad, CA).
Flow cytometry analysis
To identify human BM-MSCs, cells were harvested, washed, and incubated with PE anti-CD45, PE anti-CD73, FITC anti-CD90, and FITC anti-CD105 (BioLegend, San Diego, CA) for 30 min at 4°C. The phenotype of mice BM-MSCs was analyzed using PE anti-CD14, PE anti-CD45, PE anti-CD44 (BD Pharmingen, San Diego, CA), and APC Anti-CD166 (eBioscience, San Diego, CA). Cells were washed twice and resuspended with precooled PBS. Stained cells were measured by flow cytometry on Accuri C6 (BD Biosciences, Franklin Lakes, NJ).
Animal treatment
Male BALB/c mice, 6–8 weeks old, were purchased from Beijing Huafukang Bioscience Company and housed in a specific pathogen-free circumstance in the Animal Experiment Center of Wuhan University/Animal Biosafety Level-III Laboratory, and the experiments were performed in compliance with the Guide for the Care and Use of Medical Laboratory Animals (Ministry of Health, People's Republic of China, 1998).
In this study, we initiated a mouse model of acute HBV infection using a pUC18 vector containing 1.3-fold overlength HBV genome (pHBV1.3). BM-MSCs (1 × 106) were injected into mice (n = 6/group) through the tail vein, 24 h before hydrodynamic injection with pHBV1.3 or pUC18 (10 μg) within 6 s. Subsequently, mice were sacrificed after 4 or 7 days. The serum samples were collected to detect the levels of ALT, hepatitis B surface antigen (HBsAg), and hepatitis B envelope antigen (HBeAg) with a spectrophotometric measurement of absorbance values at 450 nm and HBV DNA with quantitative PCR. Meanwhile, the liver tissues were harvested for histological, real time PCR (RT-PCR), and western blot analysis.
H-E staining and immunohistochemistry
The liver tissues were treated with 4% paraformaldehyde for 24 h, embedded in paraffin, cut, and affixed to 6-micron slides. For hematoxylin-eosin staining, slides were deparaffinized and cell cytoplasm was stained with Hematoxylin, whereas nuclear was stained with Eosin to determine morphological changes. For immunohistochemistry, liver sections were treated with 3% hydrogen peroxide and blocked with 5% bovine serum albumin, then incubated sequentially with anti-HBc antibody (Sigma, St. Louis, MO), biotin-labeled secondary antibody, avidin–biotin complex, and 3, 3’-diaminobenzidine solution. Finally, the sections were counterstained with Hematoxylin.
Detection of ALT, HBsAg, HBeAg, and HBV DNA
ALT activity was measured according to the supplier's protocol for OD510. HBsAg and HBeAg were measured with a commercial Enzyme-Linked Immunosorbent Assay (ELISA) Kit (Kehua Bioengineering, Shanghai, China). HBV DNA were quantitated using the PCR Kit (Liferiver Biotechnology, Shanghai, China).
Real-time RT-PCR
Total RNA was extracted from cells or liver tissues in TRIzol reagent (Invitrogen, Carlsbad, CA), and 2 μg of RNA was reverse transcribed into complementary first-strand complementary DNA using Moloney Murine Leukemia Virus Reverse Transcriptase (Invitrogen, Carlsbad, CA). The reverse transcription products were amplified by a CFX 96 Detection System (Bio-Rad, Hercules, CA) using the SYBR Green Mix (Applied Biosystems). The primer sequences used for the amplification are shown in Supplementary Table S1 (Supplementary Data are available online at
Western blot analysis
The liver tissues were lysed in radioimmunoprecipitation assay (RIPA) buffer with dithiothreitol, cocktail, and phenylmethanesulfonyl fluoride. The protein samples were then separated by 12% sodium dodecyl sulfonate-polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane (GE Healthcare, Waukesha, WI). The membrane was then blocked with 5% skim milk and incubated with rabbit polyclonal anti-NKG2D antibody (1:200, Santa Cruz, CA) and rabbit monoclonal anti-β-actin antibody (1:6,000; Proteintech, Wuhan, China). Peroxidase-conjugated goat anti-rabbit IgG antibody (1:40,000; Sigma) was used as a secondary antibody and the immunoblot signals were detected with enhanced chemiluminescence substrate (Millipore, Billerica, MA).
Cell cultures
Human BM-MSCs were collected from healthy donors for BM transplantation by Wuhan Hamilton Biotechnology Co. Ltd. Informed consent was obtained for each subject. This study was undertaken in accordance with ethical regulations imposed by the Chinese legislation. All the cell culture media contained 100 U/mL penicillin and 100 μg/mL streptomycin.
Human BM-MSCs were maintained in DMEM supplemented with 20% FBS. Murine BM-MSCs were maintained in α-MEM supplemented with 15% FBS, 0.1% 2-mercaptoethanol. Murine liver-derived MNCs were cultured in RIPA 1640 medium containing 10% FBS. YTs, a human NK cell line, were cultured in RIPA 1640 medium containing 15% FBS. YTs were incubated in medium alone or with TGF-β1 (1 ng/mL, 10 ng/mL; R&D Systems, Minneapolis, MN) for 48 h. YTs or MNCs were cocultured with BM-MSCs alone or in combination with anti-TGF-β1 neutralizing antibody (10 μg/mL; R&D Systems) for 48 h.
Cytotoxicity assay
Cell cytotoxicity was determined by the Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan) assay. K562 cells or Yac-1 cells were used as target cells (T), and YTs or the liver MNCs were used as effector cells (E), respectively. Target cells were incubated with effector cells at E:T = 1:5. After 24 h, 10 μL of CCK-8 reagent was added to each well and incubated for 4 h at 37°C. The production of blue formazan crystals was determined by spectrophotometric measurement of absorbance values at 450 nm. Specific lysis (%) was calculated as follows: [1–(ODE+T–ODE)/ODT] × 100%.
Statistical analysis
Data were presented as mean ± standard error of mean and analyzed by Student's test or analysis of variance (ANOVA) when appropriate (P < 0.05 was considered as significant).
Results
BM-MSCs attenuate liver injury induced by hydrodynamic injection with pHBV1.3
To explore the potential function of BM-MSC treatment during the HBV infection, we first isolated and cultured BM-MSCs from male BALB/c mice. These cells were purified by their adherence to the cell culture dish and identified for the phenotype positive for CD44 and CD166, but negative for CD14 and CD45 (Fig. 1B). Flow cytometric analysis indicated that these spindle-shaped cells have characteristic MSC phenotype.
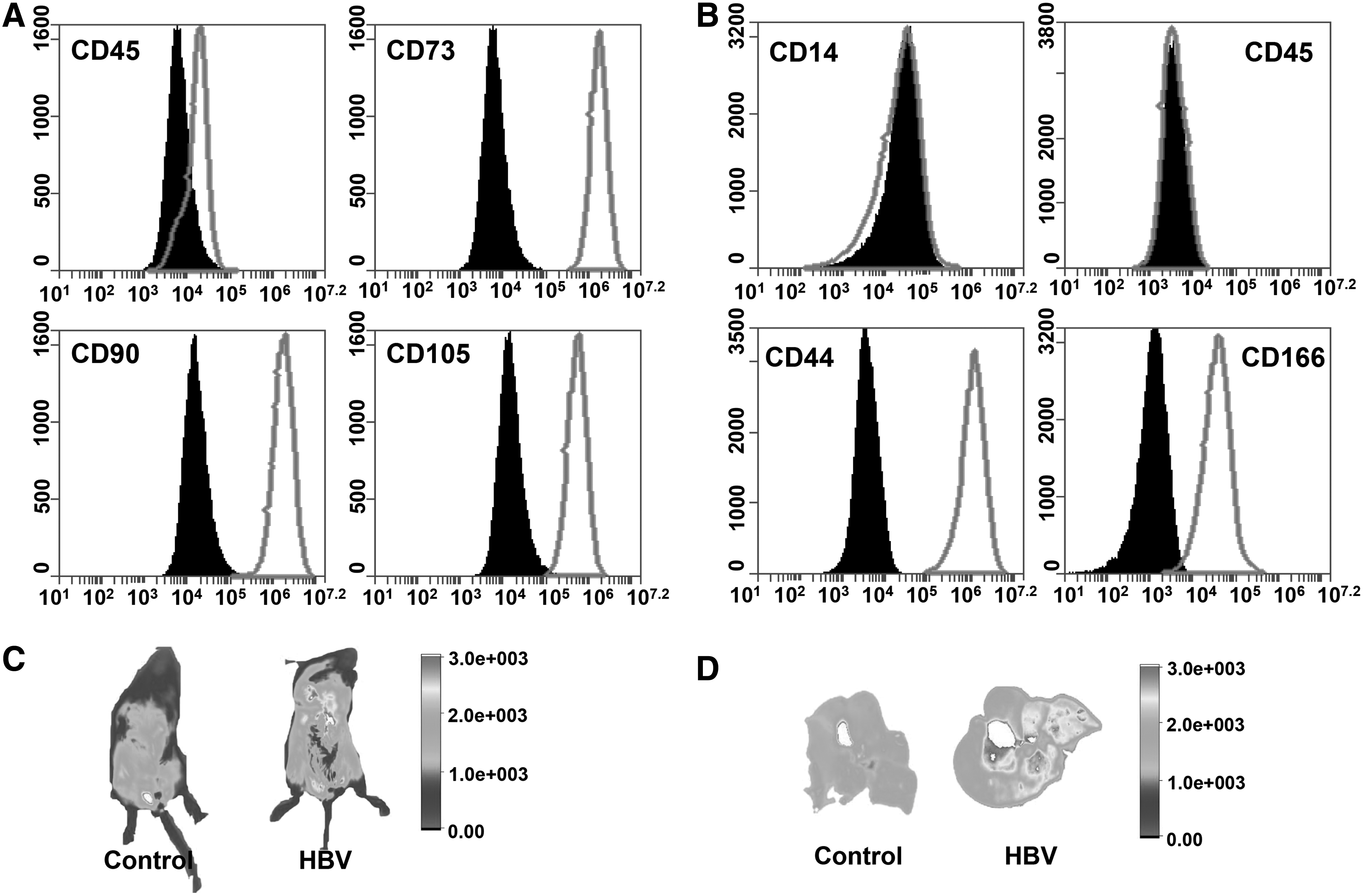
BM-MSCs are accumulated in the liver during acute HBV infection. Identification of human BM-MSCs
To determine whether BM-MSC administration could reduce HBV-induced liver injury, we used a mouse model of acute HBV infection caused by hydrodynamic injection with pHBV1.3. BALB/c mice were treated with tail vein injection of 1 × 106 BM-MSCs, 24 h before hydrodynamic injection with pHBV1.3 or pUC18 (10 μg) within 6 s. To track the location of BM-MSCs, these cells were predyed with CellTracker CMFDA at a concentration of 20 μM. We found that fluorescent signals from pHBV1.3-injected mouse were remarkably stronger than the controls, and that the majority of these signals were from the liver. To confirm whether the BM-MSCs could migrate to the injured liver caused by pHBV1.3 injection, we isolated the livers from mice. Indeed, the fluorescence images showed that these BM-MSCs were almost accumulated in the injured liver (Fig. 1C).
Next, we tested the effect of BM-MSCs on HBV-induced liver injury. We observed an elevated ALT activity in the serum of pHBV1.3-injected mice for 4 or 7 days. However, administration of BM-MSCs decreased serum ALT levels (Fig. 2A, B). In addition, H-E staining showed that inflammatory cells infiltrated to the injured locus upon HBV infection, whereas BM-MSC treatment attenuated the infiltration of inflammatory cells (Fig. 2C).
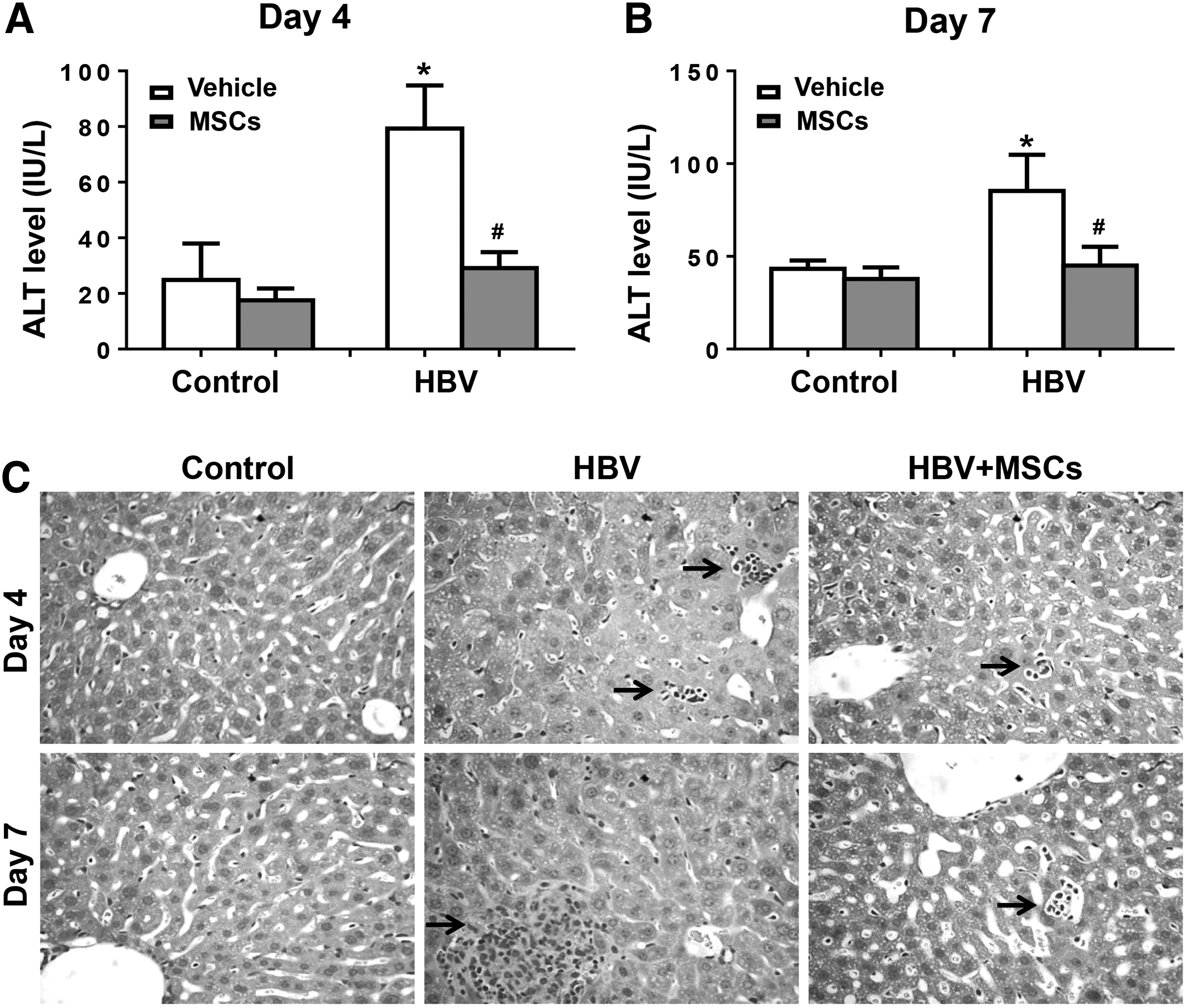
BM-MSCs attenuate liver injury during acute HBV infection. BALB/c mice were preinjected with BM-MSCs or PBS. The following day, mice were hydrodynamic injected with pHBV1.3 or pUC18 and sacrificed for additional 4 days
We also analyzed the effect of BM-MSCs on cytokine expression in our model. As shown in Figure 3A and B, pHBV1.3 injection markedly promoted the expression of interleukin-1β, interleukin-6, tumor necrosis factor-α, and chemokine (C-C motif) ligand 3. However, BM-MSC administration reduced the expression of the above cytokines. In addition, there was a dramatical decrease of TGF-β1 expression in the liver 4 or 7 days after pHBV1.3 plasmid injection, whereas TGF-β1 expression was increased by administration of BM-MSCs (Fig. 3A, B). Taken together, these results demonstrated that BM-MSC treatment ameliorates liver damage caused by pHBV1.3 injection.
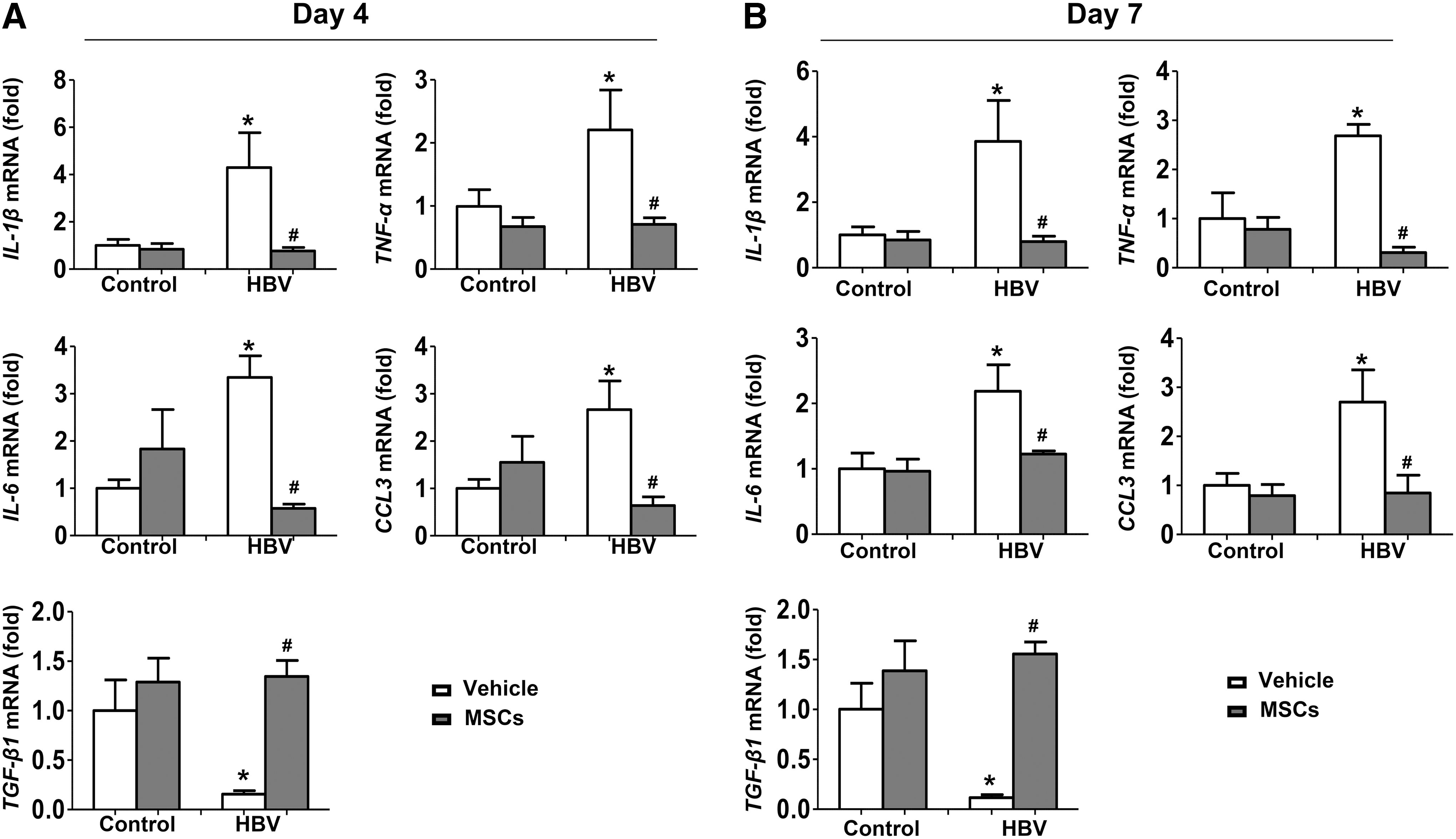
BM-MSCs reduce liver inflammation during acute HBV infection. BALB/c mice were preinjected with BM-MSCs or PBS, 24 h before hydrodynamic injection of pHBV1.3 or pUC18. After 4 days
BM-MSCs decrease NKG2D expression on NK cells in vitro and in vivo
NK cells have been shown to play an important role in controlling HBV infection [13]. Previous studies have shown that MSCs could mediate immunosuppression on various immune cells [8,11,14]. Therefore, we hypothesized that BM-MSCs could modulate the function of NK cells during HBV infection. As NK cells express a repertoire of activating and inhibitory receptors to determine their function [15,16], we investigated whether the expressions of NKG2A, NKG2C, and NKG2D could be regulated by BM-MSCs.
As shown in Figure 4A, there was no significant difference in the expressions of NKG2A and NKG2C on liver NK cells cocultured with BM-MSCs for 24 or 48 h compared with the controls. However, NKG2D expression was significantly suppressed on liver NK cells after coculture with BM-MSCs. Moreover, we carried out the same coculture experiments by using a human NK cell line (YTs) and human BM-MSCs. Flow cytometric analysis showed that these human BM-MSCs were positive for CD73, CD90, and CD105, but negative for CD45, thus have a typical MSC phenotype (Fig. 1A). Similarly, human BM-MSCs markedly suppressed NKG2D expression on YTs (Fig. 4B).
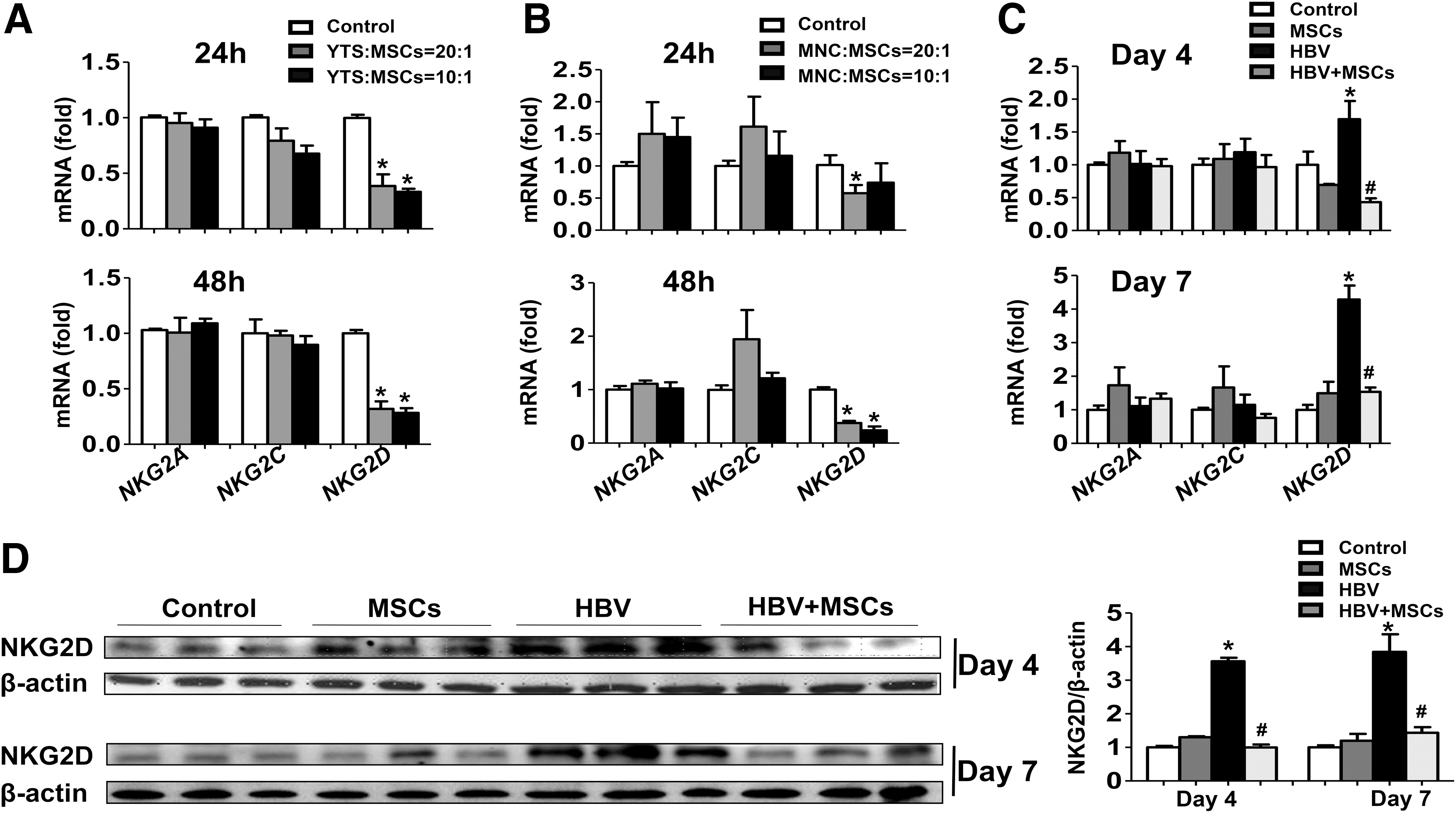
BM-MSCs decrease NKG2D expression on NK cells in vitro and in vivo.
To further confirm this issue, we analyzed NKG2D expression in the liver followed by BM-MSC treatment. Hydrodynamic injection with pHBV1.3 induced a significant increase of NKG2D expression in the liver. However, administration of BM-MSCs suppressed the elevation of NKG2D expression, as determined by real-time RT-PCR and western blot analysis (Fig. 4C, D). Collectively, these results indicated that BM-MSC treatment was associated with the decrease in the expression of NKG2D on NK cells in vitro and in vivo.
BM-MSCs decrease cytotoxicity of NK cells
The multiple functions of NK cells, such as cytotoxicity can be induced by interaction between activating or inhibitory receptors with their ligands [17].
To confirm whether liver NK cells cocultured with BM-MSCs has a defect in cytotoxic activity, cell cytotoxicity was evaluated by measuring the lysis of a mouse lymphoma cell line Yac-1 cells. As expected, the cytotoxicity was significantly suppressed in the liver NK cells cocultured with BM-MSCs, compared with control (Fig. 5A). Similarly, we confirmed this issue by using YTs and human BM-MSCs to perform the same coculture experiment, and we chose a human chronic myelogenous leukemia cell line K562 as target cells. As shown in Figure 5B, human BM-MSCs also suppressed cytotoxicity of YTs. These results suggested that the reduced expression of NKG2D by BM-MSCs might result in a defect in killing target cells.
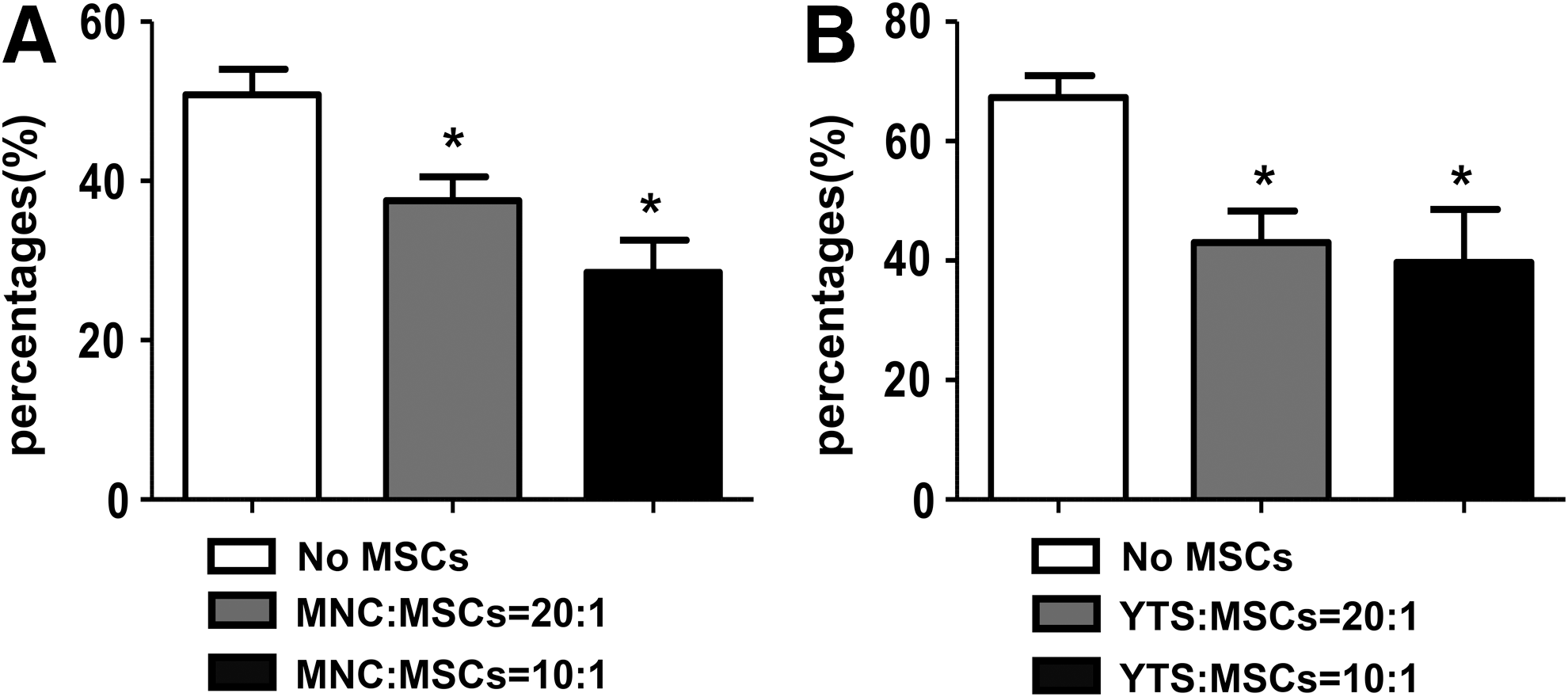
BM-MSCs suppress cytotoxicity of NK cells in vitro. The effect of BM-MSCs on MNCs or YTs cell-mediated cytotoxicity was measured by CCK8 assay.
BM-MSC-derived TGF-β1 suppresses NKG2D expression on NK cells
Previous studies have shown that soluble TGF-β1 is closely associated with the reduction in NKG2D during chronic HCV [18] and HBV infection [6].
To investigate whether BM-MSC-derived TGF-β1 regulated NKG2D expression on NK cells, we first measured the expression of NK receptors after TGF-β1 stimulation. Clearly, NKG2D, but not NKG2A, NKG2C, and NKG2E expressions, was significantly downregulated by TGF-β1 stimulation (Fig. 6A). Furthermore, we used anti-TGF-β1 antibody to block TGF-β1-mediated regulation of NKG2D expression. As shown in Figure 6B, there was a dramatical decrease in NKG2D expression on liver NK cells or YTs cells cocultured with BM-MSCs, whereas TGF-β1 blockade restored NKG2D expression. These results suggested that BM-MSC-derived TGF-β1 contributes to BM-MSC-mediated suppression of NKG2D expression on NK cells.
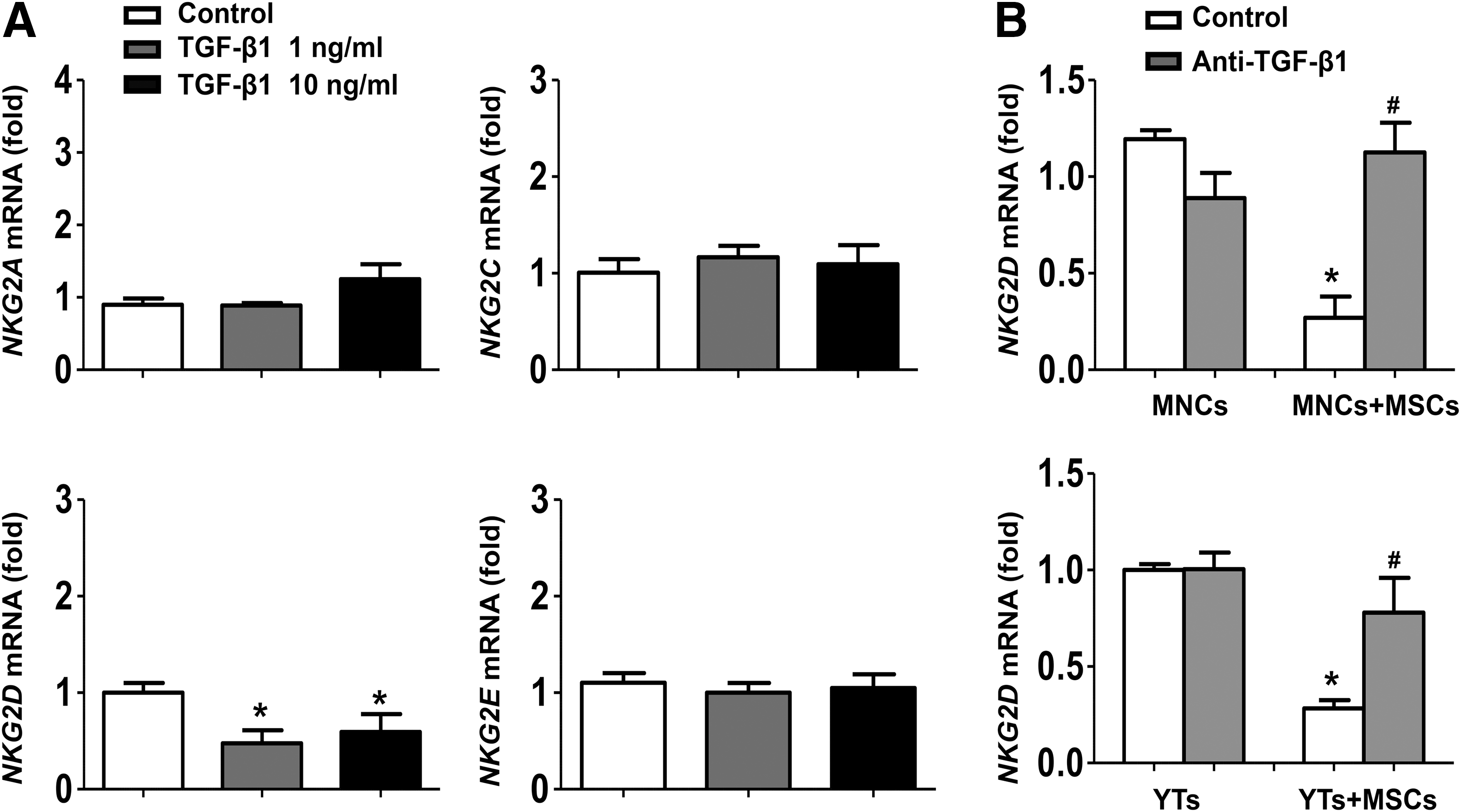
BM-MSC-derived TGF-β1 suppresses NKG2D expression on NK cells.
BM-MSC treatment enhances HBV gene expression and replication in vivo
Innate immunity and adaptive immunity are well known to cooperate with each other to eliminate the invaded pathogenic microorganisms. We tested the final outcome of virus by measuring the titers of HBsAg, HBeAg, and HBV DNA in mouse sera (Fig. 7). PCR results showed that the HBV titers decreased to a low level 4 and 7 days postinjection. However, administration of BM-MSCs helped the virus titers to maintain a higher level (Fig. 7A). Moreover, immunohistochemical staining showed that BM-MSCs could increase hepatitis B core antigen expression in mouse livers (Fig. 7C). These results revealed that the levels of HBV gene expression and replication are notably enhanced by BM-MSC administration.
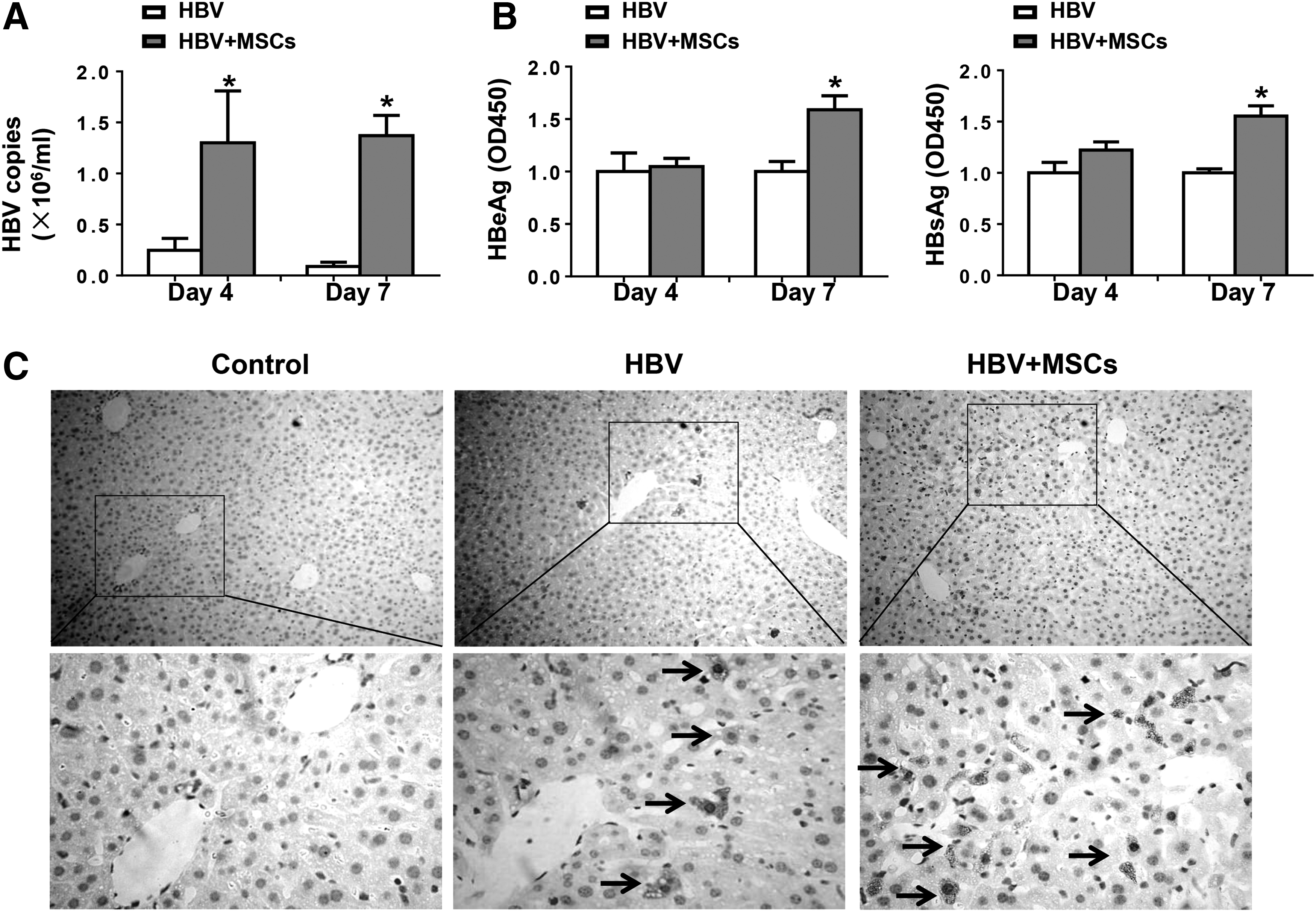
BM-MSC treatment enhances HBV gene expression and replication in vivo. BALB/c mice were injected with PBS or MSCs, 24 h before hydrodynamic injection with pHBV1.3 or pUC18. After 4 or 7 days, mice were sacrificed. Serum and liver tissues were collected.
Discussion
Accumulating evidence has shown that BM-derived cells can be used as an efficient therapeutic strategy against many diseases, including chronic liver diseases [19 –21]. Among the various types of BM-derived cells, MSCs have drawn most attention, not only for their multidifferentiation capacity, but also the immunoregulatory functions on other immune cells.
In this study, we found that BM-MSCs were recruited to the damaged liver and alleviated the liver injury caused by hydrodynamic injection with pHBV1.3. Furthermore, TGF-β1 secreted by BM-MSCs suppressed the cytotoxicity activity of NK cells through inhibiting NKG2D expression on NK cells. Finally, BM-MSC administration led to a marked increase of HBV proteins and DNA levels in the mouse sera and livers. Taken together, these results demonstrate that BM-MSC treatment suppresses NKG2D expression on NK cells, contributing to an alleviation of the liver injury caused by pHBV1.3 injection. Interestingly, the levels of HBV gene expression and replication are enhanced by BM-MSC administration.
NK cells are enriched in lymphocytes within the liver. They play a critical role in the defense against viral infection and regulation of virus-specific T cell response [22]. Several studies have suggested a role of NK cells in the control of viral hepatitis, liver fibrosis, and liver tumorigenesis [7]. NK cells can lyse the virus-infected cells, induce apoptosis of target cells, and release inflammation cytokines through cell–cell contact or secreting cytokines. However, the excessive activation of NK cells could lead to liver inflammation and injury [23,24]. To better understand the function of NK cells during acute HBV infection, we used an acute HBV infection mouse model by hydrodynamic injection of pHBV1.3. We observed an elevation of ALT levels and mild inflammation in the livers of pHBV1.3-injected mice compared with the control.
The function of NK cells is regulated by a series of signals from activating and inhibitory receptors on the surface and their interaction with corresponding ligands [25]. In this study, we analyzed the expression of NK receptors on NK cells. Our data showed that the activating receptor NKG2D, but not NKG2A, NKG2C, and NKG2E expression, was significantly increased after pHBV1.3 injection, suggesting that the liver injury during acute HBV infection may, at least in part, result from the excessive activation of NK cells.
Previous reports have shown that there are high expressions of the inhibitory receptor NKG2A in active CHB patients. Blockade of NKG2A can markedly augment NK cell cytotoxicity from active CHB patients and blockade of NKG2A in HBV-carrier mice can reverse NK cell tolerance, suggesting that NKG2A might be a potential target to treat CHB [26]. However, our results showed that there is no change in the expression of NKG2A in acute HBV infection mouse model, which might be due to the different mouse genetic background. Indeed, BALB/c mice initiate a more intensive immune response against HBV, and viral replication persists for a shorter time compared with that in C57BL/6 mice [27].
The interaction of activating receptor NKG2D and its ligand on NK cells promote the secretion of IFN-γ, which finally results in severe liver injury in a polyI:C-induced fulminant hepatitis model [28]. In a fulminant hepatic failure induced by murine hepatitis virus strain 3, Fas/FasL and NKG2D/NKG2D ligand pathway play an important role in the final liver failure [29]. Together with our results, suppressing the excessive activation of NK cells can be regarded as an efficient strategy to ameliorate liver injury.
It is well known that MSCs have been used in the regenerative medicine for many years due to their ability to differentiate into various cell types [30,31], but recent studies have also shown that MSCs have immunosuppressive effect on many immune cells, including T, B, NK, NKT cells, and macrophages [32 –35]. MSCs have nonselective immunosuppressive capacity on CD4+ and CD8+ T cell proliferation by arresting cell division [36 –38]. MSCs can also reduce proliferation and antibody secretion in B lymphocytes [14], and inhibit the maturation, differentiation, and antigen presentation of monocyte-derived dendritic cells [39]. Moreover, MSCs can suppress NK cell proliferation, cytolytic activity, and cytokine production [10,40].
In this study, we found that BM-MSCs were recruited to the injured liver induced by pHBV1.3 injection, which is consistent with the previous reports, which demonstrated that MSCs can migrate to sites of tissue damage and inflammation [41 –43]. It should be noted that the inherent MSCs from the recipient mice may also be recruited to the injured liver, which cannot be excluded from this study. Importantly, our results suggest that BM-MSC treatment suppresses the activity of NK cells through downregulation of NKG2D expression on NK cells, thereby attenuating the immune-mediated liver injury.
Interestingly, as a consequence of the reduced cytotoxicity activity of NK cells, treatment with BM-MSCs enhances HBV gene expression and replication. This is consistent with the previous report that Tregs protect the liver from damage at the cost of prolonging virus clearance in transgenic HBV infection model [44]. Therefore, we speculate that viral immune escape and persistent infection always come along with the liver tissue damage. This should be taken into consideration in the clinical treatment with these immunosuppressive cells. The mechanism by which the virus evades host immune system and persists a longer time needs to be addressed in future studies.
MSCs can modulate the surrounding immune cells through the secretion of soluble factors or cell–cell contact. In CHB patients, the immune tolerance of NK cells is caused by the immunosuppressive IL-10 and TGF-β1, which might be immunotherapeutic target to restore the capacity of NK cells [45]. In addition, during HCV infection, TGF-β1 decreases NKG2D expression on NK cells, resulting in impaired cytolytic activity and lower IFN-γ production [18]. During the immune tolerance phase of HBV infection, there is a similar mechanism which helps the virus evade the host immune responses [6,45].
Our data showed that BM-MSCs downregulated NKG2D expression on NK cells, whereas blocking TGF-β1 could restore NKG2D expression to normal levels in in vitro coculture experiments. Moreover, BM-MSC administration enhanced TGF-β1 expression, which was decreased in the pHBV1.3-injected mice. These results suggest that BM-MSC-derived TGF-β1, at least in part, contributes to the immunomodulation of NK cells in our model. It should be noted that TGF-β1 might also be produced by other cell types than MSCs, after interaction with MSCs in vivo, which cannot be excluded from the current study.
Previous studies have shown that MSCs suppress the proliferation and cytotoxicity activity of NK cells through secreting soluble factors, indoleamine-2, 3-dioxygenase, and prostaglandin [10]. A possible explanation of this finding might be that the complex microenvironment in the physiological conditions and a number of soluble factors/cytokines other than TGF-β1 might participate in the inhibition signal at the same time. More recently, TGF-β1 was shown to inhibit the HBV replication through the recruitment of RNA exosome to viral RNP complex in vitro [46]. This discrepancy may be explained by the fact that BM-MSC-derived TGF-β1 promotes HBV expression and replication through exerting an immunosuppressive effect on NK cells, but not directly acting on the virus.
In summary, our findings demonstrate that BM-MSCs exert immunomodulatory effect on liver NK cells and limit immune-mediated liver injury during acute HBV infection by suppressing NK cell activity. Meanwhile, our data suggest that BM-MSCs influence innate immunity during viral infection at the cost of prolonging virus clearance, which needs to be considered in the clinical use of BM-MSCs.
Footnotes
Acknowledgment
This work was supported by a grant from the National Natural Science Foundation of China (no. 81301427).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.