
Abstract
Spinal root injuries result in newly formed glial scar formation, which prevents regeneration of sensory axons causing permanent sensory loss. Previous studies showed that delivery of trophic factors or implantation of human neural progenitor cells supports sensory axon regeneration and partly restores sensory functions. In this study, we elucidate mechanisms underlying stem cell-mediated ingrowth of sensory axons after dorsal root avulsion (DRA). We show that human spinal cord neural stem/progenitor cells (hscNSPC), and also, mesoporous silica particles loaded with growth factor mimetics (MesoMIM), supported sensory axon regeneration. However, when hscNSPC and MesoMIM were combined, sensory axon regeneration failed. Morphological and tracing analysis showed that sensory axons grow through the newly established glial scar along “bridges” formed by migrating stem cells. Coimplantation of MesoMIM prevented stem cell migration, “bridges” were not formed, and sensory axons failed to enter the spinal cord. MesoMIM applied alone supported sensory axons ingrowth, but without affecting glial scar formation. In vitro, the presence of MesoMIM significantly impaired migration of hscNSPC without affecting their level of differentiation. Our data show that (1) the ability of stem cells to migrate into the spinal cord and organize cellular “bridges” in the newly formed interface is crucial for successful sensory axon regeneration, (2) trophic factor mimetics delivered by mesoporous silica may be a convenient alternative way to induce sensory axon regeneration, and (3) a combinatorial approach of individually beneficial components is not necessarily additive, but can be counterproductive for axonal growth.
Introduction
A
Different neurotrophic factors [glial-derived neurotrophic factor (GDNF), neurotropin (NT)-3, and Artemin] can entice injured dorsal root axons to successfully penetrate the DRTZ glial scar and establish functional connections in the spinal cord [11 –13]. Regrowth of sensory fibers through the transected dorsal root was also achieved with the help of olfactory ensheathing cells forming a growth-permissive tissue bridge through the DRTZ glial scar [14,15].
We recently showed that human embryonic stem cell (hESC)-derived spinal cord progenitors assist growth of sensory axons into the spinal cord, accompanied by partial recovery of sensory functions after the complete avulsion of the dorsal root [16]. In this study, we test if mesoporous silica particles loaded with peptide mimetics (MesoMIM) of GDNF (Gliafin) and ciliary neurotrophic factor (CNTF) (Cintrofin), which affect sensory axon growth [12,17,18], also assist sensory axon regeneration. If so, a combined approach with implantation of human spinal cord progenitors [human spinal cord neural stem/progenitor cells (hscNSPC)] and MesoMIM could have additive effects, stimulation of sensory axon regeneration through the delivery of trophic factor peptide mimetics and modification of the dorsal root–spinal cord interface by implanted hscNSPC.
Materials and Methods
Animals
All animal experiments were approved by the Uppsala Local Ethics Committee for Animal Experimentation, as required by Swedish Legislation and in accordance with European Union Directives. Experiments were performed on athymic, immunodeficient male adult nu/nu (NMRI-Foxn1nu/Foxn1nu) mice (n = 27, Mollegaard, Denmark).
Transplantation of human fetal spinal cord neurospheres and implantation of peptide mimetics
Preparation of human embryonic tissue was described previously [19]. Mesoporous silica with an average particle size of 12 μm and average pore size of 20 nm were loaded with peptide mimetics like described before [20]. Compared to the relatively large native growth factors, smaller growth factor mimetics have preferable properties for the loading and release by mesoporous silica. In this study, the previously characterized peptide mimetic for GDNF (Gliafin, 153- ETMYDKILKNLSRSR-167; UniProtKB entry No. Q07731, [21]) or the peptide mimetic for CNTF (Cintrofin, 148-DGGLFEKKLWGLKV-161; UniProtKB entry No. P26441, [22]) was loaded into mesoporous silica. For transplantation purposes and in vitro assays, equal amounts of mesoporous silica loaded with either Gliafin or Cintrofin were used and their combined use is from here on referred to as MesoMIM.
All animals were 8 weeks old at the time of the surgical procedure. The surgical procedure of dorsal root avulsion (DRA) injury was described in detail previously [16]. In short, after induction with 4% isoflurane in 1:1 medical air:medical O2, animals were maintained at 2.5% isoflurane. The skin, muscles, and fascia of the lower back was dissected to expose the vertebrae corresponding to the left L3–L5 dorsal root entry points. After laminectomy, the dura was opened to expose the underlying dorsal roots. Topical analgesia (Xylocaine) was applied on the spinal surface over the L3–L5 dorsal roots, which were subsequently gently pulled from the spinal cord with a pair of forceps so that the rootlets ruptured at the root entry zone.
After DRA, animals received ∼50 neurospheres (100–350 μm in diameter; ∼340 000 cells) (DRA + hscNSPC), neurospheres in equivalent numbers and size with ∼20 μg MesoMIM (DRA + hscNSPC + MesoMIM), MesoMIM alone (DRA + MesoMIM), no treatment DRA, or animals were left intact (naive). After DRA with or without treatment, the roots were repositioned and the muscles and skin sutured using single silk (6.0 and 4.0 respectively) sutures. Animals were allowed to recover in a warm cage and maintained on subcutaneous Buprenorphine injections for 3 days. Animals were analyzed after 1 week (DRA + hscNSPC; n = 4 and DRA + hscNSPC + MesoMIM; n = 4) or 2 months (naive; n = 3, DRA; n = 3, DRA + MesoMIM; n = 5, DRA + hscNSPC; n = 4, and DRA + hscNSPC + MesoMIM; n = 4). Only animals with complete avulsions and surviving grafts were included in the study.
Transganglionic tracing
After postoperative survival times of 1 week or 2 months, mice were anesthetized and 3 μL of a 1% solution of cholera toxin B subunit (CTB) was injected into the left sciatic nerve as previously described [16]. Three days after tracer injection, animals were reanesthetized and perfused, and L3–L5 spinal cord tissue was collected and processed for immunohistochemistry and analysis.
Immunohistochemistry
Animals were terminally anaesthetized, perfused, and 14-μm-thick serial coronal cryosections were prepared according to previously described protocols [16]. Sections were incubated with mixed primary antibodies overnight at 4°C (for the list of used antibodies, see Table 1). The anti-CTB antibody was incubated for 4 h at room temperature.
DCX, doublecortin.
Microscopy
Images for CTB and glial fibrillary acidic protein (GFAP) quantification were taken using a Plan-Apochromat 10 × objective (NA 0.45) attached to a Nikon Eclipse E800 epifluorescence microscope equipped with a Nikon DXM1200F CCD camera. Overlays of multichannel pictures were prepared with Fiji ImageJ2.0.0-rc-2 [23].
All other immunolabeled sections were analyzed using a Zeiss LSM700 confocal laser scanning microscope. Single images were captured using an LD LCI Plan-Apochromat 20 × objective (NA 0.8); for orthogonal projection, images were taken with a Plan-Apochromat 63 × objective (NA 1.4). Z-stacks were taken with an optical slice thickness of 0.25 μm at an interval of 0.25 μm. Tile-scanned images were captured using a Plan-Apochromat 20 × objective (NA 0.8). Bright field images of in vitro-cultured hscNSPC were taken using a Plan-Apochromat 10 × objective (NA 0.25) attached to a Nikon Eclipse TS100 epifluorescence microscope equipped with a Nikon DS Fi1 camera.
Image processing and statistical analysis
Cell counts and measurements of immunoreactivity were performed in a blinded manner on transverse sections, collected throughout the entire length of the transplant. The serial sections were collected on slides in identical order (section 1 to slide 1, section 2 to slide 2, and so on, and then in the second round, section 11 again to slide 1, section 12 to slide 2, and so on) until a total of 5 sections were present on a single slide. Then, the same procedure was used to collect the next 50 sections on the next 10 slides.
To analyze the differentiation of the transplanted cells, every 10th slide, encompassing every 10th, 20th, 30th, 40th, and 50th section of the spinal cord with the transplant, was immunolabeled for HuNu together with one of the phenotypical markers [SOX2, OLIG2, huKi67, GFAP, MAP2, or doublecortin (DCX)]. This places the minimal distance between sections that are immunolabeled with the same combination of antibodies at 140 μm and makes their analysis more representative, because accidently counting the same cell twice is now highly unlikely.
For cell counting, confocal laser scanning microscope images were taken at 20 × magnification, and all HuNu-positive cell nuclei and those dually labeled with the marker used for analysis were manually counted throughout the region encompassing the entire dorsal horn and the area of engraftment. Data were recorded using the Fiji ImageJ2 plug-in “Cell Counter” pooled per animal and for further analysis, grouped and averaged by the two experimental conditions.
The data were analyzed using the Mann–Whitney U test. One week after transplantation, the proportion of SOX2+-/HuNu+-, OLIG2+-/HuNu+-, and Ki67+-/HuNu+-labeled cells was calculated from the total number of HuNu+ cells. Two months after transplantation, the proportion of GFAP+-/HuNu+-, MAP2+-/HuNu+-, DCX+-/HuNu+-, OLIG2+-/HuNu+-, and SOX2+-/HuHu+-labeled cells was calculated in the same way. The data from the hscNSPC and hscNSPC + MesoMIM groups with regard to the distribution of GFAP, MAP2, DCX, OLIG2, and SOX2 expression were analyzed using a two-way analysis of variance (ANOVA). In addition, 2 months after transplantation, HuNu+ cells, which had migrated away from the area of engraftment into the ipsilateral ventral horn and contralateral side, were counted.
The area of CTB immunoreactivity in the dorsal horn was analyzed for every 10th slide of the lumbar levels L3–L5 2 months after surgery in all experimental groups. For quantification, images were preprocessed in Fiji ImageJ2 by subtracting the background by using tissue autofluorescence of the 488 nm channel and the implemented rolling ball algorithm, followed by setting a manual threshold for CTB immunoreactivity. The dorsal horn was outlined in the raw image and transferred onto the thresholded image, the total area of CTB immunoreactivity in the ipsilateral dorsal horn was measured, and differences in the total area of immunoreactivity were determined using a Kruskal–Wallis analysis of variance followed by Dunn's multiple comparison test. All data were normalized using the mean CTB+ area of three CTB-injected naive animals quantified identically like the experimental groups.
GFAP and NG2 immunoreactivity at the site of DRA 2 months after injury was analyzed for each condition in every 10th slide and for an area encompassing the full length of the transplant. For quantification, images were preprocessed in Fiji Image2 by using the implemented rolling ball algorithm set to a size of 10 pixels and the contrast enhanced to a saturation of 0.35. The site of DRA injury and hscNSPC engraftment was outlined and defined as region of interest, followed by setting a threshold for GFAP+ and human GFAP+, or NG2+ staining.
To be able to analyze the injury-induced murine glial scar alone, separately from the GFAP signal of the transplanted cells, two different antibodies against GFAP were used. The first was immunoreactive against murine and human GFAP and the second targeted only a human-specific epitope. This allowed us to subtract the human-specific GFAP+ area from the overall GFAP+ area, resulting in the isolated GFAP+ area of the murine glial scar. Sections dually immunolabeled with both GFAP antibodies and appropriate secondary antibodies to restrict their signals to two distinct color channels were imaged and processed in Fiji Image2. The human GFAP immunoreactive area in the first color channel was automatically thresholded using Otsu's method and subtracted from the total GFAP+ area in the second color channel, followed by automatic thresholding using Tsai's method [24,25], both thresholding techniques are implemented in Fiji Image2. The resulting thresholded area represented the GFAP+ area of the isolated murine glial scar.
Due to individual differences in the diameter and morphology of the spinal cord, the area of the isolated murine glial scar was divided by the total area of the spinal cord. Both the isolated murine glial scar GFAP+ area and NG2+ area were analyzed using a one-way ANOVA followed by Dennett's multiple comparison test against the DRA group. All data were normalized using the mean GFAP+ or NG2+ area of three naive animals, which were quantified in the same manner as in the experimental groups.
All statistical analyses were performed using GraphPad Prism 5.04. The confidence interval was stated at the 95% confidence level, placing statistical significance at P < 0.05.
hscNSPC in vitro differentiation assay
hscNSPC were maintained as free-floating neurospheres as described before [26]. For in vitro differentiation assays, hscNSPC neurospheres were seeded on 0.0001% poly-l-ornithine (Sigma) followed by 20 μg/mL laminin (Sigma)-precoated tissue culture plastic in two variants: (i) in differentiation medium (consisting of DMEM/F12, Neurobasal at 1:1 (Invitrogen) supplemented with 1 × B27, 1 × N2, and 1% MEM nonessential amino acids (Invitrogen)) (ii) in differentiation medium supplemented with MesoMIM (200 ng/mL Gliafin and 200 ng/mL Cintrofin). Cells were cultured for 5 and 8 days followed by RNA extraction using the Qiagen RNeasy Mini Kit (Qiagen) following the manufacturer's recommendation. cDNA synthesis using the High-Capacity RNA-to-cDNA kit (Invitrogen) was performed according to the manufacturer's protocol.
Quantitative reverse transcription polymerase chain reaction analysis
All primers were designed using Beacon Design 8 (Premier Biosoft) and purchased from Thermo Fischer Scientific (see Table 2). The qRT-PCR master mix contained 2 × DreamTaq Buffer (Thermo Scientific), 0.2 μL 20 mM dNTP, 0.05 μL of both forward and reverse primer (100 pmol/μL), 0.5 μL 10 × SYBRGreen (1:10,000, diluted in TE buffer, pH 7.8, Invitrogen), 1 μL DMSO (Sigma), and 0.08 μL DreamTaq polymerase (5 U/μL, Thermo Scientific). 2 μL of cDNA diluted to 30 ng/μL was used and the final volume was adjusted to 20 μL/well with sterile water. Every sample was run in triplicates and negative controls were included on each plate. An iCycler real-time detection instrument (Bio-Rad Laboratories, Sweden) with the following conditions was used: initial denaturation for 3 min at 95°C followed by 55 cycles of 10 s at 95°C, 30 s at 41°C–56°C (optimal temperature for each primer pair), and 30 s at 72°C. Thereafter, a melting curve was performed for 81 cycles with 10 s intervals and an incremental temperature increase of 0.5°C per cycle, starting at 55°C. MyIQ (Bio-Rad Laboratories, Sweden) software was used to obtain CT values for all samples.
Primers marked with “a” were used as housekeeping genes.
CNTF, ciliary neurotrophic factor; GDNF, glial-derived neurotrophic factor.
Primer efficiency was calculated for each run using LinRegPCR software [27], followed by Grubb's test (GraphPad software, USA) to remove outliers in efficiency calculations. The GeNorm protocol [28] was used to check stability of the housekeeping genes and then used as normalization factors for each sample. The values were then corrected to one and mean CT values (±SEM) were plotted. For statistics, at least four independent experiments were analyzed per all primer pair.
Results
hscNSPC transplant integration and differentiation
hscNSPC showed immunoreactivity for the neural progenitor cell markers SOX2and OLIG2 before transplantation (Supplementary Fig. S1a; Supplementary Data are available online at
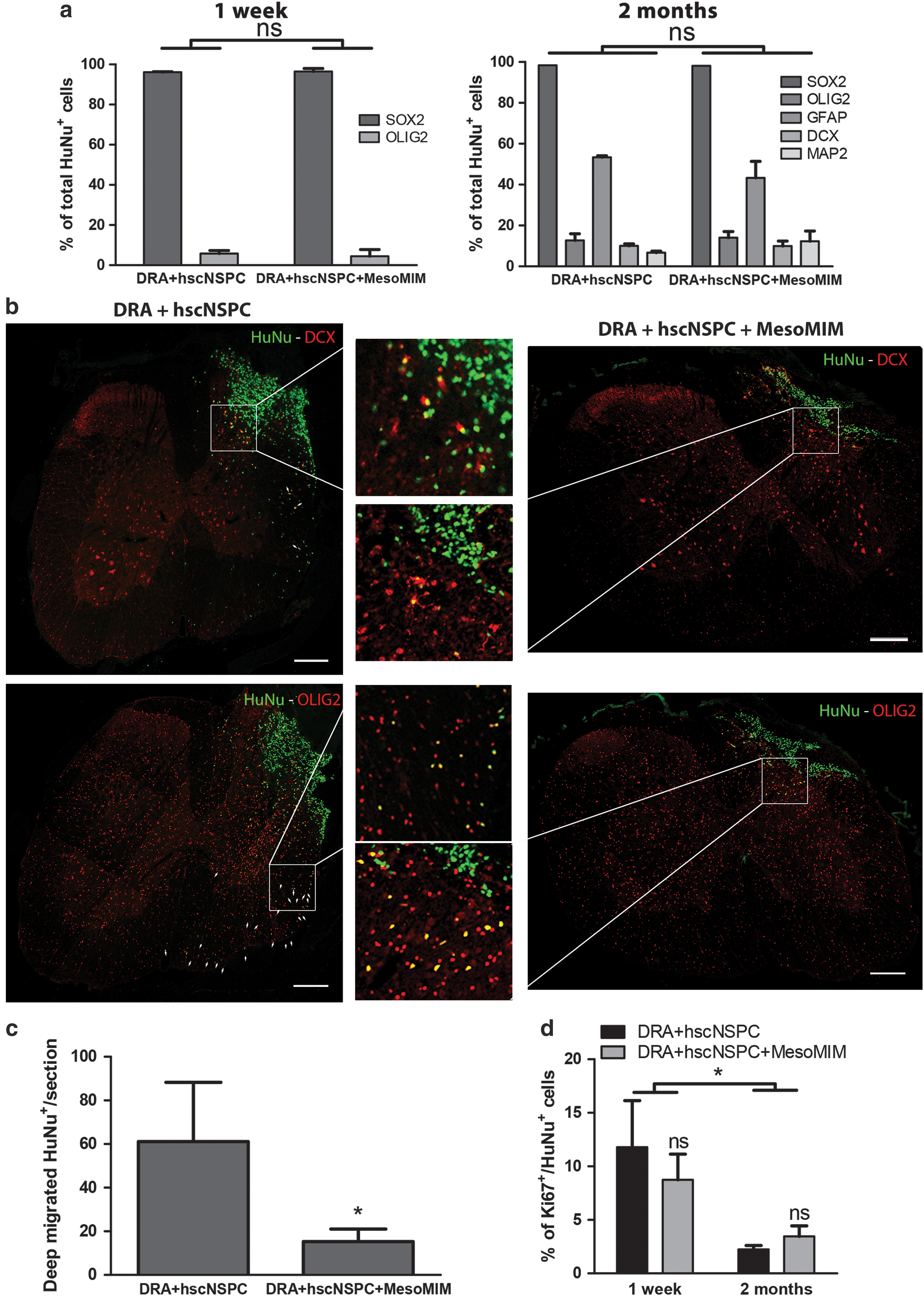
Coimplantation of mimetic-loaded silica particles altered migration, but not differentiation of hscNSPC transplanted to the site of DRA.
After 2 months, 22.8 ± 6.1% of the initially transplanted number of cells was found in the hscNSPC group and 12.2 ± 4.8% in the hscNSPC + MesoMIM group. hscNSPC continued to express SOX2 (98.4 ± 0.7%) and showed slightly increased expression of OLIG2 (12.0 ± 3.2%). At the graft site, hscNSPC displayed signs of glial cell differentiation indicated by GFAP expression in 53.3 ± 0.7% of all HuNu+ cells and differentiation toward neurons indicated by the expression of DCX in 10.0 ± 1.0% and MAP2 in 6.7 ± 0.8% of hscNSPC (Supplementary Fig. S1c), while maintaining expression of SOX2 (Supplementary Fig. 1d).
Coimplantation with MesoMIM reduced the proportion of GFAP+ cells (43.3 ± 8.1%) and increased the proportion of MAP2+ cells (12.3 ± 5.0%), but did not alter the differentiation pattern of other hscNSPC markers (SOX2+: 98.1 ± 0.9%; OLIG2+: 14.0 ± 4.1%; DCX+: 10.0 ± 2.4%). The overall expression pattern showed no statistical difference between both groups for the two analyzed time points (P > 0.05, Fig. 1a).
Two months after surgery, transplants of the hscNSPC + MesoMIM group were primarily confined to the site of injury, whereas hscNSPC transplanted alone were detected in the ipsilateral and contralateral side of the spinal cord. In both groups, few migrated cells expressed the immature migratory neuron marker DCX, whereas the majority expressed the neural progenitor cell marker OLIG2 (hscNSPC 93.0 ± 2.6% and hscNSPC + MesoMIM 94.3 ± 3.2%, Fig. 1b). HuNu-labeled hscNSPC migrated to a significantly lesser extent when they were coimplanted with MesoMIM (P < 0.05, Fig. 1c).
Human cells expressed the proliferation marker Ki67 in both groups (hscNSPC; 7.9 ± 3.2% and hscNSPC + MesoMIM; 8.7 ± 2.4%) 1 week after transplantation. Significantly fewer Ki67-positive human cells (hscNSPC; 2.2 ± 0.4% and hscNSPC + MesoMIM; 4.2 ± 1.1%) were found at 2 months after transplantation (P < 0.05, Fig. 1d and Supplementary Fig. 1e).
Taken together, coimplantation of MesoMIM with hscNSPC resulted in a reduction of hscNSPC migration, but did not affect hscNSPC differentiation at detectable levels. To corroborate these findings in vitro, hscNSPC were cultured alone or together with MesoMIM for 5 and 8 days and the expression of differentiation associated genes was analyzed using qPCR. In line with the in vivo findings, cultured hscNSPC spread over the culture dish, whereas hscNSPC cultured together with MesoMIM showed minimal migration (Fig. 2a). hscNSPC cultured alone or together with MesoMIM showed small differences in their expression of markers of undifferentiated hscNSPC (SOX2, OLIG2, and Nestin) on day 8 and more mature hscNSPC (β-3 Tubulin, MAP2) on days 5 and 8 (Fig. 2b). In addition, qPCR analysis revealed a trend towards increased expression of GDNF and CNTF on day 5 and significantly reduced expression of GDNF on day 8 in the MesoMIM-treated group (P < 0.05, Fig. 2c).
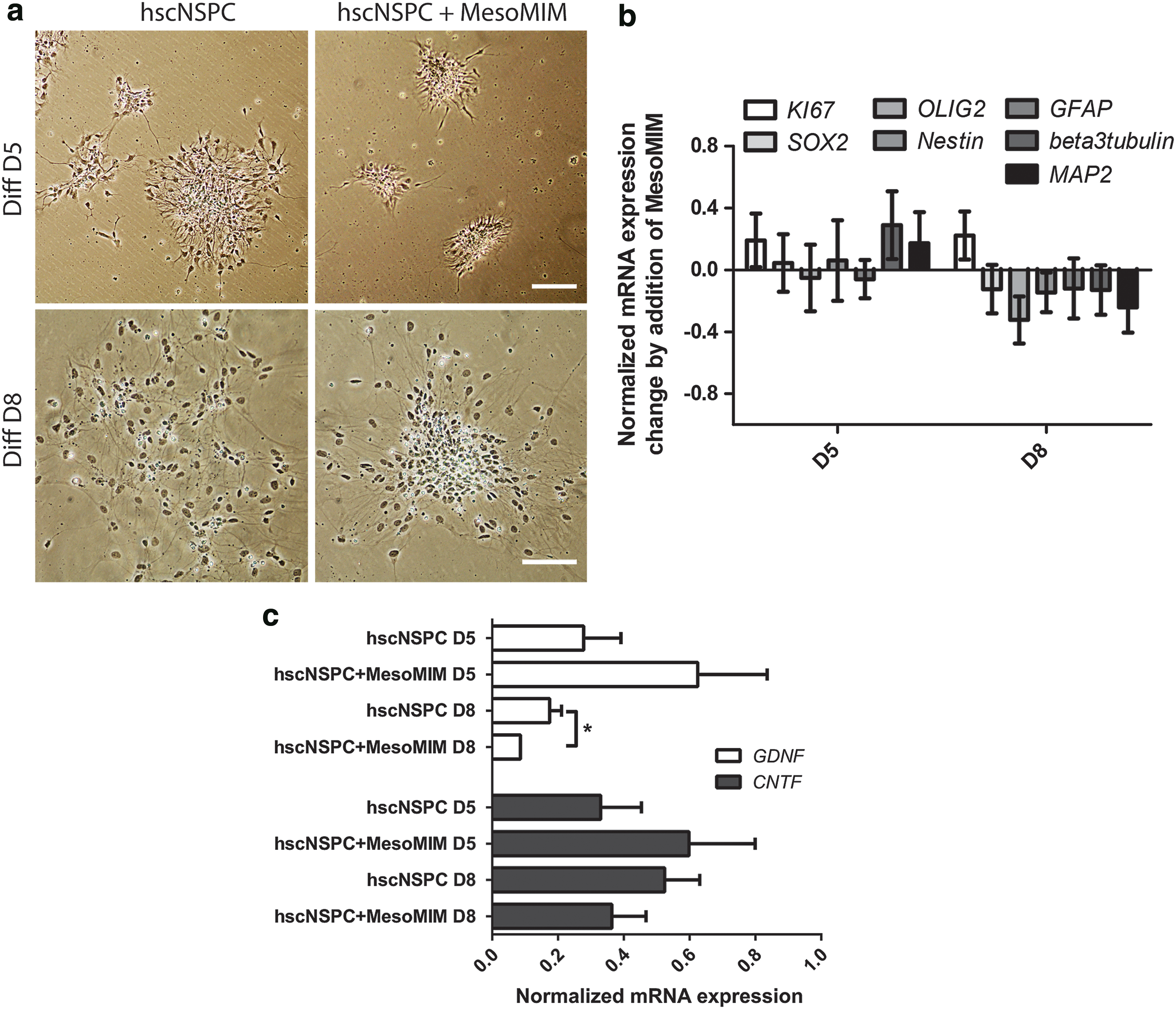
Mimetic-loaded silica particles led to diminished migration of hscNSPC and reduced expression of GDNF during in vitro differentiation of hscNSPC.
Transganglionic tracing of sensory fibers
One week after transplantation, some CTB-positive fibers were detected inside the spinal cord of animals that received hscNSPC transplants (n = 3) (Fig. 3a, asterisks, arrowheads indicate basal lamina outlined by expression of laminin) and HuNu+ cells were found inside the spinal cord as well as outside the basal lamina in proximity of the CTB+ dorsal root stump (Fig. 3a).
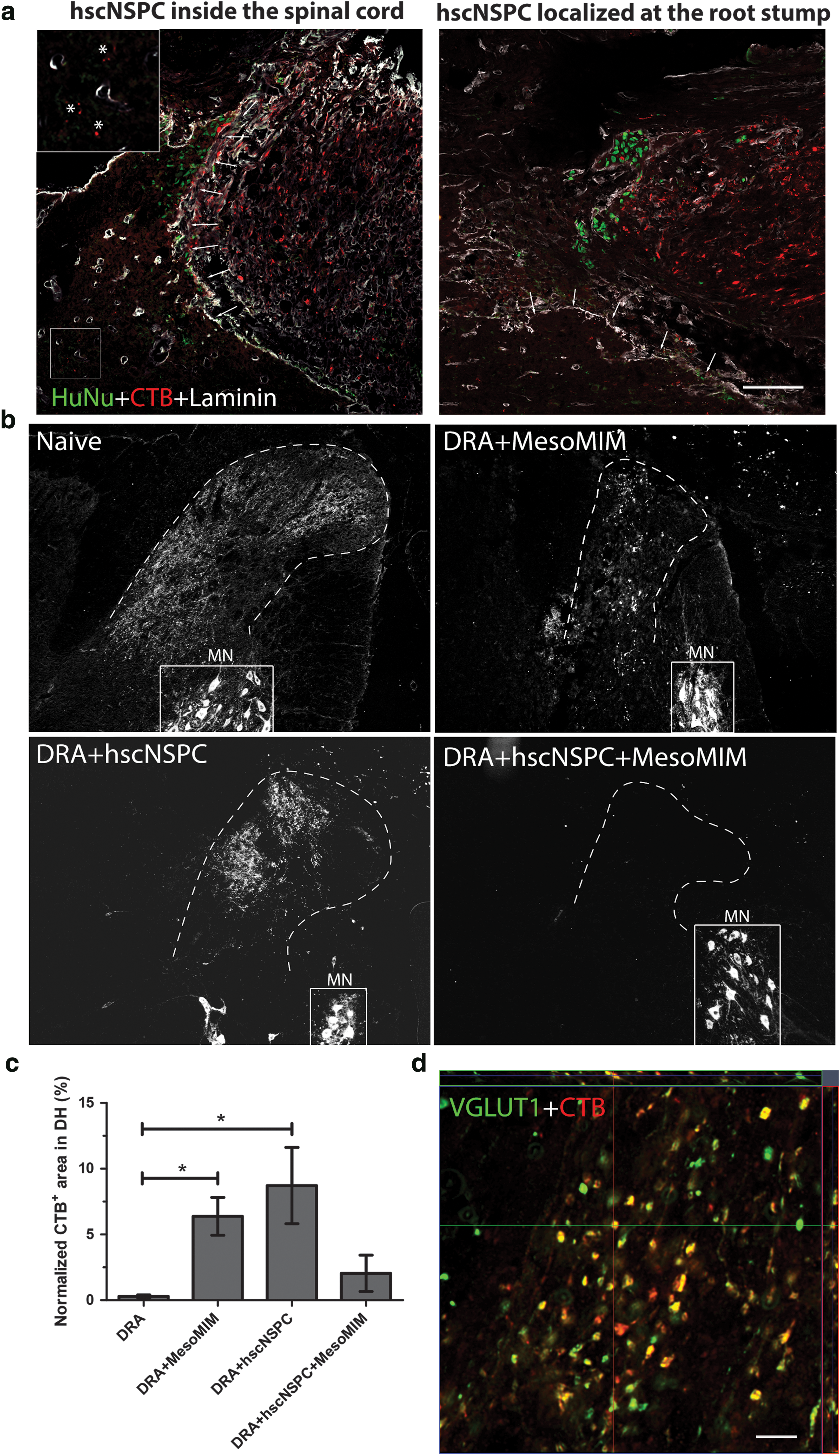
Implantation of mimetic-loaded silica particles alone and hscNSPC alone led to growth of CTB-labeled sensory fibers into the dorsal horn.
Two months after transplantation, CTB labeling was absent from the dorsal horn of animals after DRA (n = 3); animals treated with MesoMIM alone showed CTB labeling in the dorsal horn (n = 5). In animals receiving hscNSPC alone, CTB labeling was slightly more extensive (n = 4), whereas CTB labeling was absent from all animals except one receiving hscNSPC + MesoMIM (n = 4) (Fig. 3b). Ventral horn motor neuron pools were consistently labeled with CTB in all groups, demonstrating successful transport of injected CTB to the spinal cord (Fig. 3b). The CTB+ area in the dorsal horn was significantly increased in DRA + MesoMIM and DRA + hscNSPC groups when compared to DRA (P < 0.05, Fig. 3c). This indicates that both treatments, hscNSPC and MesoMIM, support sensory axon ingrowth into the injured spinal cord. CTB-labeled axons in the dorsal horn expressed the glutamatergic marker protein VGLUT1, in line with the dorsal root ganglion (DRG) origin of ingrowing axons [29].
Modification of the glial scar after hscNSPC transplantation
Injury to the spinal cord results in the formation of a nonpermissive glial scar composed of hypertrophic astrocytes and NG2+ glia [30]. Two months after DRA, the ipsilateral dorsal horn showed markedly increased GFAP immunoreactivity. Implantation of MesoMIM did not alter GFAP expression at the site of injury. In contrast, hscNSPC contributed to the GFAP immunoreactivity in the ipsilateral dorsal horn. Subtraction of the human GFAP signal from the total GFAP signal showed reduced murine GFAP (total GFAP–human GFAP) immunoreactivity in the hscNSPC group, but not in the hscNSPC + MesoMIM group (Fig. 4a).
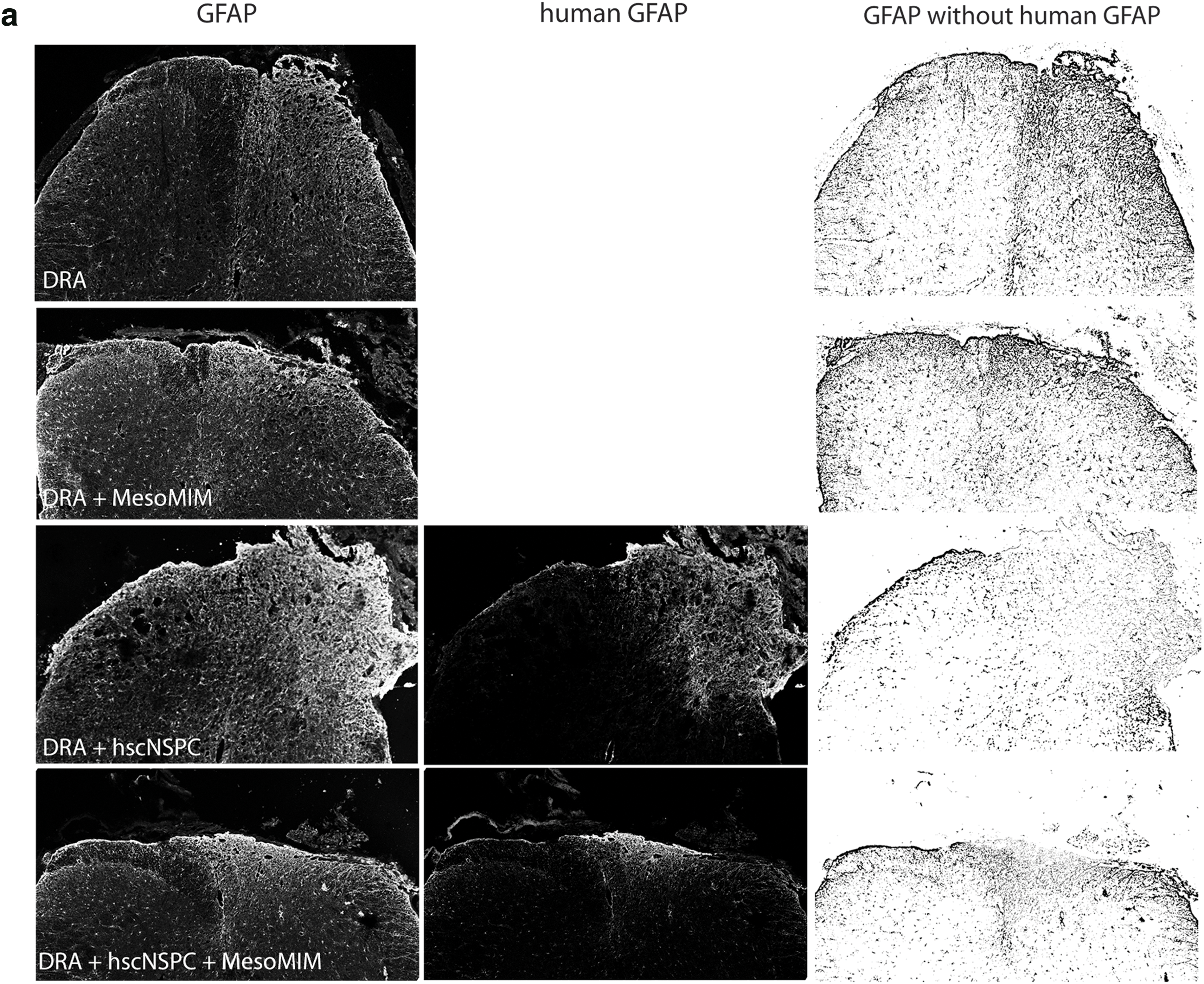
Transplantation of hscNSPC, but not mimetic-loaded silica particles reduced glial scar formation at the site of DRA.
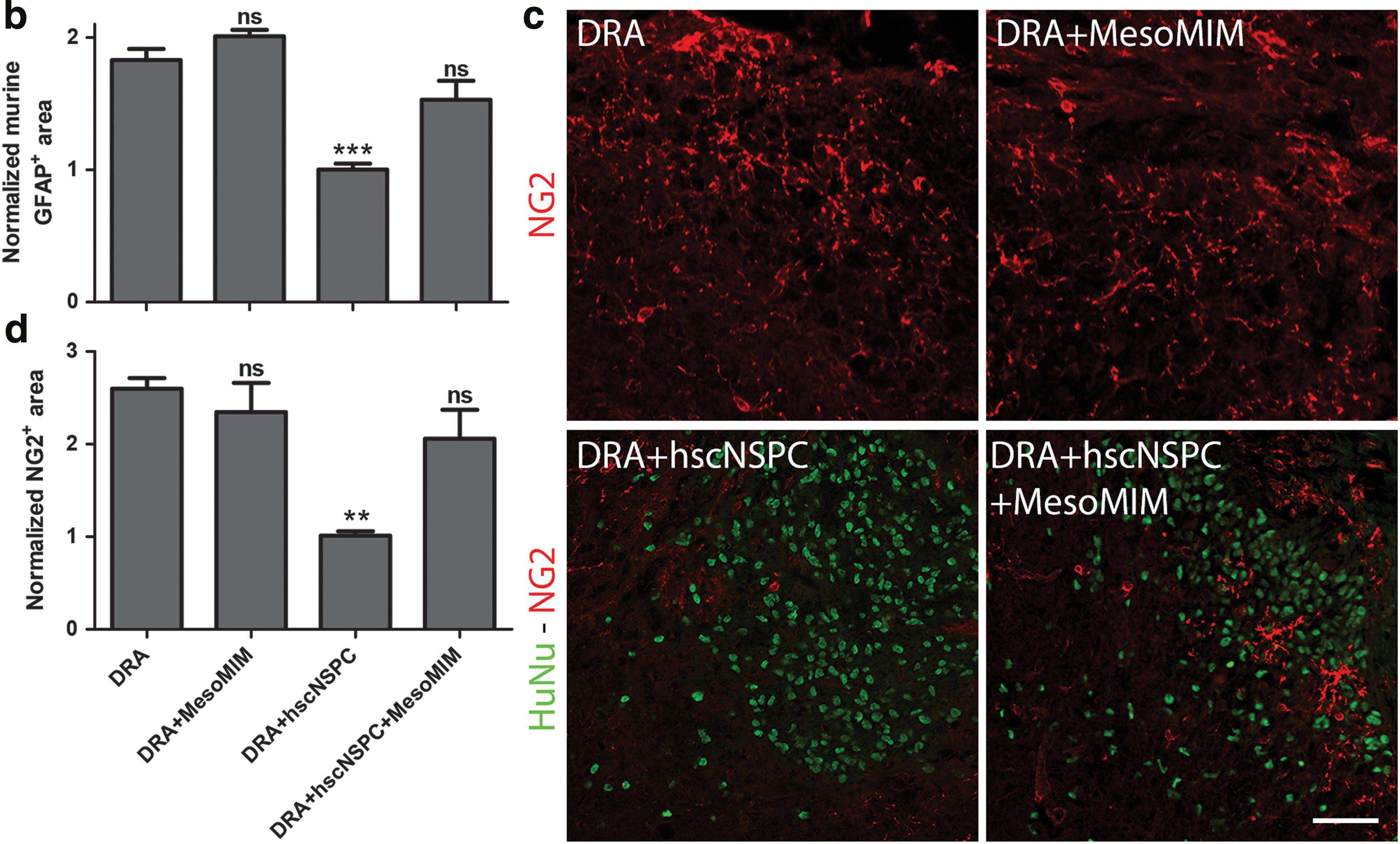
DRA increased the murine GFAP+ area by 1.8-fold compared to naive animals. Animals that received hscNSPC alone exhibited reduced murine GFAP immunoreactivity compared to DRA (P < 0.001) and were similar to the levels found in naive animals (naive 1 ± 0.08 and DRA + hscNSPC 1 ± 0.05, Fig. 4b).
DRA also led to an increase of NG2 at the site of injury, and no reduction of NG2 could be detected when MesoMIM or hscNSPC + MesoMIM was implanted (Fig. 4c). Only hscNSPC alone resulted in significantly lower levels of NG2+ immunoreactive area (P < 0.01), close to the level found in naive animals (naive 1 ± 0.16 and DRA + hscNSPC 1.01 ± 0.05, Fig. 4d). This indicates that engrafted hscNSPC reduced the accumulation of murine GFAP and NG2 at the site of injury, MesoMIM had no effect on glial scar formation, and this positive effect of hscNSPC was diminished by coimplantation with MesoMIM.
Discussion
DRA injuries remain a serious problem for affected patients. In this study, we show that transplantation of hscNSPC neurospheres derived from human fetal spinal cord assist the growth of host sensory axons into the spinal cord by providing a growth-permissive tissue bridge, similar to hESC-derived spinal cord progenitors [16]. Our findings also indicate that local delivery of peptide mimetics of GDNF (Gliafin) and CNTF (Cintrofin) using mesoporous silica particles (MesoMIM) assist growth of sensory fibers from the peripheral to the central nervous system after DRA, although less effectively. Interestingly, the combination of these treatments failed to support regeneration of sensory fibers. Our results and proposed underlying mechanisms for sensory regeneration are summarized in the scheme presented in Figure 5.
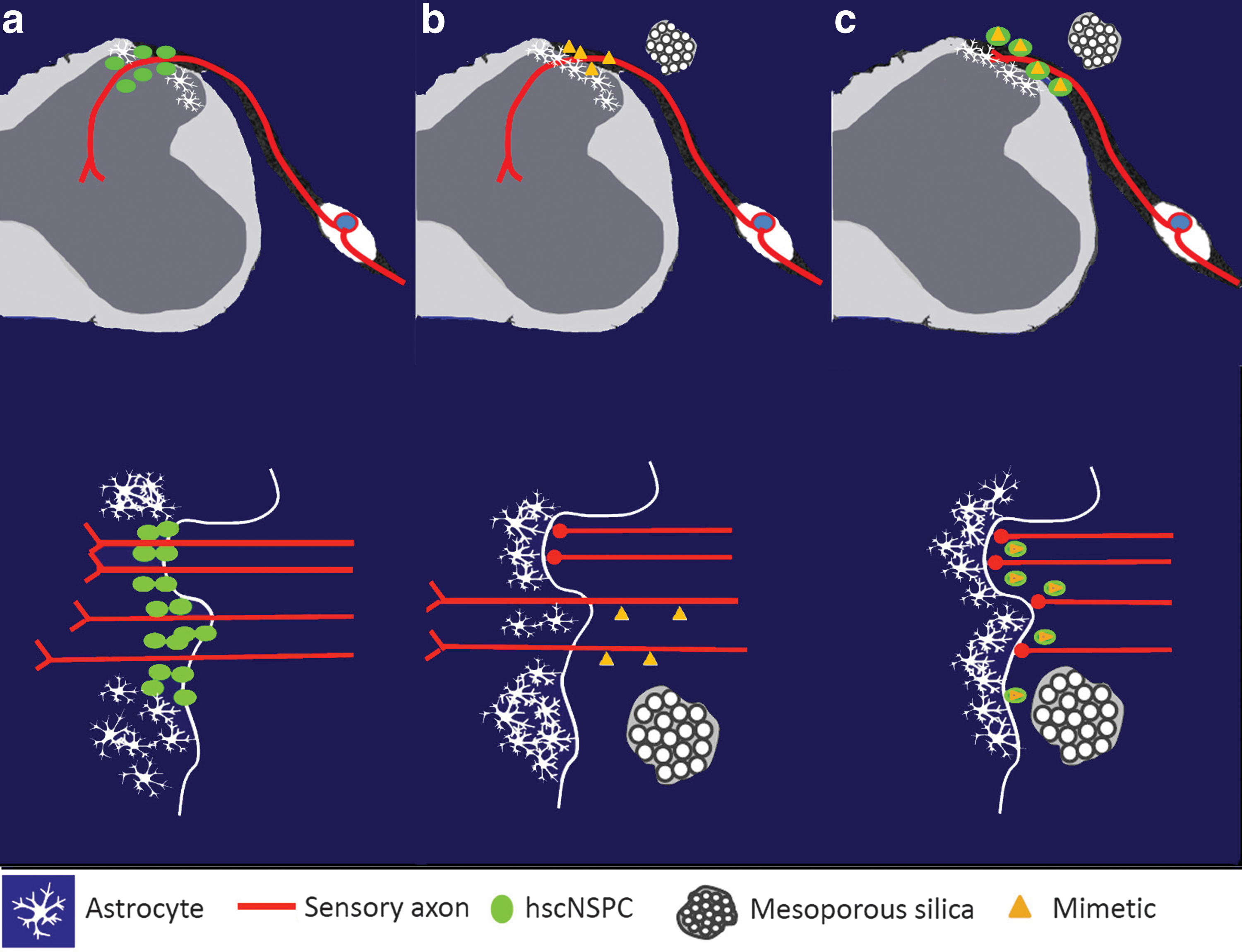
Scheme summarizing the results and proposed underlying mechanisms for sensory regeneration. The upper panel represents transverse sections through the spinal cord segment and the corresponding dorsal root ganglia. The lower panel represents the DRTZ area where the regenerating sensory axons enter the spinal cord.
Local or systemic administration of growth factors positively influences regeneration of sensory axons [12,31 –33]. The neurotrophin family member nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and NT-3 act on distinct subpopulations of dorsal root ganglion cells. Local administration of NGF or NT-3, but not BDNF, to crushed dorsal roots resulted in limited spinal cord ingrowth of subtypes of sensory axons [12,31 –33]. A similar application led to ingrowth of a broader population of sensory axons [12], and systemic treatment with the GDNF family member Artemin was shown to induce functional sensory regeneration into the spinal cord after a dorsal root crush [11]. The mechanism underlying this ingrowth appears to be an increased regenerative capacity of injured dorsal root ganglion cells rather than growth-permissive modifications at the DRTZ.
The cytokine CNTF is a major inducer of the gp130/JAK/STAT3 signaling pathway and an important part of the regenerative program in sensory neurons [17,18]. Therefore, we chose to study the effect of peptide mimetics for GDNF (Gliafin) and CNTF (Cintrofin) delivered by mesoporous silica. Not unexpectedly, acute treatment with these agents supported sensory fiber regeneration despite the fact that this treatment did not modify glial scar formation at the DRTZ. These findings suggest that mimetic-loaded silica particles allow sensory fibers to grow into the spinal cord before the emergence of a growth inhibiting glial scar [13]. Previous studies showed successful delivery of Gliafin and Cintrofin in vivo to coimplanted stem cells, induced differentiation, and increased axonal growth toward MesoMIM [20].
Despite the less efficient effect on sensory axon regeneration by MesoMIM compared to the extent of ingrowth observed in animals receiving hscNSPC transplants, our experiments demonstrate that mesoporous silica has the potential to deliver neuroregenerative agents in DRA and spinal cord injury. Mesoporous silica is applied locally, minimizing the off-target effects on uninjured tissue, and can be designed for the release of desired agents and defined concentrations for a predetermined period of time [34]. Novel trophic factor mimetics for other promising neurotrophic factors like Artemin or NT-3 and/or the use of retinoic acid receptor agonist, which was shown to promote sensory regeneration and to reduce astrogliosis in DRA [35], offer opportunities for improved therapeutic efficacy.
Similar to human spinal cord progenitors derived from hESC [16], hscNSPC also formed a cellular bridge across the dorsal root–spinal cord interface and markedly attenuated the injury-induced glial scar. This raises the questions which specific cell types and/or which stage of differentiation of the implanted cells may be beneficial for sensory axon regeneration. Olfactory ensheathing cells and immature astrocytes implanted to the injured spinal cord also support regeneration of sensory axons through the DRTZ [15,36]. While these outcomes were achieved in less severe injury models, it shows the ability of immature glial cells to interact and intermingle with host astrocytes, suggesting that this modification in glial scar formation is necessary for successful sensory ingrowth.
Transplanted human spinal cord progenitors are able to migrate over long distances inside the spinal cord and replace host astrocytes [37]. Compared to untreated hscNSPC transplants, the MesoMIM-treated transplants showed a significant reduction of migration and an inability to form a bridge at the DRTZ permissive for axonal growth. In vitro studies confirmed that treatment with MesoMIM reduces stem cell migration and their expression of GDNF. The ability of neural stem/progenitor cells to secrete neurotrophic factors [38 –40] is believed to contribute to the stimulation of axonal outgrowth following spinal cord injury [41 –44]. Reduced neurotrophic factor secretion might, thus, contribute to the reduced regenerative potential of hscNSPC when coimplanted with MesoMIM.
In our previous studies with mouse ESC-derived neural progenitors, MesoMIM strongly increased survival and induced rapid differentiation of the transplanted cells [20]. In this study, we did not detect significant differences in the expression pattern of a panel of neural cell differentiation markers between MesoMIM-treated and MesoMIM-untreated hscNSPC transplants. These data suggest that the inability of hscNSPC to migrate and form a growth-permissive bridge is not the result of enhanced stem cell differentiation in the MesoMIM-treated transplants, but primarily a consequence of impaired migratory capacity.
Transplanted neural stem cells are characterized by a remarkable ability to migrate toward degenerating CNS regions and have the potential to contribute to the development of new therapeutic approaches [45 –49]. Targeted migration of neural stem cells is primarily dependent on cues provided by the lesion [45,47,50,51], suggesting that locally released trophic factor mimetics created an attractive microenvironment for transplanted hscNSPC, as was previously shown for MesoMIM -treated mouse ESC-derived transplants [20]. This attractive local microenvironment “trapped” the hscNSPC at the transplantation site and prevented them from forming a growth-permissive bridge.
For the regenerating fibers, however, the establishment of functional connections seems to be the prioritized physiological goal. The release of trophic factors by MesoMIM, when implanted alone, serves as a cue to facilitate this intrinsic/cell-autonomous axonal growth toward spinal cord targets. When cotransplanted, hscNSPC are trapped by MesoMIM and thereby deplete the trophic factor pool. As a consequence, any subsequent sensory axon regeneration fails.
Footnotes
Acknowledgments
We gratefully acknowledge the support with cell cultures and immunohistochemistry by Svitlana Vasylovska and qPCR by Tanya Aggarwal and Mikaela Eriksson. The study was supported by the Swedish Research Council (Project No 20716), Stiftelsen Olle Engkvist Byggmästare and Signhild Engkvist's Stiftelse.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.