
Abstract
Spermatogonial stem cells (SSCs) provide the foundation for spermatogenesis and fertility throughout the adult life of a male. Genetic manipulations of SSCs combined with germ cell transplantation present a novel approach for gene therapy and production of genetically modified animals. However, the rarity of SSCs within mammalian testes remains an impediment to related applications, making in vitro expansion of SSCs a prerequisite. Nevertheless, long-term culture systems of SSCs from large animals have not been established yet. In this study, we developed an optimized in vitro culture condition for porcine undifferentiated spermatogonia. The germ cells were isolated and enriched from 7-day-old porcine testes by an optimized differential planting. We tested different feeder layers and found that neonatal autologous Sertoli cells acted better than the SIM mouse embryo-derived thioguanine- and ouabain-resistant (STO) cell line and adult Sertoli cells. The effects of several growth factors were also investigated. Using neonatal Sertoli cells as feeder and Dulbecco's modified eagle medium: nutrient mixture F-12 (DMEM/F12) culture medium supplemented with 10% KSR and four cytokines, the undifferentiated spermatogonia can proliferate in vitro for at least 2 months without loss of stemness. The expression of SSC markers indicated that the cultured cells maintained SSC expression profiles. Moreover, xenotransplantation and in vitro induction showed that the long-term cultured cells preserved the capacity to colonize in vivo and differentiate in vitro, respectively, demonstrating the presence of SSCs in the cultured cells. In conclusion, the conditions described in this study can support the normal proliferation of porcine undifferentiated spermatogonia with stemness and normal karyotype for at least 2 months. This culture system will serve as a basic refinement in the future studies and facilitate studies on SSC biology and genetic manipulation of male germ cells.
Introduction
G
SSCs are unique adult stem cells that transmit genetic information to subsequent generations. Moreover, transplantation of SSCs into recipient testes yields donor SSC-derived spermatogenesis [6 –10]. Thus, genetic manipulations of SSCs combined with germ cell transplantation present a novel approach for gene therapy and production of genetically modified animals [11]. In addition, SSCs provide an ideal model to elucidate the mechanisms for stem cell self-renewal and differentiation [3]. The ratio of SSCs has been estimated as 1/3,500 in the testes of adult mice [12], and thus the rarity of SSCs within mammalian testes remains an impediment to transplantation and other related applications that require sufficient numbers of SSCs, making in vitro expansion of SSCs a prerequisite [13].
Glial cell line-derived neurotrophic factor (GDNF) is the initially identified growth factor required for SSC self-renewal [14,15]. Based on it, Kanatsu-Shinohara et al. [16] and Brinster and colleagues [17] have established the long-term culture systems for murine SSCs. In addition, several growth factors, including bFGF [17], IGF1 [18], and CSF1 [19], are also beneficial to proliferation of male germline stem cells in vitro. At present, effective culture condition of SSCs have been established in mice [16,17,20 –22], hamsters [23], rats [24], rabbits [25], and recently in tree shrew [26].
Nevertheless, stable long-term culture systems of SSCs from large animals, including boars, have yet been developed. It seems that SSCs from large domestic animals hold some unique characteristics since the established culture systems developed for rodent SSCs could not support the long-term proliferation of SSCs from livestock species [13,27,28]. Zheng et al. found that EGF and bFGF have a positive effect on porcine gonocyte-derived colonies [13]. In addition, the number of gonocyte-derived colonies was significantly higher in the presence of 1% rather than 10% fetal bovine serum (FBS) [13,29]. Furthermore, the effect of culture temperature was also investigated, and the conclusion was that low temperature is beneficial to the proliferation of SSCs [30]. Subsequently, the same group reported that GATA4-positive feeder cells could support the maintenance of porcine primitive male germ cells [31]. Despite the achievements, it is noticeable that in their studies, the starting cells were not enriched SSCs [13,30 –32]. Consequently, the efficient system for isolation and cultivation of porcine undifferentiated spermatogonia has not been established.
Therefore, the aim of this study was to optimize the conditions for propagation of porcine undifferentiated spermatogonia in vitro. Specifically, multiple feeder layers, serum-free supplement, and cytokine cocktails have been investigated systematically. With this optimized culture condition, porcine undifferentiated spermatogonia can proliferate for at least two months.
Materials and Methods
Animals
Testis samples were obtained from 4- to 7-day-old PIC piglets when they were castrated in Besun farm and transported in Dulbecco's phosphate-buffered saline (DPBS) to the laboratory within 2 h. All experimental procedures were approved by Northwest A&F University's Institutional Animal Care and Use Committee.
Digestion of testis tissue
The collected testes were washed thrice with DPBS, cut into pieces, and digested with collagenase type IV (2 mg/mL; Invitrogen) and DNase I (0.1 mg/mL; Sigma) in Dulbecco's modified eagle medium: nutrient mixture F-12 (DMEM/F12; Hyclone) for 20 min, at 37°C. The separate fragments of seminiferous tubules were collected by natural sedimentation for 5 min and washed with DPBS thrice to remove interstitial cells. Erythrocytes were eliminated using a lysis buffer (Sigma-Aldrich). The tubules were treated with 0.25% Trypsin-EDTA (Gibco) for 5 min, at 37°C. To obtain the single cell suspension, the dispersed testicular cells were filtered through a 40-μm mesh. The single cell suspension was pelleted by centrifugation at 300g for 5 min and resuspended in DMEM/F12 supplemented with 2% (v/v) FBS (Gibco).
Enrichment of undifferentiated spermatogonia
To enhance the purity of undifferentiated spermatogonia, we improved the system of differential plating. The cell suspension was plated into 10-cm plastic culture dishes with 2 × 107 cell/dish and cultured in a CO2 incubator at 37°C. After 6 h, weakly adhering cells were gently washed down from the dishes and transferred into a new dish, and then cultured in CO2 incubator at 35°C overnight. The next day, the germ stem cell population was harvested by repeatedly pipetting the added PBS over the surface area of the dish, while the somatic cell monolayer was still bound to the dish. The suspension containing undifferentiated spermatogonia was collected and utilized for further experiments.
Feeder layer preparation
SIM mouse embryo-derived thioguanine- and ouabain-resistant (STO) cells and porcine Sertoli cells from neonatal testes and adult testes were used as feeder cells. To prepare Sertoli cells, isolated testicular cells were plated into a dish, maintained in DMEM/F12 medium with 5% FBS, and incubated for 1 h in a CO2 incubator at 37°C. After discarding nonadherent cells, the adherent Sertoli cells were cultured for three to four passage. To prepare feeder cell monolayers, Sertoli cells at P3–P4 and STO cells were mitotically inactivated by treatment with mitomycin C (10 μg/mL) for 3 h followed by extensive washing in DPBS.
Cell culture
The enriched cells were seeded with 5 × 105 cell/well in six-well dishes on feeder layers (Sertoli cells or STO) in a CO2 incubator at 35°C under BM (basal medium): DMEM/F12 medium with 100 IU/mL penicillin, 100 mg/mL streptomycin, 1 ×
Based on the aforementioned observations, the freshly enriched cells were seeded with 5 × 105 cell/well on neonatal Sertoli cell feeder cells in six-well dishes in a CO2 incubator at 35°C under different culture conditions, including BM supplemented with 10 ng/mL GDNF, KSR+: BM supplemented with 10% serum-free supplement KSR (Gibco) and 10 ng/mL GDNF, GGb: BM supplemented with 10% KSR, 10 ng/mL GDNF, 20 ng/mL GFRA1, and 10 ng/mL bFGF, GGI: BM supplemented with 10% KSR, 10 ng/mL GDNF, 20 ng/mL GFRA1, and 20 ng/mL IGF1, GbI: BM supplemented with 10% KSR, 10 ng/mL GDNF, 10 ng/mL bFGF, and 20 ng/mL IGF, and bIE: BM supplemented with 10% KSR, 10 ng/mL bFGF, 20 ng/mL IGF1, and 10 ng/mL EGF. Colony formation was compared between the groups.
Based on the results, the BM supplemented with 10% KSR and four growth factors (GDNF, GFRA1, bFGF, and IGF1 at the aforementioned doses) was used for long-term culture of undifferentiated spermatogonia on juvenile Sertoli cell feeder layer. The cultured cells were passaged to new feeder layers every 5–7 days using 0.05% trypsin-EDTA (Gibco).
Immunocytofluorescense
The purity of the enriched undifferentiated spermatogonia was estimated by using anti-UCHL1 (1:100; Santa Cruz Biotechnology). The cultured cells were subjected to immunocytofluorescense to detect expression of MGSC markers, including goat anti-UCHL1 (1:100; Santa Cruz Biotechnology), goat anti-PLZF (1:100; Santa Cruz Biotechnology), and goat anti-GFRA1 (1:100; Santa Cruz Biotechnology). Cells were fixed with 4% paraformaldehyde for 20 min and washed with PBS. The samples were incubated with 3% BSA in PBS containing 0.1% Triton X-100 for 2 h at room temperature and incubated with one of the primary antibodies at 4°C overnight. The samples were washed thrice with PBS and incubated with donkey anti-goat (FITC/TR-conjugated) secondary antibody (1:400; Santa Cruz Biotechnology) for 1 h at 37°C. After washing thrice with PBS, the cells were counterstained with 4,6-diamidino-2-phenylindole (DAPI, 1:1,000; Beyotime) for 5 min, mounted with 50% glycerol, and viewed with Olympus IX71 (Olympus) inverted fluorescence microscope.
Quantificational real-time polymerase chain reaction
Total RNA was extracted by Trizol reagent (Invitrogen). Approximately, 1 μg of total RNA was reversely transcribed using the transcriptor first strand complementary DNA (cDNA) synthesis kit (Roche). FastStart Universal SYBR Green Master (Roche) was used for real-time quantitation of messenger RNA (mRNA) levels using an IQ5 (Bio-Rad Laboratories). Specific primers for polymerase chain reaction (PCR) amplification of the genes mentioned in this study are shown in Table 1. Data were analyzed using the comparative Ct-method, with GAPDH serving as reference genes.
Western blotting
Total protein was extracted from the freshly isolated undifferentiated spermatogonia and the undifferentiated spermatogonial colonies using the RIPA Kit (HAT) according to the manufacturer's instructions. Approximately, 50 μg of protein was separated by electrophoresis in 8%–12% sodium dodecyl sulfate–polyacrylamide gel and transferred to a polyvinylidene fluoride (PVDF) membrane (Bio-Rad Laboratories).
The membranes were blocked by incubation in 5% nonfat milk for 1 h followed by incubation overnight with primary antibodies as follows: PLZF (1:1,000 dilution; Santa Cruz Biotechnology), GFRA1 (1:1,000 dilution; Santa Cruz Biotechnology), THY-1 (1:1,000 dilution; Santa Cruz Biotechnology), UCHL1 (1:2,000 dilution; Santa Cruz Biotechnology), LIN28 (1:2,000 dilution; Abcam), and beta-Actin(1:5,000, Sigma). The membranes were washed in Tis-buffered saline with Tween 20 and incubated with donkey anti-goat IgG-HRP (1:2,000; CWBIO), donkey anti-rabbit IgG-HRP (1:2,000; CWBIO), or goat anti-mouse IgG-HRP (1:5,000; CWBIO). Signal was detected using Immobilon Western chemiluminescent HRP Substrate (Millipore).
Karyotype analysis
The cultured cells isolated from the feeder cells were plated in a 12-well dish coated with laminin (LN) and incubated overnight with BM containing 0.2 μg/mL cocamide. Cells were harvested as single cell suspensions and metaphase spreads were prepared as previously described [33]. The slides were stained with Giemsa (Sigma) or DAPI (Beyotime) and mounted with 50% glycerol. The number of chromosomes was calculated manually.
Xenotransplantation assay
Xenotransplantation assay was used to probe the biological characteristics of the cultured cells as previously described [13]. To devastate endogenous germ cells, the recipient mice were treated with alkylating agent busulfan (40 mg/kg) 4 weeks before transplantation as described by Ogawa et al. [34]. The cultured porcine cells were labeled with fluorescent dye PKH26 (PKH26 Red Fluorescent Membrane Linker; Sigma).
In brief, cells were washed by DPBS and centrifuged at 400g for 5 min. The cell pellets were resuspended with Diluent C to prepare a 2× cell suspension. The 2× cell suspension was rapidly added into an equal volume of 2× PKH26 dye solution (4 × 10−6 M) to get a final concentration of 2 × 10−6 M for PKH26 and 107 cells/mL. The staining was stopped by adding an equal volume of serum after 3 min of incubation. The labeled cells were washed by DPBS thrice to ensure removal of unbound dye. The cell pellets were resuspended in DMEM/F12 containing 0.04% trypan blue with a concentration of 107 cells/mL (cell viability ≥80%). Approximately, 10 μL of the labeled cell suspension (107 cells per mL) was injected into the seminiferous tubules of each recipient testis. The same amount of labeled Sertoli cells from 7-day-old piglets (at P3–P4) were injected as the negative control.
Approximately 8 weeks after transplantation, recipient mouse testes were collected. The tunica albuginea was removed and the seminiferous tubules were gently dissected and analyzed under a dissecting microscope. Donor germ cells were identified by the specific PKH26 red fluorescent dye. A cluster of germ cells was defined as a colony when a cluster either consists of more than 10 cells or is at least 50 μm in length.
In vitro induction by retinoic acid
For in vitro induction, the undifferentiated spermatogonial colonies were passaged on adult Sertoli cells with 10 nM all-trans retinoic acid (RA; Sigma-Aldrich) and 10% FBS in the culture medium. After the 24-h treatment, total RNA was extracted and analyzed by quantitative RT-PCR.
Statistical analysis
All experiments were repeated at least thrice. Multiple comparisons were analyzed by one-way analysis of variance (ANOVA) followed by the least significant difference (LSD) model of SPSS 17.0. All data were presented as mean ± standard error of the mean (SEM) and differences were considered significant at P < 0.05.
Results
Enrichment of undifferentiated spermatogonia
Differential plating was optimized to enrich undifferentiated spermatogonia. The overall purity of undifferentiated spermatogonia was assessed by staining the spermatogonial marker UCHL1 (Fig. 1B, C). The FACS analysis showed that the ratio of undifferentiated spermatogonia was improved significantly from 10.50% ± 3.13% to 72.66% ± 2.96% (P < 0.001). Next, the enriched cell population was utilized for further experiments.
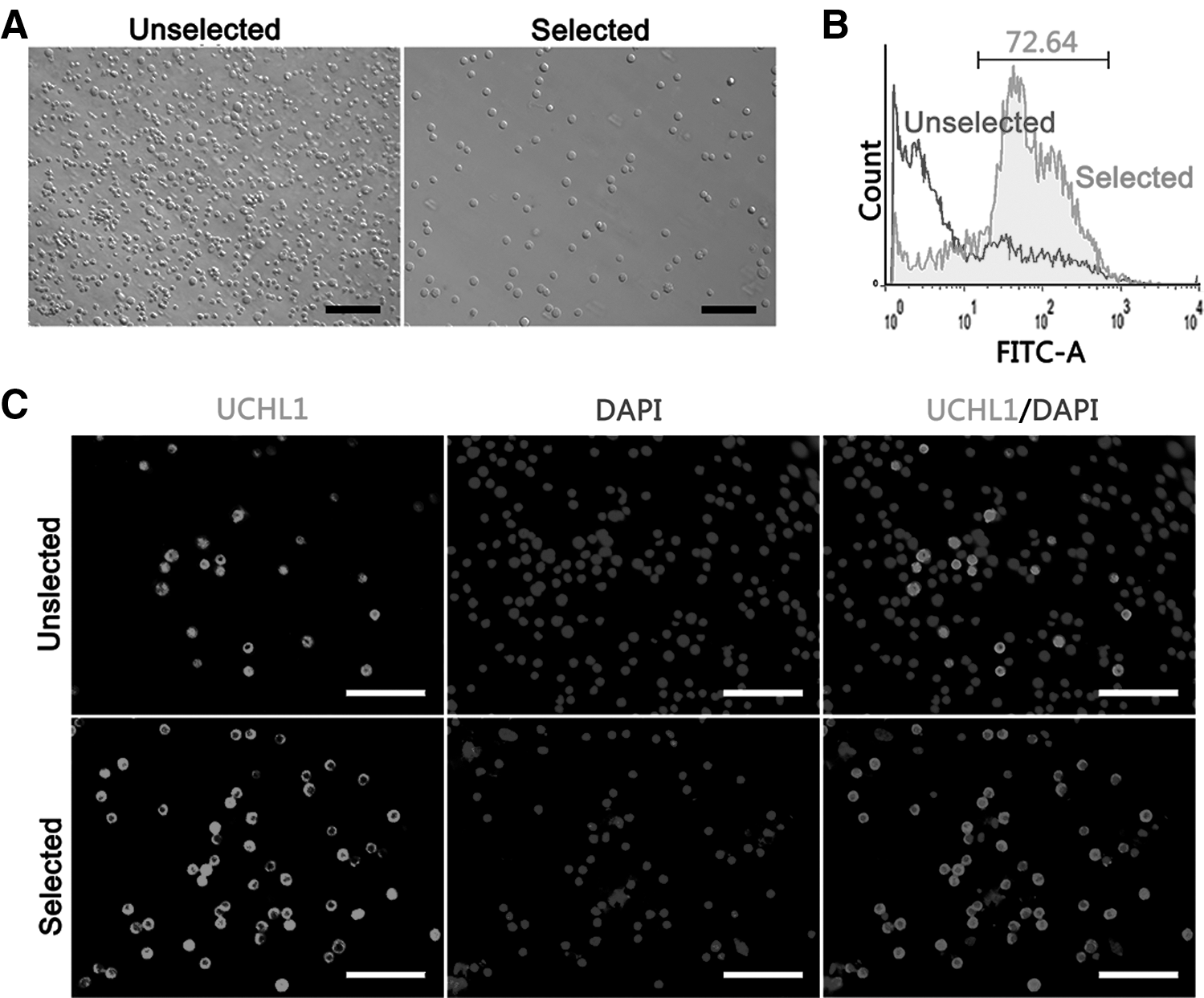
Assessment of enriched undifferentiated spermatogonia. To ascertain the efficiency of purified porcine undifferentiated spermatogonia, an unselected cell population and a selected cell population were examined, respectively.
Effects of serum-free supplement KSR on germ cell culture
To determine the optimal condition for undifferentiated spermatogonia, we first compared the effects of different feeder layers. The enriched undifferentiated spermatogonia were seeded on mitotically inactivated neonatal, adult Sertoli cells or STO feeder layer. As shown in Fig. 2A, the percentage of nonadhered cells in the STO feeder layer groups (26.44% ± 1.48%, P < 0.01) was significantly higher than those in the Sertoli feeder layer group (14.56% ± 1.73% for neonatal and 11.68% ± 1.19% for the adult Sertoli cell group) after 24 h of incubation.
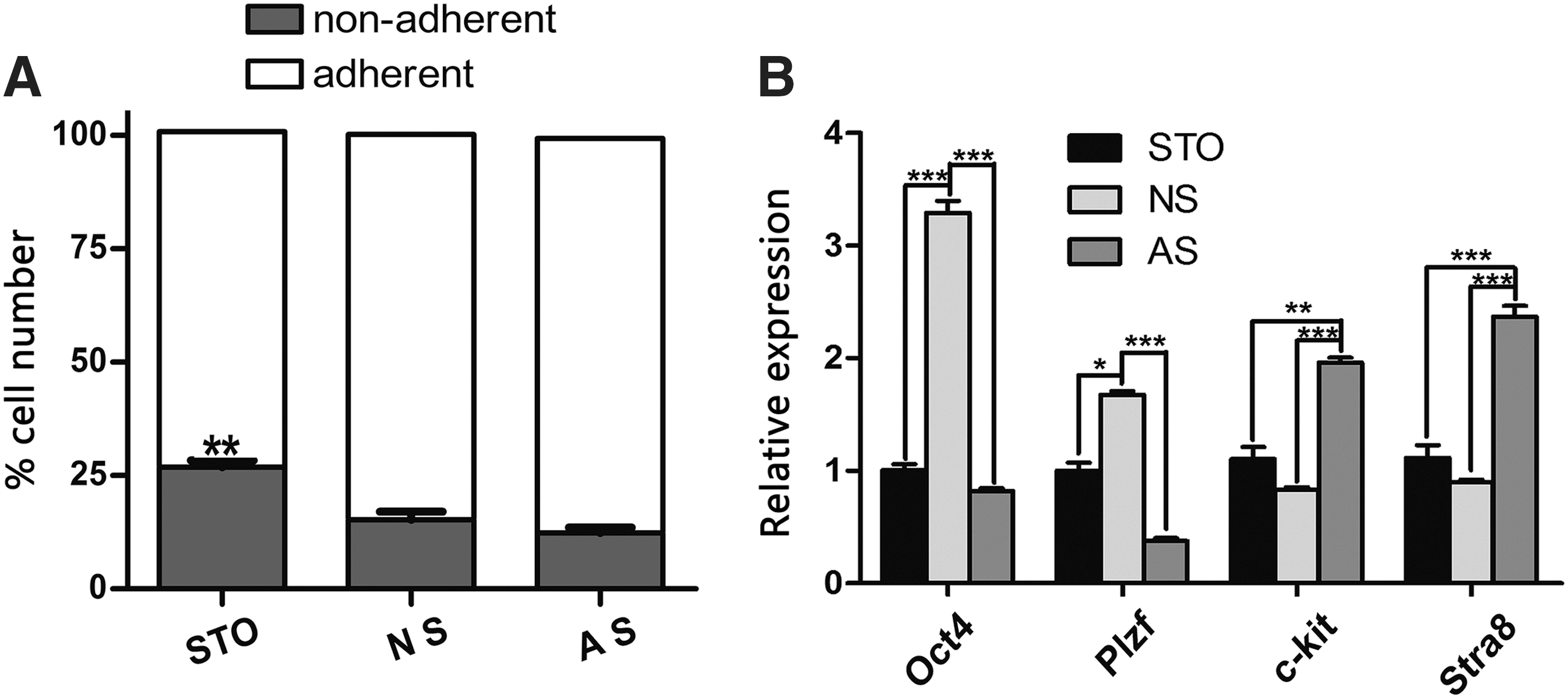
Effects of different feeder layers on in vitro maintenance of undifferentiated spermatogonia.
To characterize the cultured cells on different feeder layers, we detected the expression levels of SSC-related genes (Oct4, Plzf, c-kit, and Star8) by 1 week of culture. The expression of the differentiation markers (c-kit, P < 0.01; Stra8, P < 0.001) was upregulated, and the germline stem cell markers (Oct4, P < 0.001, and Plzf, P < 0.05) were downregulated in the adult Sertoli cell feeder group compared to the other feeder layer groups. The expression of Oct4 and Plzf in neonatal Sertoli feeder group was significantly higher than that in the STO or adult Sertoli cell group (Fig. 2B).
Effect of serum-free supplement KSR on germ cell culture
To optimize the culture condition for the survival and self-renewal of undifferentiated spermatogonia, we added KSR to the BM supplemented with 1% FBS and GDNF (10 ng/mL). After 2 weeks of culture, the number of undifferentiated spermatogonia in the medium containing KSR (10%) showed a 2.55 ± 0.16-fold (P < 0.01) increase compared to that of the control (without KSR, 1.12 ± 0.09-fold; Fig. 3A). Interestingly, the expression of genes that promote cell proliferation was higher in the 10% KSR treatment group (Cdk2, P < 0.001; Cdk4, P < 0.001; and PCNA, P < 0.01, Fig. 3B). Conversely, Cdkn2b (a G1-S inhibitor, P < 0.05) was downregulated in the 10% KSR group (Fig. 3B).
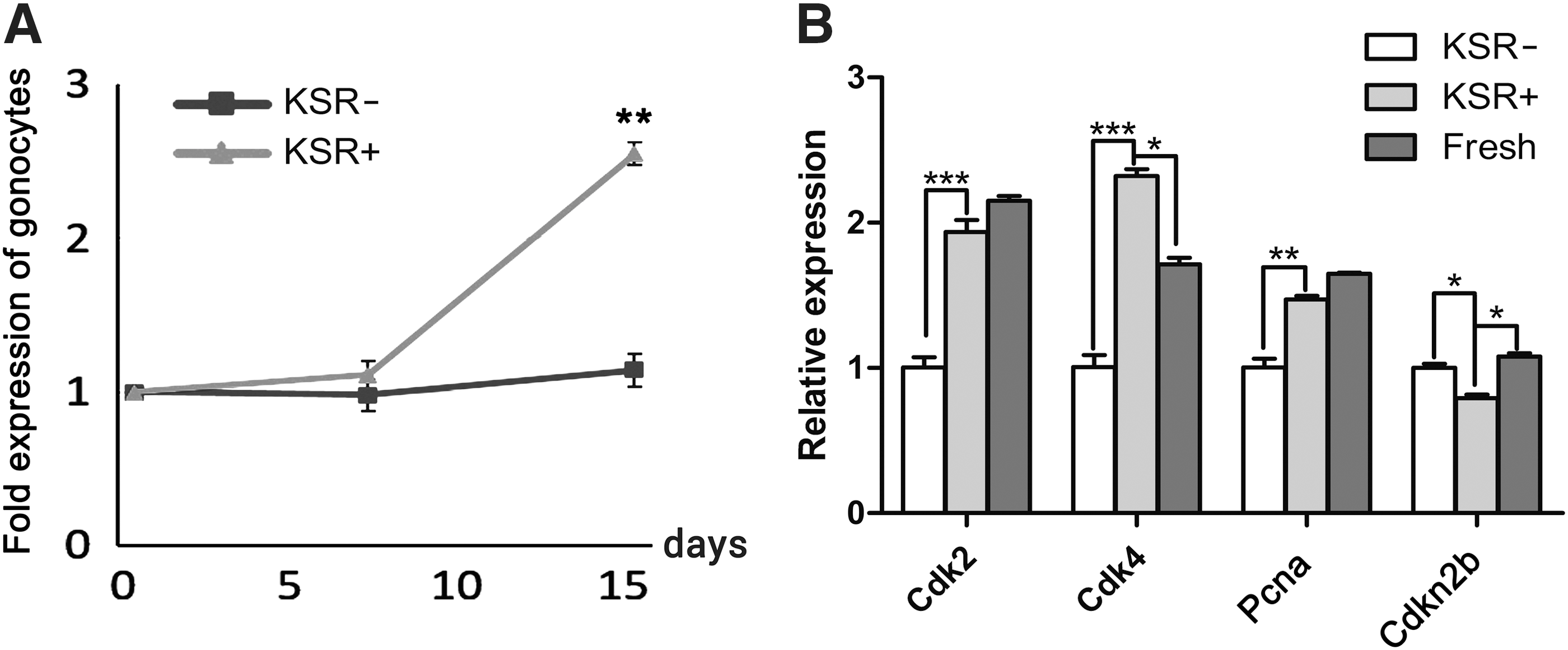
Proliferation of undifferentiated spermatogonia under KSR supplemented medium.
Cell culture with different combinations of growth factors
Next, we tested the effects of different combinations of three growth factors on the survival of undifferentiated spermatogonia during the primary culture (Fig. 4A). Colony formation was observed on days 5–8 of in vitro culture. No difference among groups could be observed during this period. However, after 3 weeks of culture, the mean number of colonies developed in groups GGb (94.19 ± 4.67) and GGI (106.78 ± 4.15) were significantly higher than that in other groups (GbI, 62.54 ± 2.50; bEI, 38.70 ± 3.06; Fig. 4Ba; P < 0.001). The average dimensions of colonies were 2,937.42 ± 119.06 μm2 in the GGb group, 2,117.70 ± 67.26 μm2 in the GGI group, 2,168.70 ± 70.75 μm2 in the GbI group, and 1,758.50 ± 105.40 μm2 in the bEI group (Fig. 4Bb).

Colony formation on different combination of growth factors.
Long-term culture of undifferentiated spermatogonia in the GGbI-supplemented medium
Based on the above observations, a medium supplement with GDNF, GFRA1, bFGF, and IGF1 was utilized to support the long-term culture of undifferentiated spermatogonia. To simulate the temperature of the scrotum, the culture temperature was reduced from 37°C to 35°C. In these conditions, undifferentiated spermatogonia were maintained for up to 8 weeks, with passaging to new feeder layers every 5–7 days. Colonies appeared at all passages, indicating the expansion of undifferentiated spermatogonia. Colonies with unclear borders consisted of multiple cells, and showed the typical morula-like morphology, reminiscent of the previously established murine SSC culture (Fig. 5A).
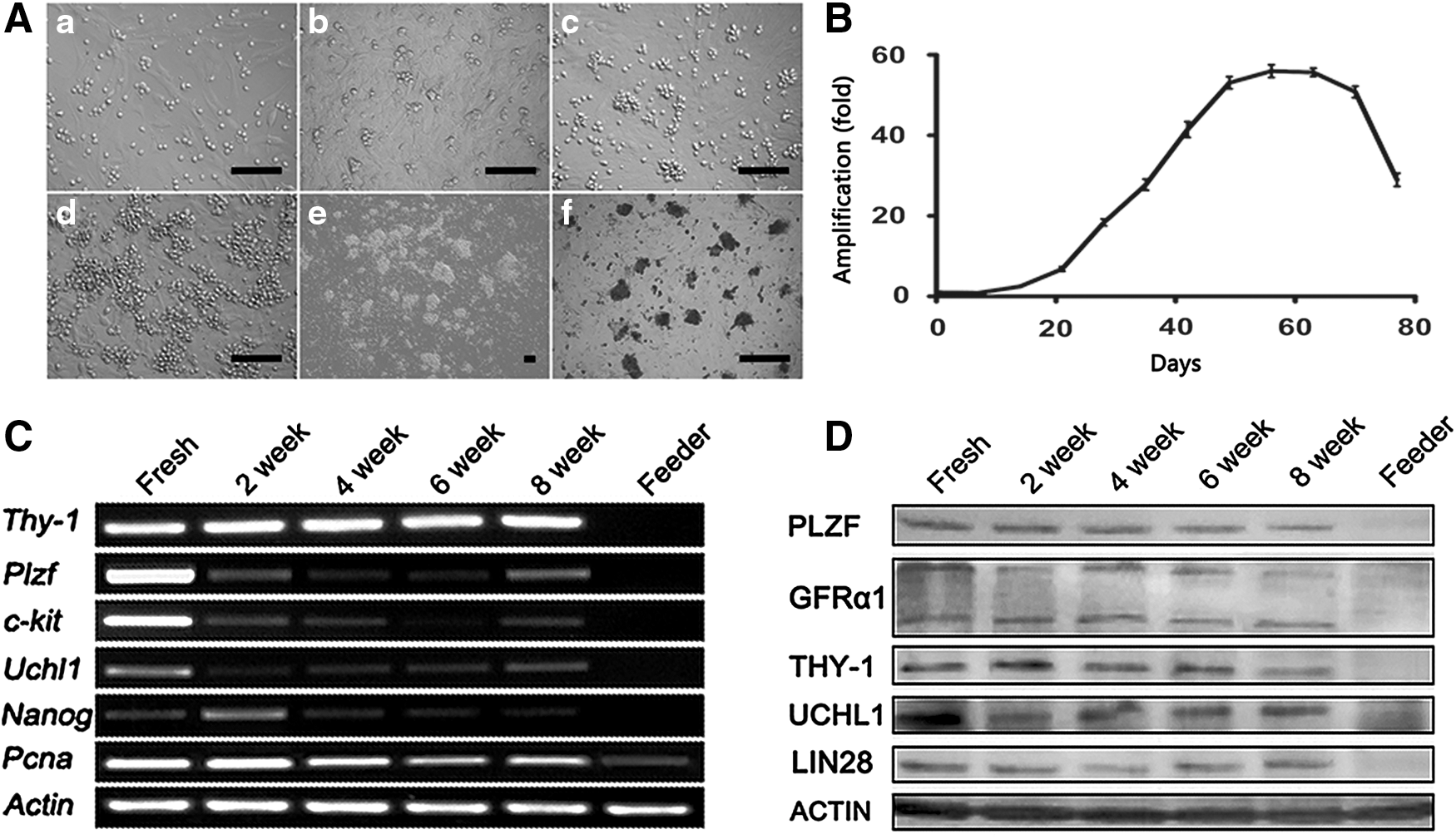
Long-term culture of undifferentiated spermatogonia in medium supplemented with KSR, GDNF, GFRα1, bFGF, and IGF1 on neonatal Sertoli feeder layer.
The number of UCHL1-positive cells on different days was detected (n = 3 different culture examined), and the propagation dynamics is shown in Fig. 5B. The cells showed a robust proliferation after 3 weeks of culture, and the number of undifferentiated spermatogonia increased 56-fold in 2 months. However, the proliferation came to a standstill after 2 months of cultivation. Then the number of UCHL1-positive cells experienced a rapid decline, concomitant with pronounced cell differentiation.
Characterization of undifferentiated spermatogonial colonies
The derived colonies at different time points of culture were isolated from feeder cells and used for RT-PCR and western blot analysis. The samples were collected at 0, 2, 4, 6, and 8 weeks postcultivation. Plzf, Uchl1, Thy-1, and Nanog were expressed throughout the culture period, and the expression of proliferating cell nuclear antigen (Pcna) was also detected during this period (Fig. 5B). Western blot assay confirmed the expression of PLZF, GFRA1, THY-1, UCHL1, and LIN28 in colonies at the protein level (Fig. 5C).
To examine the phenotype of the cultured cell, the cells after 45 days of culture were transferred to LN-coated plates for immunostaining. Germ cell clumps formed in about 7 days. Immunocytofluorescence analysis showed that the colonies were positive for GFRA1, PLZF, and UCHL1, indicating an undifferentiated spermatogonial phenotype (Fig. 6). Moreover, the chromosomal analysis indicated that the majority of cells (81.27% ± 3.62%) after 54 days of cultivation exhibited normal karyotype (38 chromosomes; Fig. 7).

Immunocytochemical staining of colonies derived from cultured cells with GFRα1, PLZF, and UCHL1 on day 52 in vitro culture. Scale bars = 100 μm.
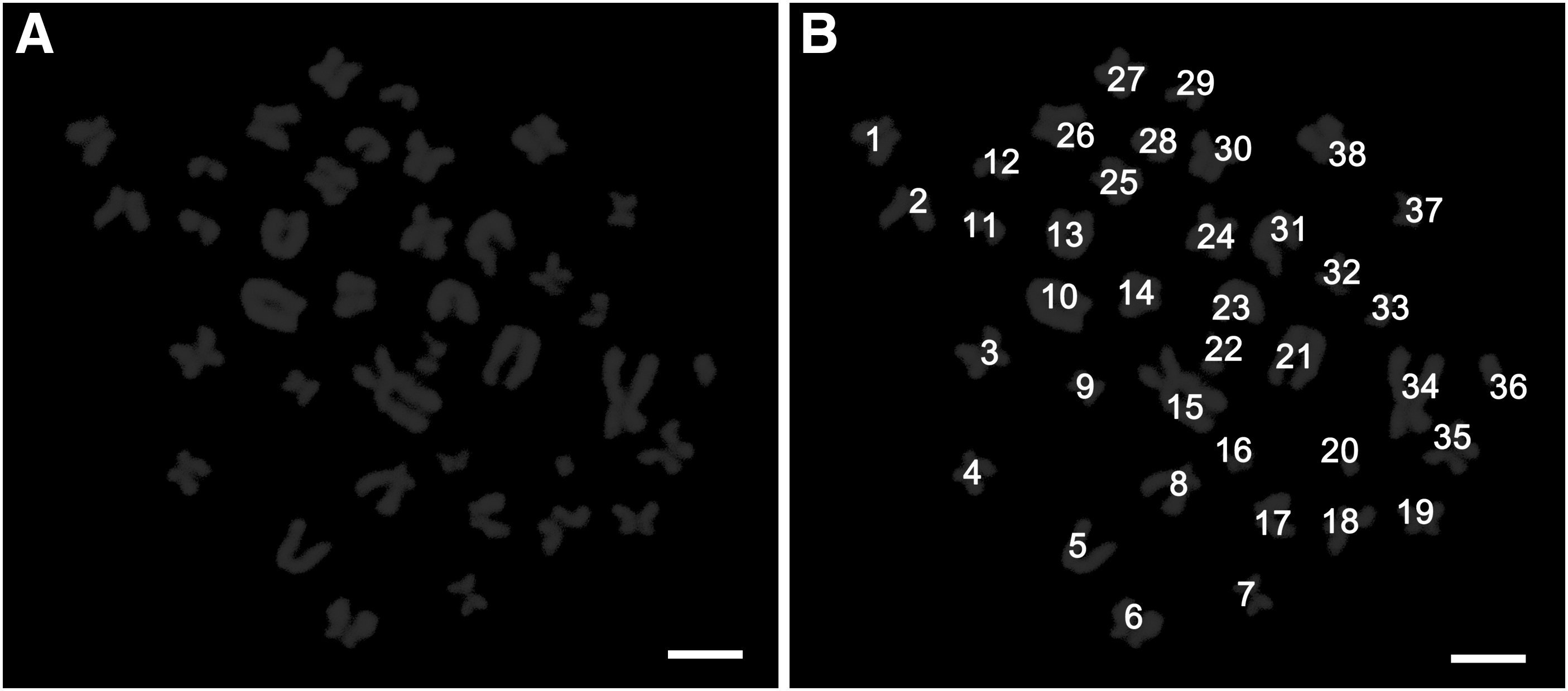
Chromosomal analysis of cultured cells.
Stem cell potential of the cultured cells
To substantiate the stem cell potential of the cultured cells, the undifferentiated spermatogonial colonies were isolated and dissociated to single cell suspension and transplanted into the seminiferous tubules of the busulfan-treated recipient mouse testes. To visualize the donor cells, prelabeling with a red fluorescent dye PKH26 was performed. The PKH26-labeled cells colonized in recipient seminiferous tubules 8 weeks posttransplantation (Fig. 8A, B), and 84. 67 ± 12.51 colonies were produced per 105 transplanted cells (n = 6). These results indicated that the cells cultivated in vitro for 2 months still have stem cell characteristics.
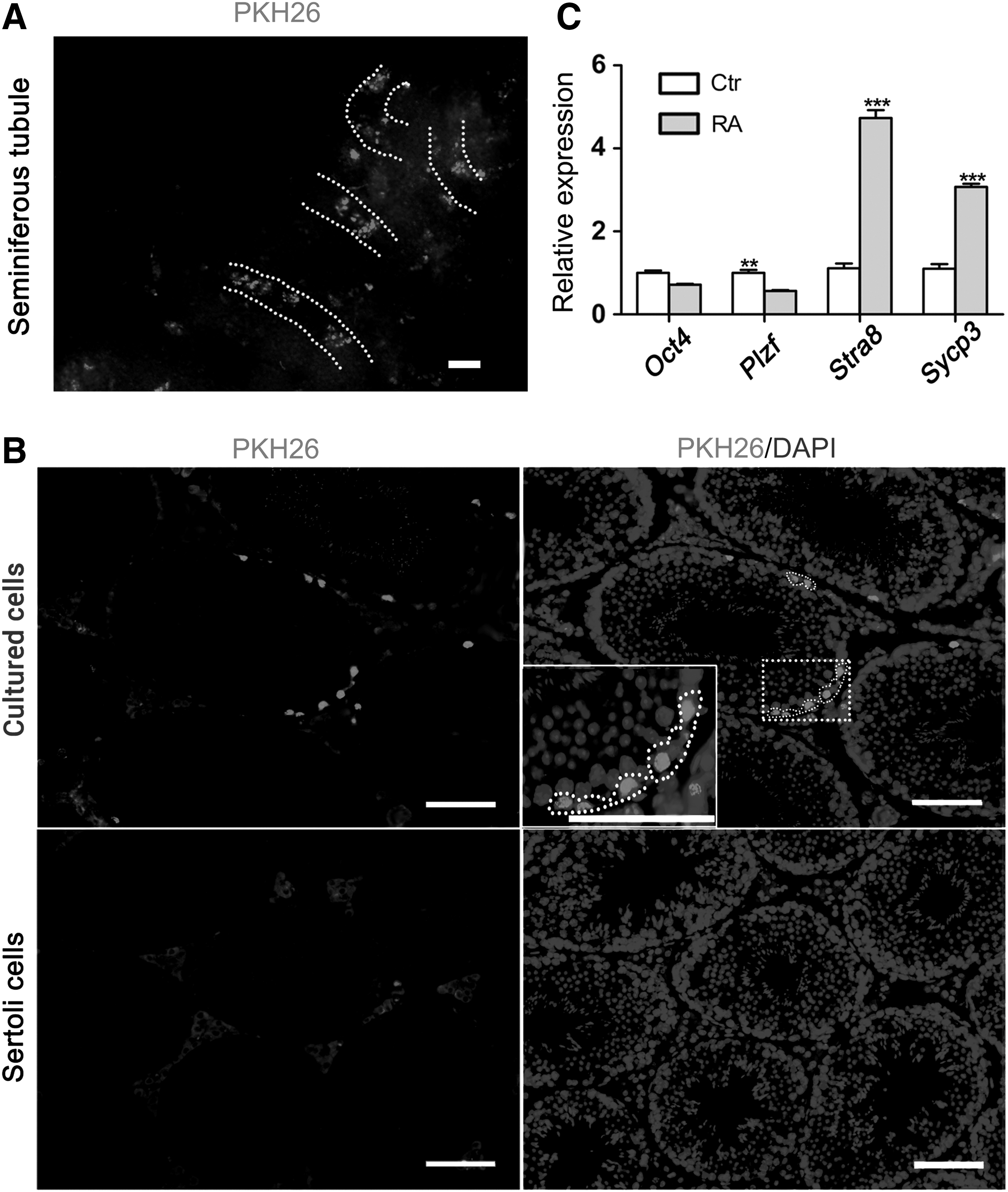
Xenotransplantation and in vitro induction by RA.
We also investigated if the cultured cells could be induced to differentiation in vitro. To this end, we exposed the cells (after 56 days of culture) on the adult Sertoli cell feeder to RA. After 24 h of RA induction, the expression level of Stra8 and Sycp3 was significantly increased (P < 0.001), whereas Plzf was significantly downregulated (P < 0.01; Fig. 8C), indicating that the cultured cells still have the potential of differentiation and initiation of meiosis.
Discussion
The pig (Sus scrofa) is an economically important livestock species. It is also being increasingly exploited as a main nonrodent model for biomedical and pharmacological research because of its high similarity to humans in anatomy and physiology [35]. Genetic manipulations of SSCs combined with germ cell transplantation present a novel approach for gene therapy and production of genetically modified animals. However, few attempts have been done to develop porcine SSC culture in vitro, a prerequisite for related applications due to the rarity of SSCs within mammalian testes. SSCs are the exceptional adult stem cells that can transmit genetic information into next generations. Thus the combination of SSC transplantation with genomic editing techniques could be applied to cure infertility or eliminate genetic diseases. Moreover, SSCs can transform to pluripotent stem cells under appropriate culture conditions [36 –42]. Establishment of long-term culture systems to support the continuous proliferation of SSCs in vitro is crucial to the fulfillment of their great potential in biomedicine and agriculture.
Although long-term culture of rodent SSCs has been developed [16,17,20 –24], it is not the case for those from large animals. The lack of fitting feeder cells for subculture is one of the bottlenecks for the development of the culture systems. Our previous study showed that undifferentiated spermatogonia could survive for 1 month in vitro when cocultured with autologous Sertoli cells [13], which suggests that the endogenous Sertoli cells could be a good option. A recent article showed that Sertoli cells are critical for the support of tree shrew SSC expansion [26]. In this study, we compared the effects of neonatal and adult Sertoli cells and STO cells for feeder layers. We found that undifferentiated spermatogonia exhibited better affinity as neonatal and adult Sertoli cells in comparison with STO cells. The expression of differentiation markers c-kit and Stra8 in the germ cell-derived colonies forming on adult Sertoli cells was much higher than that on neonatal Sertoli or STO cells, indicating that adult Sertoli cells may induce germ cell differentiation. It was also reported that the colony number, diameter, and beta1-integrin expression were higher when Sertoli cells were serving as feeders, compared to STO cells [43]. The observation obtained in this study is consistent with the recently published work by Lee et al. [31] in which they subcultured the germ cell-derived colonies with GATA4- positive testis cells. Moreover, using autologous Sertoli cells as a feeder layer could eliminate pathogens from other species. Therefore, we chose the neonatal Sertoli cells as feeder layers for further optimization.
Our previous study showed that high concentration of serum displayed detrimental effects on porcine undifferentiated spermatogonial proliferation [13]. The similar phenomenon has also been found in cultures of SSCs from goats [44] and bovines [29], indicating that a reduced concentration of serum could be essential for the maintenance of stem cell activity [13,28]. On the other hand, alternatives for serum could be studied. The serum-free supplement KSR, originally developed for human [45,46] and mouse embryonic stem (ES) cells [47] and pig EGCs [48], has been successfully used in serum-free culture for mouse SSCs [21]. As shown in those articles, KSR contains the agents that promote self-renewal and maintenance of pluripotency [20]. This study demonstrated that the KSR-supplemented medium was effective for expansion of porcine undifferentiated spermatogonia. Albumax, a lipid-rich bovine serum albumin and a component of KSR, has been demonstrated to be necessary for SSC self-renewal in vitro through a lipid-related signal [20 –22]. It has also been shown that KSR promotes self-renewal and blocks mitotic arrest and differentiation [49]. Further studies are needed to determine whether KSR contains other beneficial components, and, if any, their regulation on SSC proliferation or differentiation.
Increasing evidences have demonstrated that appropriate cytokines are essential for long-term culture of SSCs [13,17 –19,25,32,50]. Compared to other growth factors, GDNF is indispensable for the proliferation and maintenance of rodent and human SSCs in culture [24]. In mice, Kubota et al. reported that the addition of soluble GFRα1 is crucial to the successful culture of SSCs from mouse strains apart from DBA/2 [17]. Our previous study [13] and another report [32] failed to reveal beneficial effects of GDNF on the expansion of porcine undifferentiated spermatogonia colony. Interestingly, this study clearly demonstrates that the addition of GDNF in combination with GFRα1 is superior to that without GFRα1. Lee et al. [30] have reported that GFRα1 is expressed on the surface of porcine undifferentiated spermatogonia. Given the literature and these data, it is plausible that GDNF alone is inefficient for the in vitro maintenance of porcine SSCs, but the addition of GFRα1 intensifies GDNF signals, thereby enhancing the formation of SSC colonies. As mentioned by Aponte et al. [28], other possible attributes could be related to the age, species, cell density, and somatic cell population.
In addition to GDNF, evidences available indicate that bFGF [50,51] and IGF1 [18] are important for murine stem cell behavior. Nevertheless, the functions of bFGF and IGF1 for SSCs in large animals remain largely elusive. Similar to the results found in humans [52], in this study, we found that bFGF addition yielded a larger size of colonies, which indicates that bFGF stimulates the increase of colony diameter. Recent studies have demonstrated that bFGF relies on MAP2K1 activation [51] and the coaction with GNDF signaling pathways [50]. Another growth factor IGF1 has been shown to contribute to the derivation of pluripotent cells from male germ cells [18]. Collectively, GDNF, GFRα1, bFGF, and IGF1 were used for refinement of the long-term culture system.
Using Sertoli cells as feeder layers and the culture medium supplemented with KSR and a cytokine combination of GDNF, GFRα1, bFGF, and IGF1, we could propagate porcine undifferentiated spermatogonia for over 2 months with a normal karyotype. More importantly, we observed consistent colony formation throughout all passages, with all of them showing typical grape-like morphology comprising multiple germ cells, different from the previously reported ES cell-like colonies. The morphology of colonies is similar to that from mice [16,17], rats [24], hamsters [23], rabbits [25], bovines [53], and tree shrews [26]. The expression of genes Plzf, Uchl1, Nanog, and Oct4 in the derived colonies indicated that the cultured cells maintained SSC markers. Western blot and immunocytochemistry analysis further verified the expression of SSC markers, such as GFRα1, UCHL1, PLZF, THY-1, and LIN28 in the cultured cells.
Recently, a study reported that neonatal pig germ cell-derived colonies can be maintained for 2 months with GATA4-expressing testicular somatic cells [31]. However, that study failed to perform a functional assay for cultured cells. The establishment of male germ cell transplantation can provide a functional assay to characterize SSCs [54]. The functional SSCs could relocate to the basement membrane and regenerate the donor SSC-derived spermatogenesis in host testes after transplantation [8,54]. However, for pigs, the methodologies for generating suitable recipient males require further development because injection of chemotoxic drugs with doses that can thoroughly eliminate endogenous germ cells is usually lethal to the recipient large animals, including boars.
Therefore, we xenotransplanted the cultured cells into the sterile recipient mouse testes that were pretreated with busulfan to eliminate the endogenous germline, but have an intact niche. We found that the cells after 2 months of in vitro culture still preserve the capacity to colonize in recipient seminiferous tubules, indicative of the stem cell capacity. Meanwhile, RA induction showed that the cells after 56 days of culture still had the potential of differentiation and initiation of meiosis. These findings indicate the presence of functional SSCs in the cultured colonies. Finally, in line with a recent report about the long-term culture of bovine undifferentiated spermatogonia [53], we observed that differentiation and apoptosis outweighed cell proliferation (data not shown), resulting in the decline in the total cell number after 2 months of cultivation.
This irreversible phenomenon makes room for future improvement. In this regard, several issues need to be taken into consideration. First, elucidating various signaling pathways underlying porcine spermatogenesis and further identifying critical factors involved in porcine SSC self-renewal and differentiation will definitely help to improve the in vitro culture conditions. Robust omic methodologies such as high-throughput RNA sequencing could be adopted to unravel the transcriptomic profile of different stages of male germ cells in vivo, as well as early and late passages of cells in vitro, as shown in previous studies in mice [55] and the recent report in tree shrew [26]. Second, the identification of bona fide markers for porcine SSCs and development of methodologies for further enrichment of porcine SSCs could enhance the efficiency of SSC culture. Third, it is necessary to uncover the milieu of growth factors secreted by the Sertoli cells, peritubular myoid cells, and Leydig cells.
In conclusion, this study describes an optimized culture system that can maintain porcine undifferentiated spermatogonia with stemness and normal karyotype for at least 2 months. This culture system will serve as a basic refinement in future studies and facilitate studies on SSC biology and genetic manipulation of male germline stem cells.
Footnotes
Acknowledgments
This study was supported, in part, by National Basic Research Program of China (973 program; 2014CB943100), the National Natural Science Foundation of China (grant nos. 31272439, 31230048), and the Doctoral Program Foundation of Higher Education of China (grant no. 20130204110017).
Author Disclosure Statement
No competing financial interests exist.