
Abstract
Stem cell-based therapies have demonstrated great potential for the treatment of cardiac diseases, for example, myocardial infarction; however, low cell viability, low retention/engraftment, and uncontrollable in vivo differentiation after transplantation are still major limitations, which lead to low therapeutic efficiency. Biomaterials provide a promising solution to overcome these issues due to their biocompatibility, biodegradability, low/nonimmunogenicity, and low/noncytotoxicity. The present study aimed to investigate the impacts of keratose (KOS) hydrogel biomaterial on cellular viability, proliferation, and differentiation of c-kit+ human cardiac stem cells (hCSCs). Briefly, hCSCs were cultured on both KOS hydrogel-coated dishes and regular tissue culture dishes (Blank control). Cell viability, stemness, proliferation, cellular morphology, and cardiac lineage differentiation were compared between KOS hydrogel and the Blank control at different time points. We found that KOS hydrogel is effective in maintaining hCSCs without any observable toxic effects, although cell size and proliferation rate appeared smaller on the KOS hydrogel compared to the Blank control. To our surprise, KOS hydrogel significantly promoted vascular smooth muscle cell (VSMC) differentiation (∼72%), while on the Blank control dishes, most of the hCSCs (∼78%) became cardiomyocytes. Furthermore, we also observed “endothelial cell tube-like” microstructures formed by differentiated VSMCs only on KOS hydrogel, suggesting a potential capability of the hCSC-derived VSMCs for in vitro angiogenesis. To the best of our knowledge, this is the first report to discover the preferred differentiation of hCSCs toward VSMCs on KOS hydrogel. The underlying mechanism remains unknown. This innovative methodology may offer a new approach to generate a robust number of VSMCs simply by culturing hCSCs on KOS hydrogel, and the resulting VSMCs may be used in animal studies and clinical trials in combination with an injectable KOS hydrogel to treat cardiovascular diseases.
Introduction
C
c-kit+ human cardiac stem cells (hCSCs) are resident cardiac stem cells that exhibit basic stem cell properties (self-renewing, clonogenic, and multipotent) as well as an ability to differentiate into all cardiovascular lineages [4,5]. Although the functional contribution of c-kit+ hCSCs to in vivo cardiomyocyte (CM) turnover is one of the centrally debated issues concerning their regenerative potential [3,6], plentiful preclinical and clinical studies have provided enormous evidence to demonstrate their in vitro abilities of cardiac lineage differentiation [ie, CMs, endothelial cells (ECs), and vascular smooth muscle cells (VSMCs)] as well as in vivo capacity to improve cardiac function following transplantation [3 –8]. Regardless of differences between in vitro and in vivo studies of c-kit+ hCSCs, the major challenges described above still remain to be solved for all stem cell-based therapies [2,5,7,8].
Biomaterials possess several beneficial attributes, such as biocompatibility, biodegradability, low or nonimmunogenicity, and low or noncytotoxicity, to overcome the above issues [9]. Keratin is a natural biomaterial that has all of the above beneficial properties. More importantly, this structural protein can be readily isolated from various tissues such as the hair and skin of animals or humans and can be easily produced in various formats (film, sponges, and hydrogels) [10]. Over the last few decades, knowledge of keratin and its derivatives has significantly increased, especially in regard to their ultrastructure, molecular and cell biology, physiological and pathological roles, as well as their practical applications in drug delivery and cellular/tissue engineering in regenerative medicine [11]. Due to its low or nonimmunological response, keratin does not induce an immune response, even in allogeneic or xenogeneic transplantations [10,12].
In vitro studies have shown that keratin can significantly enhance the colony-forming efficiency of human mesenchymal stem cells [13], regulate differentiation and proliferation of blastemal cells [14], and modulate adhesion, proliferation, and differentiation of adipose-derived stem cells [15]; and it was even suggested as a potential stem cell marker [16]. However, to date, no studies have been reported regarding the utilization of keratose (KOS) hydrogels to culture c-kit+ hCSCs. The only in vivo study using a keratin-based hydrogel in repairing heart infarction is the rat model reported by Shen et al. [17], in which direct injection of keratin into infarct hearts was shown to significantly promote angiogenesis, preserve cardiac function, and attenuate ventricular remodeling compared to the vehicle control. Thus, it is important to understand the basic impacts of keratin on hCSC proliferation and differentiation.
In the present study, we aim to examine whether KOS hydrogel is capable of maintaining hCSC culture and proliferation and how KOS hydrogel affects cardiac lineage differentiation compared to regular cell culture dishes (Blank control). Various techniques, including live/dead cell viability assay, cell proliferation assay, real-time polymerase chain reaction (PCR), western blot, and immunocytochemistry, were used in the study. Our long-term goal is to investigate the feasibility of using c-kit+ hCSCs in combination with KOS hydrogels as a new platform for stem cell differentiation and/or for biomaterial-based stem cell therapy to treat cardiovascular diseases.
Materials and Methods
Reagents
All chemicals used in the present study are listed below unless otherwise stated elsewhere: 30% bis-acrylamide clarity ECL-Western blotting substrate and Laemmli buffer (all from Bio-Rad), 5-azacytidine (5-Aza), β-mercaptoethanol, bovine serum albumin (BSA), Bradford reagent, erythropoietin (EPO),
hCSC isolation and culture
Patient heart samples (ie, atrial appendages) were obtained as discarded tissues from local hospitals. Donor confidentiality was maintained at the hospital's request and no patient identification information or medical history was collected according to the approved protocol. A written consent agreement was obtained for collection of discarded atrial appendages by the hospital and all procedures were approved by the Institutional Review Board (IRB) of Virginia Polytechnic Institute and State University for human subject research. The isolation and culture of hCSCs were previously described [18].
Briefly, the isolated cells were cultured in a 37°C incubator (5% CO2 and 21% O2) with hCSC medium consisting of Ham's F12, 10% FBS, 10 ng/mL human b-FGF, 0.2 mM
To examine whether KOS hydrogel altered stemness, hCSCs maintained on regular Blank culture dishes were transferred on KOS hydrogel-coated dishes and continuously cultured for three passages (13–15 days in total) in hCSC medium followed by c-kit antibody staining (see the Immunocytochemistry section below), and were compared to the cells passaged only on Blank dishes.
Keratin isolation and hydrogel formation
KOS was supplied by Dr. Mark Van Dyke's laboratory at Virginia Tech. Briefly, KOS was extracted and purified from human hair fibers that had been washed with detergent, rinsed, and dried [19]. For 100 g of fibers, 2 L of a 2% (w/v) peracetic acid solution was used to oxidize protein disulfide bonds by heating it at 37°C for 14 h with gentle rotary stirring. The fibers were recovered by sieve and extracted twice with 4 L of 100 mM Tris base solution. The fibers were separated by sieve and the extract solutions combined, centrifuged to remove suspended particulates, and filtered through No. 4 Whatman filter discs (Thermo Fisher). The protein solution was purified and high-molecular-weight keratin nanomaterial was isolated by membrane filtration in a custom ultrafiltration system. The membrane cutoff used was 100 kDa and a proprietary buffer solution facilitated separation of the material of interest. After removal of low-molecular-weight contaminants, the keratin nanomaterial solution was concentrated against dilute buffer using the same 100 kDa membrane, frozen and lyophilized [19].
KOS hydrogel was made with 4.5% lyophilized oxidized keratin (w/w) in PBS 1.5% genipin (w/w) in various culture plates or coverslips. The KOS hydrogel was plated at a thickness of ∼1 mm, and let to sit for 12 h to allow the genipin to crosslink. Hydrogels that were to be used for photomicrographs were produced by making a “sandwich” between glass and plastic coverslips, so removing the glass coverslip after 12 h produced a flat surface that was ideal for cell imaging. In both cases, the hydrogels were then washed three times (5 min each) in PBS and covered with PBS for 48 h to remove any residual genipin from the hydrogels. After the PBS was removed, hCSCs were seeded on top of the KOS hydrogels.
Cell viability assay with Calcein-AM/ethidium homodimer-1 and Trypan blue
hCSCs were plated at 1.5 × 105 cells in a regular 60-mm dish (Blank control) or KOS hydrogel-coated 60-mm dish for 1, 3, and 5 days with hCSC medium. At each time point, medium was collected and cells were enzymatically detached with 10 U/mL elastase for 3 min in a 37°C incubator. All cells (detached and floating dead cells collected from the medium) were pooled together for the live/dead assay using Calcein-AM/ethidium homodimer-1 (EthD-1). Briefly, collected cells were washed once with PBS and incubated simultaneously with 2 μM Calcein-AM and 4 μM EthD-1 in a 37°C incubator for 30 min. Meanwhile, control wells were subject to a 20-min treatment with 70% methanol (dead cell controls) or without any treatments (live cell controls) and used to calculate cell viability. Following staining, cells were collected in PBS by centrifuge and then replated in a 96-well plate at a density of 1 × 104 cells per well. Six replicates of the experimental group and three replicates of the control group were used to collect data in a SpectraMax M5 plate reader (Molecular Devices).
Trypan blue was also used for evaluating cell death in a separate experimental group. In this case, cells were cultured in a 60-mm dish and elastase enzymatic detachment from the KOS hydrogel was the same as described above. The dead cells (Trypan blue positive) were counted using a hemocytometer, and cell viability (%) was obtained by dividing the number of live cells by the total number of cells. At least 1,000 cells were counted in each group and the results were plotted against passage (P) numbers (P1, P2, and P3).
EdU cell proliferation
Cells were plated at a density of 2.5 × 104 cells per well on either gelatin-coated coverslips (Blank control) or KOS hydrogel-coated coverslips in 12-well plates. Cell proliferation was examined at day 1, 3, and 5. Briefly, cells were incubated with EdU (5-ethynyl-2′-deoxyuridine) at a 1:500 dilution in hCSC medium for 18 h and then stained by following the manufacturer's protocol included in the Click-iT EdU Cell Proliferation kit. After costaining with Hoechst 33342, both the Blank control and KOS hydrogel coverslips were mounted on glass tissue slides using the Vectashield antifade mounting medium and sealed with fingernail polish. Fluorescent EdU images were taken with an Olympus IX73 microscope equipped with an XM10 camera. The images were then analyzed using ImageJ software (NIH). At least 1,000 Hoechst cells were counted on each coverslip to calculate % of EdU-positive cells over total number of cells (Hoechst nucleus staining). Statistical analysis and bar graphs were completed using Microsoft Excel 2013 and GraphPad Prism 5.
Cell morphology analysis with phalloidin staining
hCSCs were cultured at a density of 2.5 × 104 cells per well on either gelatin-coated coverslips (Blank control) or KOS hydrogel-coated coverslips for 1, 3, and 5 days. The differentiated cells were also used in this assay (see the Cardiac Lineage Differentiation of hCSCs section). For phalloidin staining, cells on the Blank control and KOS hydrogel were fixed in 4% PFA at room temperature (RT) for 30 min and washed twice in PBS (5 min each) followed by blocking in 1.5% BSA for 30 min. The resulting cells were stained with FITC-conjugated phalloidin (1:500 dilution from 1000 × stock) and Hoechst 33342 (1 μM) in PBS for 1 h at RT followed by three additional washes in PBS (5 min each). Coverslips were then mounted on tissue slides and fluorescent images were taken with the Olympus IX73 microscope mentioned above. Only cells cultured for 3 days were used for cell morphological analysis (cell area and ratio of cell length per square of cell area) by ImageJ (NIH); cells on day 1 and 5 were not selected due to the cell density being either too low or too high to obtain accurate measurements. At least 500 cells in each group were analyzed and the results were plotted as bar graphs.
Furthermore, KOS hydrogels were subject to cryosectioning to determine the cell distribution and migration within the hydrogel. To this end, hydrogel samples were fixed in 4% PFA as described above and then embedded and frozen in OCT with 30% sucrose on an aluminum block in liquid nitrogen. Sample blocks were then serially cut at a thickness of 30 μm using a rotating cryostat (Reichert Histostat; Thermo Fisher) and stained with FITC-conjugated phalloidin (1:500) and Hoechst (1 μM) in 1.5% BSA for 1 h at RT. Fluorescent images were taken using the Olympus IX73 microscope mentioned above with green (phalloidin), blue (Hoechst 33342), and red (KOS hydrogel autofluorescence) filters.
Cardiac lineage differentiation of hCSCs
hCSCs were seeded in regular 6-well or 12-well culture dishes as described above. After 2–3 days of culture, the hCSC medium was switched to cardiac differentiation medium (DM) containing 89% Ham F12 nutrient mixture, 10% FBS, 1% P/S, and 10 μM 5-Aza as described previously [18]. After the 3-day treatment with daily medium change, the differentiated cells were maintained in the above DM without 5-Aza up to 28 days with medium changed every other day. The same set of experiments as above was done to examine the effects of KOS hydrogel alone on hCSC differentiation using the same DM but without 5-Aza. In either case, the resulting cells were used for various assays listed below.
qRT-PCR (qPCR)
hCSCs were cultured in six-well plates at a density of 6 × 104 cells per well with hCSC culture medium or DM as described above. Total RNA was extracted using TRIzol reagent, and RNA concentration was determined using the NanoDrop 1000 spectrophotometer (Thermo Scientific). One microgram of total RNA was reversely transcribed in a 20 μL reaction volume using the High-Capacity cDNA Reverse Transcriptase Kit according to the manufacturer's protocol. The resulting cDNA was then diluted twofold in nuclease-free water (12.5 ng per well) and used as a template for quantitative real-time PCR analysis using Power SYBR Green PCR Master Mix in the StepOnePlus Real-Time PCR System (Thermo Fisher).
The following protocol was utilized in all experiments: a 10-min holding stage at 95°C, followed by 44 cycles at 95°C for 15 s and 60°C for 1 min, then a melting curve stage of 95°C for 15 s, 60°C for 1 min, and 95°C for 15 s. The primers are listed in Supplementary Table S1. Each target gene was examined in triplicate and the relative expression levels were analyzed using StepOne software v2.3 provided on the machine. The data are expressed as fold change relative to predifferentiation (Pre-Diff) after normalization to internal control of beta-2-microglobulin (B2 M).
Western blotting
Cells were cultured at a density of 6 × 104 cells per well in either the Blank control or KOS hydrogel-coated six-well plates using hCSC culture medium or DM as described above. For the Blank control, cell lysate was directly obtained by adding RIPA buffer, while for the KOS hydrogel-coated wells, cells were first detached using 10 U/mL elastase for 3 min and washed once with PBS to remove any residual hydrogel before being treated with RIPA buffer. Protein lysate was quantified using the Bradford assay and then mixed with 4 × loading buffer (10% β-mercaptoethanol in Laemmli buffer) at a 1:4 dilution and incubated for 5 min at 95°C. Samples were then loaded into an SDS-stacking gel and run in a Mini-PROTEAN Tetra Cell (Bio-Rad) at 100 V for 15 min followed by 150 V for 60 min. The protein was then transblotted onto a PVDF membrane using a Mini Protean Tetra Cell in an ice bath at 95 V for 1.75 h.
The transblotted membrane was blocked for 1 h at RT in 5% milk solution in TBST on a plate rocker. Membranes were immunoblotted with specific primary antibodies [c-kit, α-sarcomeric actin (α-SA), CD31, and vascular α-smooth muscle actin (α-SMA)] (Supplementary Table S2) to identify stem cells, CMs, ECs, and VSMCs, respectively. Beta-actin (β-A) was used as an endogenous control. The following optimized conditions were used in the study: 1:500 (c-kit), 1:1,000 (β-A), 1:500 (CD31), 1:1,500 (α-SMA), and 1:20,000 (α-SA). All antibodies were diluted in 5% milk solution, incubated with PVDF membranes overnight on a plate rocker at 4°C, and then washed three times (5 min each) with TBST solution. The membranes were then incubated with a mouse or rabbit secondary antibody conjugated with HRP at 1:10,000 for 2 h at RT on a plate shaker. Films were washed and treated with luminol and images were taken immediately on a Kodak Image Station 400MM.
Immunocytochemistry
Cells were plated at a density of 2.5 × 104 cells per well on either the Blank coverslips or KOS hydrogel-coated coverslips in a 12-well plate and cultured in hCSC or DM as described above. Four specific primary antibodies (c-kit, α-SA, α-SMA, and CD31) (Supplementary Table S2) were used to identify stem cells, CMs, ECs, and VSMCs, respectively.
Briefly, cells were washed once with PBS, fixed in 4% PFA at RT for 1 h, and then blocked in 1.5% BSA and 0.2% Tween20 buffer for 30 min at RT. The resulting cells were then incubated with 1:200 c-kit, 1:500 CD31, 1:1,000 α-SMA, and 1:2,000 α-SA primary antibody for 2 h at RT on a shaker, washed three times with PBS (5 min each), and stained with a secondary antibody, either goat anti-rabbit Alexa Fluor 488 or goat anti-mouse Alexa Fluor 488, at a concentration of 1:2,000 in blocking buffer. An additional set of coverslips were stained with only a secondary antibody as a negative control, and Hoechst 33342 was used in all groups to label cell nuclei.
To avoid the autofluorescent interference from the KOS hydrogel, cells on the KOS hydrogel were first detached and then cytospun on a glass tissue slide following the antibody staining mentioned above. Fluorescent images of the Blank coverslips (attached cells) and KOS hydrogel (cytospun cells) were taken using the Olympus IX73 microscope mentioned above. The percentage of positive cells for each cell type was counted and calculated against the total number of nuclei and the results were plotted as a bar graph for statistical analysis.
Statistical analysis
All experiments were repeated at least three times with more than three replicates per group. All data are shown as mean ± standard error unless otherwise stated. Student's t-test with a two-tailed distribution was used to compare two groups. One-way analysis of variance (ANOVA) followed by the Bonferroni test was used to compare three or more groups with P < 0.05 being considered statistically significant. GraphPad Prism 5 and Microsoft Excel 2013 were used for statistical analysis and plotting.
Results
KOS hydrogel maintains cell viability and stemness but reduces the cell proliferation rate of c-kit+ hCSCs
To determine whether KOS hydrogel can be used as a delivery vehicle for future stem cell-based therapy, we need to first examine the impact of KOS hydrogel on cell viability and proliferation in the following in vitro studies. Briefly, after being enzymatically detached from the Blank or KOS hydrogel-coated dishes, cells were costained with Calcein-AM and EthD-1 and then transferred into 96-well plates for analysis using fluorescent microscopy and a plate reader. The results indicate that most cells are viable after 5 days of culture (Fig. 1A, green) and only a small number of them appear dead (Fig. 1A, red). Quantitative analysis shows no statistically significant differences between the Blank (93.3% ± 0.7%, 96.4% ± 2.3%, and 94.3% ± 2.4%) and KOS groups (94.4% ± 1.2%, 93.2% ± 2.2%, and 95.4% ± 2.0%) at day 1, 3, and 5, respectively (n = 4–5, ns, Fig. 1B). Thus, KOS hydrogel is able to culture and maintain hCSCs well without toxic effects.
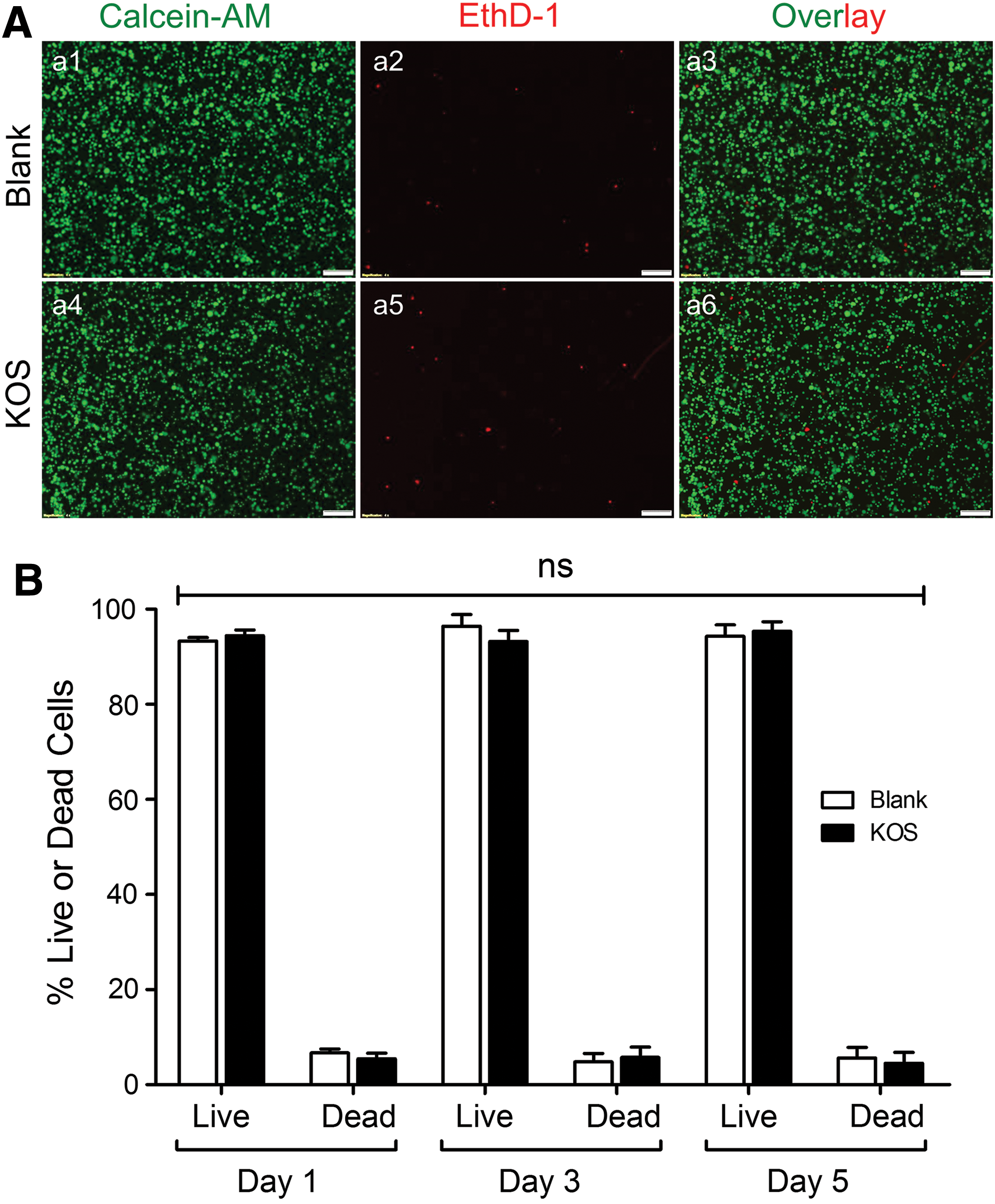
Cell viability assays using Calcein-AM and EthD-1 costaining. Cells were enzymatically collected from Blank control dishes or KOS hydrogel-coated dishes and costained with Calcein-AM and EthD-1. The stained cells were then transferred into 96-well plates for fluorescent microscopy imaging and only cells on day 5 were displayed
To determine the “long-term” effect of KOS hydrogel on cell viability, stemness (c-kit expression), and proliferation, a serial passage on KOS hydrogel was performed (Fig. 2 and Supplementary Fig. S1). Cells were cultured on the Blank control and the KOS hydrogel-coated dishes for 3–5 days to reach 80%–90% confluency before being split for the next passage on a new Blank or KOS hydrogel-coated dish. Contrast microscopy images indicate that there are no significant differences in overall cellular morphology among P1, P2, and P3 (Fig. 2A) in both the Blank and KOS culture conditions, although the imaged cells cultured on KOS hydrogel appear to be slightly more “narrow” or “bright” (which may be due to the light reflection of the KOS hydrogel). Quantitative analysis using Trypan blue staining demonstrated that the ratio of the number of cells recovered to the number of cells plated is comparable in both the Blank control (3.31 ± 0.15 in P1, 3.36 ± 0.25 in P2, and 3.21 ± 0.19 in P3) and in the KOS hydrogel culture (1.77 ± 0.14 in P1, 1.96 ± 0.15 in P2, and 1.87 ± 0.16 in P3); however, the total number of cells in the Blank control condition is significantly higher than that of the KOS hydrogel group across all passages (P1, P2, and P3) (Fig. 2B, n = 5, P < 0.001).

Effects of subpassaging hCSCs on KOS hydrogel on overall cell culture. hCSCs were cultured and passed up to one, two, and three passages (P1, P2, and P3) on KOS hydrogel and on Blank control (60-mm dishes). Representative phase-contrast images of cultures taken with a microscope on P1, P2, and P3
Meanwhile, c-kit immunostaining demonstrated that KOS hydrogel was able to maintain the stemness of hCSCs well during the three-passage period within 13–15 days at a high comparable expression level of c-kit on KOS hydrogel (90.2% ± 5.8%) versus Blank control (95.0% ± 2.9%) (Supplementary Fig. S1, n = 3, ns = no significance). These results imply that cell viability and stemness are well maintained during “long-term” culture, but the proliferation rate seems lower in KOS hydrogel than in Blank culture dishes.
To further confirm the above observation, we performed an EdU assay to quantitatively analyze cell proliferation. Briefly, for the Blank and KOS hydrogel conditions, cells were cultured on coverslips for up to 5 days. EdU staining and Hoechst nucleus costaining were performed on day 1, 3, and 5. The stained coverslips were flipped over and mounted on tissue slides and microscopy fluorescent images were taken from the bottom of the slides (to avoid and reduce autofluorescence of the KOS hydrogel) using the Olympus IX73 microscope mentioned above. As indicated from Fig. 3, there indeed appears to be significantly more EdU+ cells in the Blank (a1–a3) compared to the KOS hydrogel (a4–a6) across each time point examined (Fig. 3A). Mean percentages of EdU+ cells in the Blank (49.7% ± 2.2%, 44.7% ± 3.0%, and 46.7% ± 2.3%) are significantly higher than those in the KOS hydrogel (4.7% ± 1.0%, 20.6 ± 2.0%, and 15.1% ± 1.4%) at day 1, 3, and 5, respectively (Fig. 3B, n = 6, P < 0.001 in all groups). These findings are consistent with the relatively lower number of cells observed in Fig. 2B.
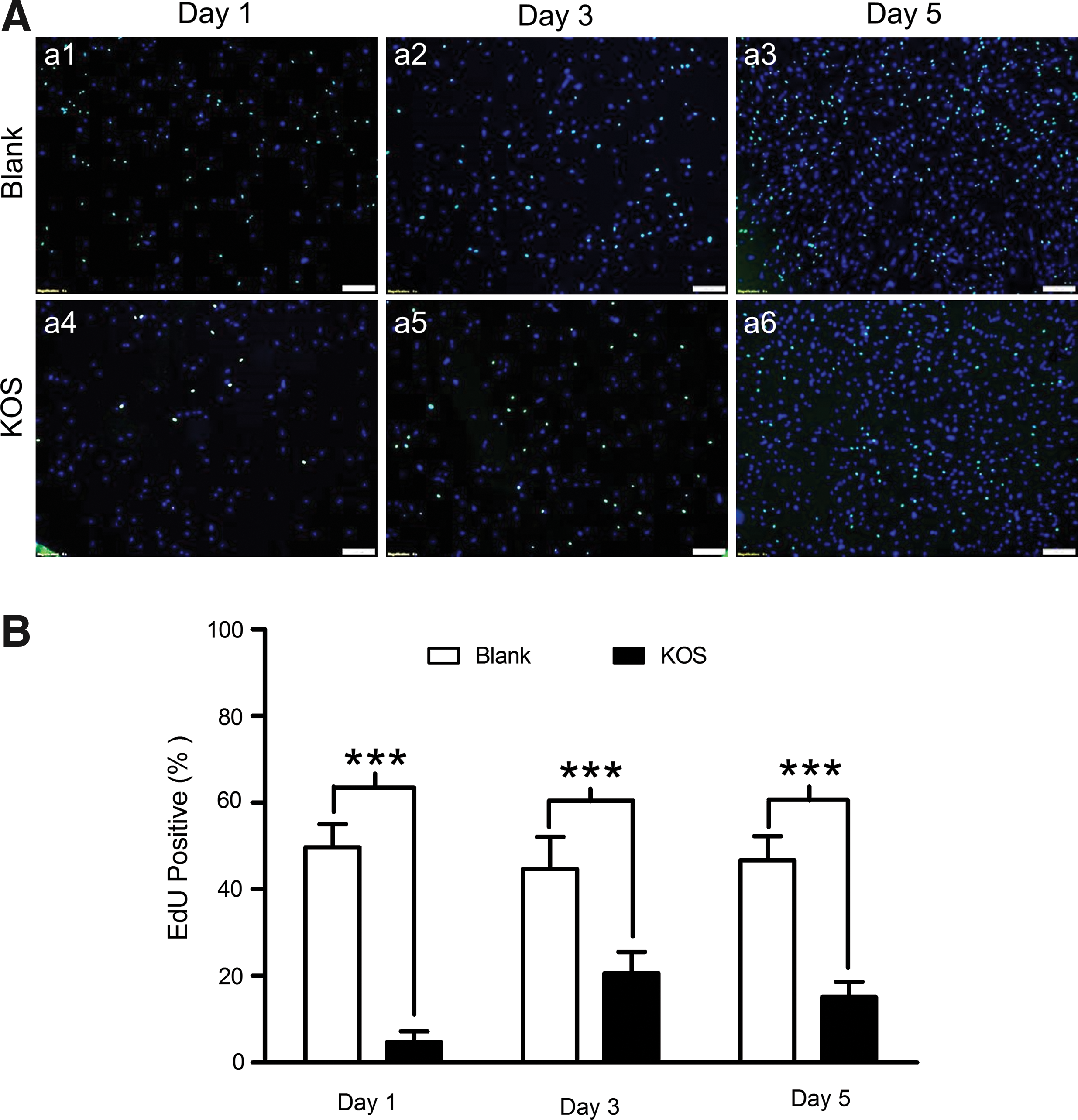
Cell proliferation assay with EdU staining. Cells were cultured on KOS hydrogel and Blank control dishes for up to 5 days. After EdU staining and Hoechst 33342 nucleus costaining, the coverslips were flipped over and mounted on microscopy slides followed by fluorescent imaging with fluorescent microscopy on days 1, 3, and 5 after cell seeding. EdU-positive nuclei in green refer to proliferative cells, while Hoechst-positive cells represent all nuclei
hCSCs do not spontaneously migrate into KOS hydrogel, but form “endothelial cell tube-like” microstructures following induced differentiation
A previous study indicates that porous biomaterials enable cells to migrate into the material [20]. To examine whether a similar migration occurs in KOS hydrogel, we seeded hCSCs on the KOS hydrogel and monitored cell location from the top view (Fig. 4A, a1–a4) of a culture dish and the side view (Fig. 4A, a5–a8) of a 30 μm cryosection of the KOS hydrogel following phalloidin/Hoechst 33342 staining. Interestingly, all cells remained on the surface of the KOS hydrogel during the course of the 5-day culture as indicated in the side view (Fig. 4A, a5–a7), although cell areas appear to become smaller and the ratio of the cell length/cell area is larger on the KOS hydrogel than on the Blank (Fig. 4B, n = 3, P < 0.01 or P < 0.001, image data not shown). To our surprise, hCSCs on the KOS hydrogel formed “endothelial cell tube-like” microstructures, similar to the “endothelial cell tube” typically formed by ECs on Matrigel-coated plates (ie, in vitro angiogenesis assay), on days 14–28 after induced differentiation (Fig. 4A, a4, a10), while cells on Blank culture dishes just appear over confluent (Fig. 4A, a9). This finding suggests that KOS hydrogel may promote angiogenesis of VSMCs differentiated from c-kit+ hCSCs.

Cell distribution and morphology on KOS hydrogel. Phalloidin
Predominant c-kit+ hCSCs become VSMCs after induced cardiac differentiation on KOS hydrogel
To investigate whether hCSCs tend to become VSMCs on KOS hydrogel compared to the Blank control, hCSCs were cultured and differentiated as described above, followed by qPCR, western blot, and immunocytochemical assays using specific primers and antibodies.
The gene expression analysis using qPCR analysis found that cardiac (NK2 homeobox 5–Nkx2.5 and cardiac troponin I), endothelial [von Willebrand factor (vWF) and vascular endothelial-cadherin (VE-cad), ie, CD144], and vascular smooth muscle [smooth muscle-myosin heavy chain (SM-MHC) & GATA binding protein 6- (GATA6)] markers showed no statistically significant differences between the Blank control and the KOS hydrogel under the basal condition (Pre-Diff, Fig. 5). However, these genes are dramatically upregulated 28 days after 5-Aza-induced differentiation both on the Blank control and the KOS hydrogel compared to the basal levels. Interestingly, the fold changes of cardiac genes were not different between the Blank and the KOS hydrogel (Fig. 5A), but endothelial markers showed statistical significance between the two culture conditions. The fold increases of vWF on the Blank control (62.3 ± 12.9) are higher than those on the KOS hydrogel (12.0 ± 2.5) (Fig. 5B, n = 4, P < 0.05) and the fold increase of VE-cad on the Blank control (7.2 ± 1.8) is lower than that on the KOS hydrogel (11.8 ± 2.4) (Fig. 5B, n = 4, P < 0.05). To our surprise, two vascular smooth muscle genes (SM-MHC and GATA6) show a robust increase with 19,517 ± 5,807-fold changes of SM-MHC and 1,226 ± 390-fold changes of GATA6 on the KOS hydrogel compared to 355 ± 80.4- and 33.4 ± 10.9-fold changes of the same genes on the Blank control (Fig. 5C, n = 4, P < 0.01 and P < 0.05), indicating that KOS hydrogel likely promotes hCSCs toward vascular smooth muscle differentiation.
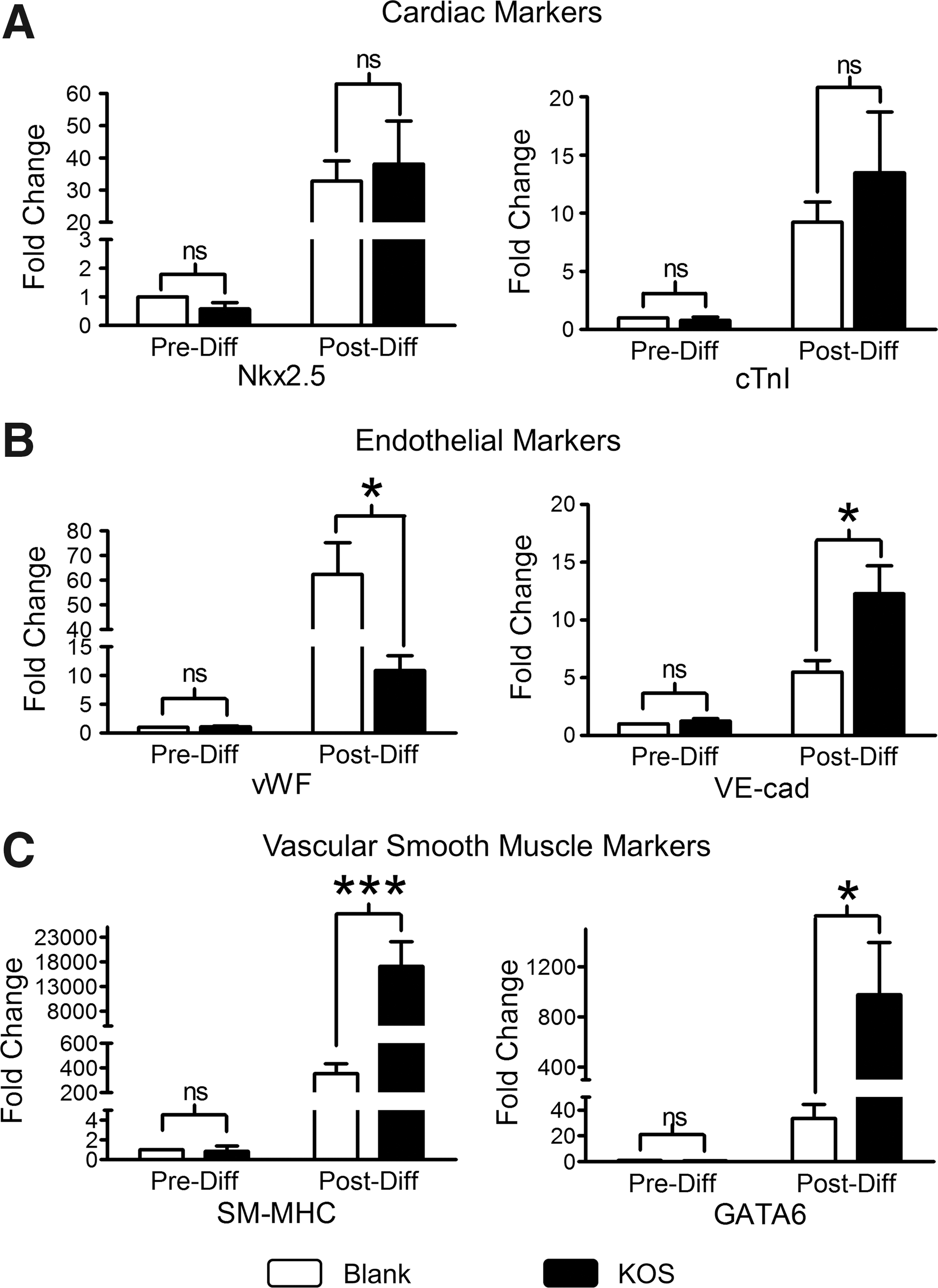
qPCR analysis of cardiac lineage markers before (Pre-Diff) and 28 days after (Post-Diff) differentiation. hCSCs were cultured on Blank or KOS hydrogel for 3–5 days followed by cardiac differentiation induced with 10 μM 5-Aza for 3 days. The differentiated cells were maintained in DM without 5-Aza up to 28 days before extraction of total RNA for qPCR analysis. Expression of cardiac (Nkx2.5 and cTnI), endothelial (vWF and VE-cad), and vascular smooth muscle (SM-MHC and GATA6) markers was plotted as fold changes (compared to Pre-Diff on Blank) and shown in Panel
Next, we examined the protein levels of stem cell and cardiac lineage markers using immunostaining with specific antibodies. Western blot results (Fig. 6) appear to agree well with those in qPCR (Fig. 5). Figure 6A shows that the stem cell marker (c-kit) is strongly expressed before differentiation (Pre-Diff), but hard to detect after differentiation (Post-Diff). Interestingly, α-SA was greatly expressed on the Blank control in contrast to the strong expressions of CD31 and α-SA on the KOS hydrogel (Fig. 6A). Quantitative analysis of these bands shows 11.54 ± 2.25-fold changes of α-SA on the Blank control and much less on the KOS hydrogel (2.99 ± 0.45) (Fig. 6B, n = 4–5, P < 0.001). In addition, there were 3.03 ± 0.37- and 3.51 ± 0.91-fold changes of CD31 and α-SMA on the Blank control compared to 6.01 ± 0.75- and 11.45 ± 1.93-fold changes on the KOS hydrogel (Fig. 6B, n = 4–5, P < 0.01, and P < 0.001), which confirms our expectation that the KOS hydrogel would enhance vascular smooth muscle differentiation of c-kit+ hCSCs.
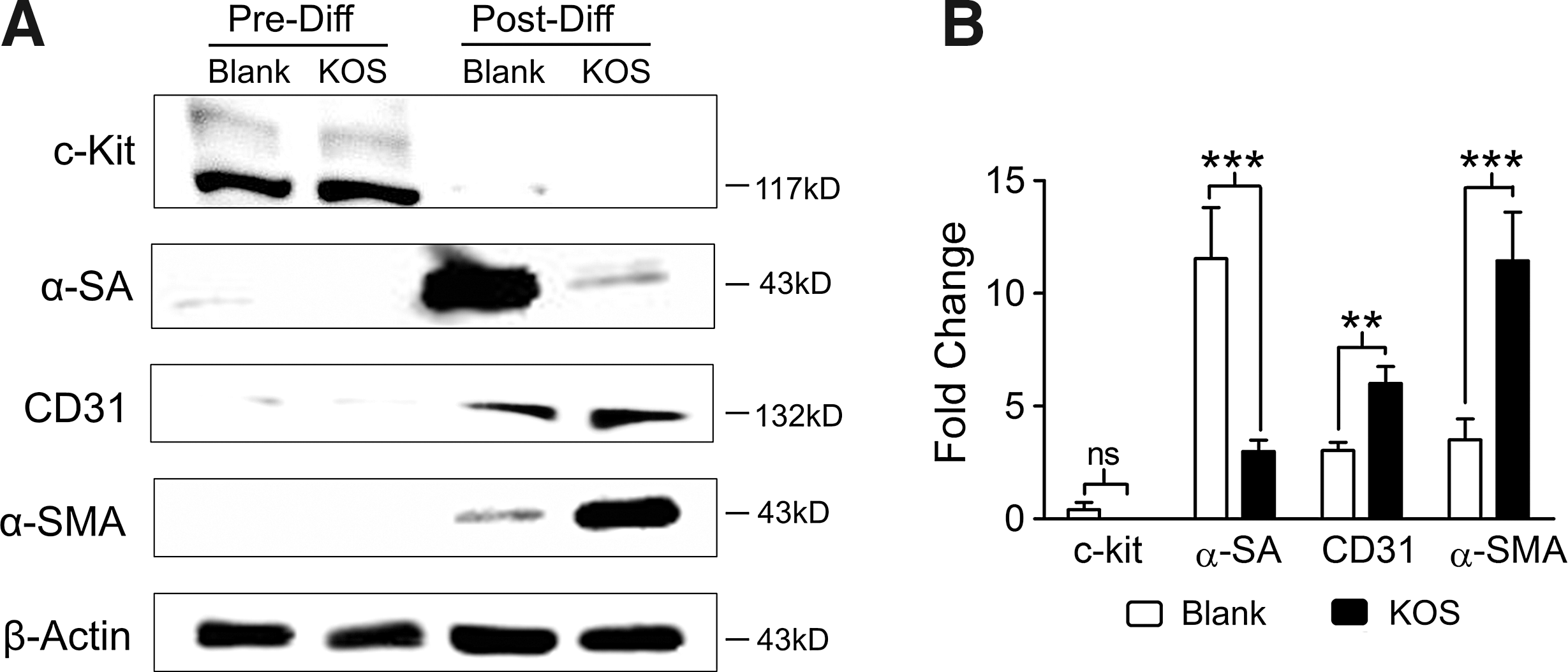
Western blot analysis of protein expression before (Pre-Diff) and after (Post-Diff) cardiac differentiation. hCSCs were cultured on Blank or KOS hydrogel for 3–5 days followed by cardiac differentiation induced with 10 μM 5-Aza for 3 days. The differentiated cells were maintained in DM without 5-Aza up to 28 days before protein extraction for western blot analysis. Protein expression of stem cell (c-kit, ie, CD117), cardiac (α-SA), endothelial (CD31), and vascular smooth muscle markers (α-SMA) was determined with specific antibodies before (Pre-Diff) and 28 days after (Post-Diff) differentiation
Finally, we used immunocytochemistry to further confirm our above findings. Due to the interference of autofluorescence from the KOS hydrogel under fluorescent microscopy, all cells on the KOS hydrogel were first detached, stained with the specific antibody, and then cytospun onto tissue slides before imaging, while cells on the Blank control coverslips were kept for staining and imaging under their “natural” morphology. As shown in Fig. 7, before differentiation, almost all hCSCs are c-kit+ on both the Blank control (95.0% ± 2.94%) and the KOS hydrogel (93.9% ± 3.25%), while the other three cardiac lineage markers (α-SA, CD31, and α-SMA) are hardly detected (α-SA: 0.5% ± 0.43% on Blank vs. 0.48% ± 0.42% on KOS; CD31: 0.38% ± 0.34% on Blank vs. CD31 0.33% ± 0.23% on KOS; α-SMA: 0.3% ± 0.23% on Blank vs. 1.27% ± 0.34% on KOS, n = 5, all are ns); however, after 28 days of differentiation, c-kit+ cells are observed on neither the Blank control (1.58% ± 0.28%) nor the KOS hydrogel (1.29% ± 0.59%) groups, and instead, the majority of c-kit+ cells became cells of cardiac lineages. Mean data show 76.85% ± 7.12% α-SA+, 6.26% ± 0.85% CD31+, and 12.45% ± 4.37% α-SMA+ cells on the Blank control dishes compared to 10.23% ± 1.1% α-SA+, 13.18% ± 2.9% CD31+, and 71.58% ± 5.25% α-SMA+ cells on the KOS hydrogel-coated dishes (Fig. 7B, n = 5, P < 0.001, P < 0.05, P < 0.001, respectively), suggesting that the Blank culture condition tends to drive hCSCs toward CM differentiation, while the KOS hydrogel mainly drives them to become VSMCs.
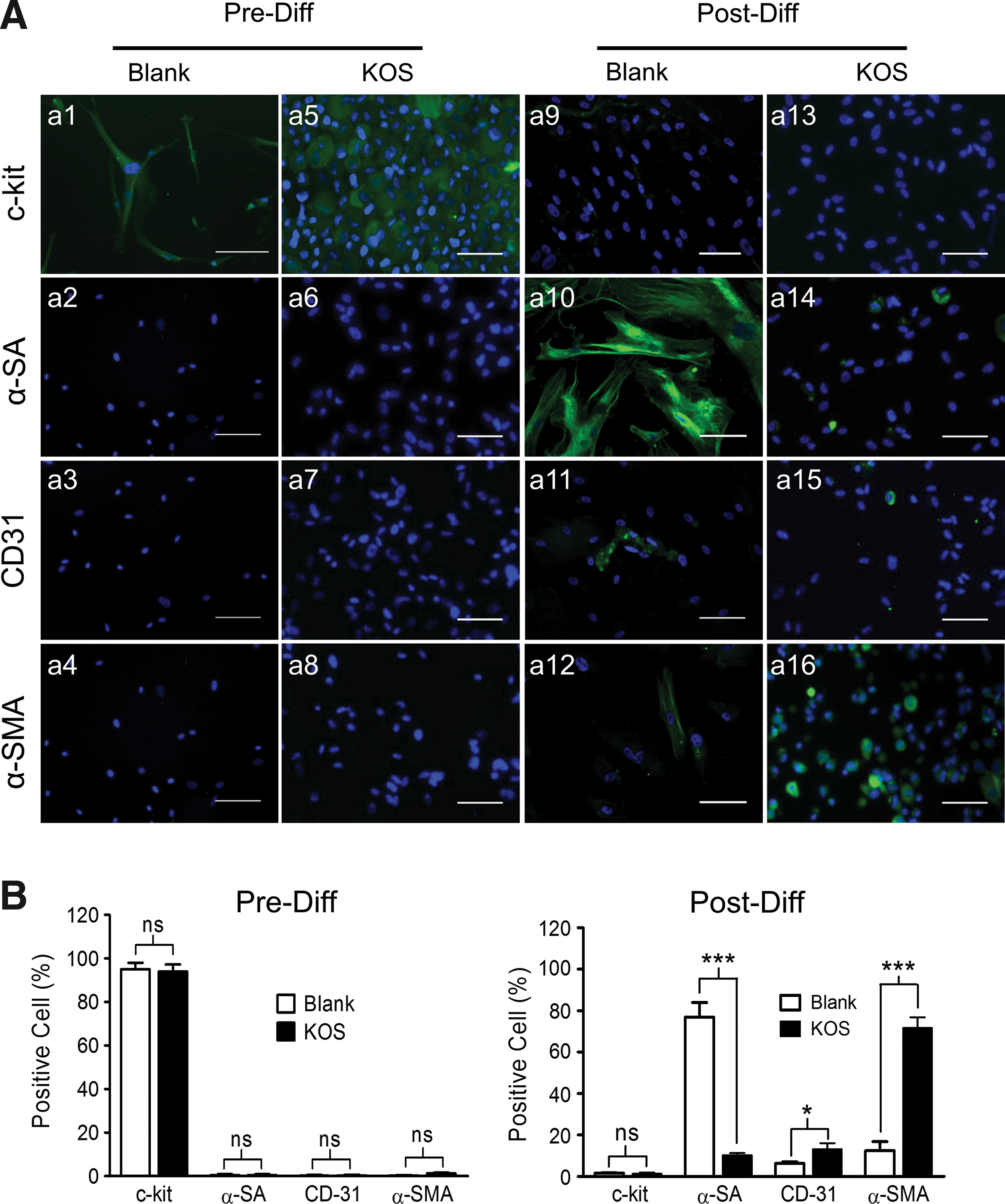
Immunocytochemical staining before (Pre-Diff) and after (Post-Diff) differentiation of hCSCs on Blank and KOS hydrogel. hCSCs were cultured on Blank or KOS hydrogel for 3–5 days followed by cardiac differentiation induced with 10 μM 5-Aza for 3 days. The differentiated cells were maintained in DM without 5-Aza up to 28 days and followed by immune staining with specific antibodies. Protein expression of stem cell (c-kit), cardiac (α-SA), endothelial (CD31), and vascular smooth muscle markers (α-SMA) was determined with specific primary antibodies before (Pre-Diff) and 28 days after (Post-Diff) differentiation and all secondary antibodies were conjugated with Alexa Fluor 488. Representative fluorescent images were taken from Blank dishes with attached cells on coverslips, while cells from KOS hydrogel were first detached and then stained before being cytospun on tissue slides for microscopy imaging
To determine whether 5-Aza is necessary for KOS hydrogel-induced VSMC differentiation, we repeated the above differentiation experiments using the DM with or without 5-Aza. The results demonstrated that hCSCs did spontaneously differentiate into VSMCs on KOS hydrogel using the DM without 5-Aza (Supplementary Fig. S2). Unexpectedly, the percentage (71.6% ± 2.23%) of VSMCs in the presence of 5-Aza is almost the same as in the absence of 5-Aza (70.5% ± 3.3%, n = 4–5, no significant difference) when differentiated on KOS hydrogel. This indicates that KOS hydrogel alone is able to differentiate hCSCs into VSMCs without 5-Aza, and the addition of 5-Aza seemed not to generate synchronic effects. In addition and as mentioned above, although “long-term” hCSCs cultured on KOS hydrogel maintained a high expression level of c-kit stem cell maker (similar to those in Blank control dish, Supplementary Fig. S1), a good portion (15.3% ± 2.7%) of hCSCs still spontaneously differentiated into VSMCs in hCSC medium, which suggests a strong biological regulatory effect of the KOS hydrogel on hCSC differentiation.
Discussion
In the present study, we investigated the effects of KOS hydrogel on cell viability, stemness, proliferation, cellular morphology, and cardiac lineage differentiation of c-kit+ hCSCs in comparison with cells cultured on regular culture dishes (Blank control). The results show that both the KOS hydrogel and the regular culture dishes are able to culture and maintain hCSCs well without any observable toxic effects nor reduction of stemness caused by the KOS hydrogel (Figs. 1, 2, and Supplementary Fig. S1); however, the cell size, ratio of cell length/size, and cell proliferation rate appear smaller on KOS hydrogel than for those on the Blank control dishes (Figs. 3 and 4). To our surprise, KOS hydrogel significantly promoted hCSCs becoming VMSCs Post-Diff (with or without 5-Aza), in contrast to those cultured on regular culture dishes, in which case most hCSCs differentiated into CMs (Figs. 5 –7) [4,8]. Furthermore, differentiated VSMCs appeared to form “endothelial cell tube-like” microstructures (Fig. 4), similar to the “endothelial cell tube” typically formed by ECs on Matrigel (ie, in tube formation or in vitro angiogenesis assay) [21].
To the best of our knowledge, this is the first report to discover the preferred differentiation of hCSCs toward VSMCs on KOS hydrogel. The innovative methodology by which cells can be recovered from KOS hydrogel through elastase dissociation makes it possible to analyze gene and protein expression without interference of keratin molecules. Our findings may provide a new and simpler approach to generate a large number of VSMCs for cell-based therapy in cardiovascular diseases.
A number of natural biomaterials, such as collagen, Matrigel, and alginate, have demonstrated the ability to promote and maintain cell viability [12,22 –25]. Keratins are naturally derived proteins that can be fabricated into several biomaterial forms, including KOS hydrogels (oxidatively derived) [26]. These materials are a potential polymeric system for several tissue engineering and regenerative medicine applications due to their ability to support cell attachment, proliferation, and migration [19].
Our results show that KOS hydrogels are capable of maintaining high cell viability and stemness after serial passaging without any observable toxic effects, which is consistent with previous studies that used either the same material or different types of stem cells [10,27,28]. Interestingly, the cell proliferation rate is reduced on KOS hydrogel compared to the Blank control dishes.
It is commonly agreed that the physical environment and/or biochemical interaction between the biomaterial and stem cells are critical mediators of cell behavior [29,30]. Cell proliferation and differentiation appear to be inversely proportional to substrate stiffness [31]. Thus, the lower stiffness of KOS hydrogel substrates and/or the potential direct interaction of keratin molecules with β-integrin or Notch1 signaling in hCSCs may play an important role in modulating hCSC proliferation and differentiation [30 –32]. Perhaps a low proliferation rate observed in the present study may reflect the physiological rate of stem cell proliferation to meet the basic needs of homeostasis under normal conditions.
An additional interesting finding in the present study is that both hCSCs (c-kit+) and hCSC-differentiated cells (CMs, ECs, and VMSCs) are unable to migrate into the KOS hydrogel during “long-term” culture and differentiation, showing “immobile” properties. Our findings appear to be different from other published studies, in which the cells (aorta smooth muscle cells and fibroblasts) are able to migrate into the porous hydrogel of various biomaterials [33,34]. The reason remains unknown, but is probably related to the pore size of the hydrogel substrate or cell type [35]. Although the pore size of our KOS hydrogel seems larger than the cells in the middle portion under the side view (Fig. 4A), the surface of the KOS hydrogel seems to form a continuous layer, which may have prevented the cells from migrating into the hydrogel. In addition, the interactions between the cells and the cell/keratin molecules may also play a role in this phenomenon [36,37].
Tube formation (also known as “in vitro angiogenesis”) is a typical in vitro Matrigel assay to evaluate the angiogenic properties of ECs or endothelial progenitor cells [38]. A surprising observation of the present study was the formation of “endothelial cell tube-like” microstructures from hCSC-differentiated VSMCs on KOS hydrogel, but not on Blank control dishes. Whether or not this “tube-formation” property of VSMCs facilitates in vivo arteriogenesis or collaterogenesis needs to be further investigated using an animal model. The underlying mechanism of the “endothelial cell-like tube” formation remains unknown, although it may be due to the in vitro angiogenesis property of the mixed small percent (∼13%) of ECs.
Numerous publications have previously demonstrated that c-kit+ hCSCs can be differentiated into three cardiac lineage cell types (ie, CMs, ECs, and VSMCs) in both in vitro and in vivo settings [4,8,39 –41]; however, whether hCSCs are able to differentiate into CMs after transplantation is still under debate [6]. In the present in vitro study, we discovered for the first time that the majority (∼72%) of hCSCs on KOS hydrogel become VSMCs after induced differentiation (independent from 5-Aza), while for the Blank control, the majority (∼77%) of cells become CMs. The KOS hydrogel seems to switch the myogenic differentiation pattern from CM on Blank to VSMCs on KOS hydrogel, whether or not 5-Aza was added in the present study. This important finding may indicate a critical role of KOS hydrogel in modulating hCSCs toward vascular smooth muscle differentiation. However, the mechanism underlying the preferred VSMC differentiation on KOS hydrogel remains to be determined.
A good number of published studies implicated that TGF-β signaling might be a cause of VSMC differentiation from various types of stem cells, such as embryonic stem cells, bone marrow mesenchymal stem cells, adipose stem cells, and multipotent adult progenitor cells [42 –45]. It has been shown that keratin is able to enhance angiogenesis or cell differentiation through upregulation of TGF-β signaling pathways [17,26,32,46]; while 5-Aza, as a demethylating epigenetic reagent, is known to be capable of promoting cardiac differentiation from various types of stem cells [47 –50]. Certainly, 5-Aza plays a critical role in the differentiation of hCSCs on Blank control, but it is not necessary for the VSMC differentiation observed on KOS hydrogel (Supplementary Fig. S2).
In the experiments where we simply induced differentiation by removing growth factors (b-FGF and EPO) from hCSCs cultured on KOS hydrogels, we discovered nearly the same percentage of VSMCs (∼71%) with or without the addition of 5-Aza. Moreover, the present study used FBS in DM, and FBS is known to contain various growth factors, including TGF-β; thus, we could not exclude the potential role of FBS-TGF-β on KOS-promoted VSMC differentiation [46]. Interestingly, platelet-derived growth factor (PDGF) and integrin signaling pathway were also found in mediating smooth muscle differentiation from pluripotent stem cells, scal-1+ stem cells, and hair follicle stem cells, probably through a mechanotransduction signaling pathway [51 –53]. Thus, the complex interactions among keratin molecules and TGF-β, PDGF, and the integrin pathway may play a critical role in the preferred VSMC differentiation [54 –56].
In conclusion, our present study demonstrates that KOS hydrogels are able to well maintain cell viability and stemness, produce “endothelial cell tube-like” microstructures (similar to the “endothelial cell tube”), and significantly promote vascular phenotypes, especially vascular smooth muscle differentiation (independent from 5-Aza), from c-kit+ hCSCs. These findings offer new methods, which are not only useful for generating a robust number of VSMCs from hCSCs by simply differentiating hCSCs on KOS hydrogel but also for providing a practical therapeutic platform with an injectable stem cell-based KOS hydrogel in treating cardiovascular diseases.
However, the present study is limited by a lack of mechanistic data. The complex biological interaction between cellular receptors and KOS hydrogel is a critical area to be further explored in understanding the cellular and molecular signaling pathways underlying the KOS-preferred VSMC differentiation. Future studies should examine the individual effects of KOS hydrogel, TGF-β, PDGF, and the integrin pathway in a defined medium (eg, KnockOut™ serum replacement to replace FBS) in the presence and absence of pathway-specific inhibitors. Studies showing a coherent and coordinated stage-specific sequence of gene and protein expression should also be performed. The transplantation of hCSC-differentiated VSMCs into ischemic animal models (eg, myocardial infarction or hindlimb ischemia) would be an important future study to determine the therapeutic roles of these cells in repairing damaged organs.
Footnotes
Acknowledgments
This work was supported by Jia-Qiang He's start-up fund and grants from the Institute for Critical Technology and Applied Science (ICTAS) at Virginia Tech (no. JFC2014_JIAHE958451025) and the Virginia's Commonwealth Health Research Board (CHRB) grant award (HE no. 208-09-16). Funding was also provided by Mark Van Dyke's start-up and departmental funds. The funders had no role in the study design, data collection and analysis, decision to publish, nor in preparation of the manuscript.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.