
Abstract
Mural cells are indispensable for the development and maintenance of healthy mature vasculature, valuable for vascular therapies and as developmental models. However, their functional plasticity, developmental diversity, and multitude of differentiation pathways complicate in vitro generation. Fortunately, there is a vast pool of untapped knowledge from in vivo studies that can guide in vitro engineering. This review highlights the in vivo genesis of mural cells from progenitor populations to recruitment pathways to maturation and identity with an emphasis on how this knowledge is applicable to in vitro models of stem cell differentiation.
Introduction
T
Due to their functional diversity, mural cells are important cells in a number of diseases and therapeutic applications. In vascular therapies, implanted mural cells can be used to enhance nascent vasculature for the repair of damaged, ischemic tissues [6]. For cellularized vascular grafts, the added smooth muscle layers can provide structure, contractility, and improved recovery [7]. Moreover, the phenotypic modulation of VSMC is often studied for its implications and potential therapies in vascular disease [5]. Mural cells are also being targeted to control the degree of vascularization in tumors [8].
Pluripotent stem cells (PSC) can provide an alternate approach to the application and study of mural cells. An often limiting factor of cell therapies is the lack of a suitable cell source, but unlike many terminally differentiated adult cells, PSC are renewable and can be guided to differentiate into any cell lineage in the body. Moreover, these PSC-derived cells are less senescent and can be superior at promoting regeneration than their in vivo counterparts [9]. With the discovery of induced PSC, PSC can now be derived from otherwise terminally differentiated adult cells [10]. Furthermore, PSC can be used as in vitro models of development. By reverse engineering the natural embryonic microenvironment, developmental pathways can be isolated from an otherwise complex system. Interestingly, many VSMC developmental pathways overlap with phenotypic modulation pathways, allowing for implications in vascular disease [5].
However, generating PSC-derived mural cells is complicated by several factors, including the following: (1) the multiple origins of mural cells, with correspondingly distinct functions, provide confounding development paths, (2) a wide range of microenvironment factors that can contribute to the development of mural cells with different combinations of factors yielding different resulting cells, (3) a lack of lineage-specific surface marker sets for either PC or VSMC, and (4) the poorly defined PC, which are difficult to identify without a physiological location.
Fortunately, there is a large amount of information from in vivo studies of embryonic origins and adult vascular progenitors, especially during embryonic development. Furthermore, studies have identified many microenvironment factors that contribute to mural development in vivo, and the maturation process has been extensively characterized throughout embryogenesis. This review will highlight information from in vivo studies that can be used toward developing better in vitro developmental models.
Origins of Mural Cells
As with all cells of the body, mural cells arise from the developing embryo where they must navigate a labyrinth of developmental paths before reaching their final destination. However, our understanding of these paths is complicated by the multiple embryonic origins of VSMC and PC, wherein fate decisions are tied to the cells’ ultimate anatomic location and function.
Embryonic origins of mural cells
The corresponding in vitro and in vivo development of mural cells begin with the embryoblast, an inner cell mass from which embryonic stem cells (ESC) are isolated. The embryoblast reorganizes into an epiblast, which derives the primitive streak (PS), a ridge-like formation that divides the organism bilaterally. Mesendoderm cells of the epiblast migrate through the anterior PS and replace hypoblast cells at the bottom of the developing embryo, forming the definitive endoderm. Migratory mesendoderm at the anterior PS also occupies between the epiblast and definitive endoderm to form the mesoderm. The remaining, nonmigratory epiblast cells default to the final germ layer, the ectoderm [11]. This process by which the epiblast gives rise to the three distinct germ layers is known as gastrulation [12]. Of these three germ layers, VSMC are produced from specific regions within both the mesoderm and the ectoderm [13].
The mesoderm spreads laterally across the embryo outward from the PS into the axial, paraxial mesoderm (PM), intermediate, and lateral plate mesoderm (LPM) regions. The mesoderm regions are specified by a bone morphogenetic protein (BMP)-4 gradient, which is most concentrated at the LPM [14]. As the organism matures, the PM forms into blocks of somite cells that line the embryo bilaterally and develop into bone and skeletal and smooth muscle, as well as various mesenchymal tissues. Meanwhile, the LPM further segregates into the cardiac mesoderm (CM), from which most heart tissue is derived, and the hematopoietic mesoderm, which gives rise to most of the vasculature. Within the LPM is a specific subset of cells that express CD309, an iconic marker for vascular progenitors that derives both endothelial cells (EC) and VSMC in vitro [15]. Within the CM, the low-concentration end of a bone morphogenetic protein-2 (BMP-2) gradient forms the proepicardium, a mesothelial lining in the heart. The concentrated side of the BMP-2 gradient transforms into the collective of cardiac and smooth muscle progenitors known as the secondary heart field [16]. All the while, the ectoderm layer initially forms two regions, the neural region and the nonneural region. Cells from the neural plate of the neural region delaminate from the neural tube and form a third region: the neural crest (NC) [17]. A BMP-2 gradient specifies these three regions with the lowest concentration at the neural plate and the highest at the nonneural regions [18]. The NC, often called the fourth germ layer for its broad multipotential, goes on to generate a wide variety of cell types, including VSMC.
Summarized in Fig. 1, the in vivo specification of these developmental regions is largely controlled by BMP, Wnt, and fibroblast growth factors (FGF), as well as transforming growth factors (TGF) that activate Activin- and Nodal-related signaling pathways. These signals often form gradients that diffuse across the developing embryo [14,16,18]. Therefore, many in vitro models reflect these in vivo studies by activating the same major signaling pathways and even mimicking ligand concentrations [19]. Because PSC tend to differentiate spontaneously and randomly, these in vitro models aimed toward lineage specification are often supplemented with inhibitors to derive pure populations of the desired cells. Of note, cell fate pathways may differ between PSC types even under the same differentiation conditions as demonstrated by in vitro specifications between human and mouse ESC (mESC) [20].
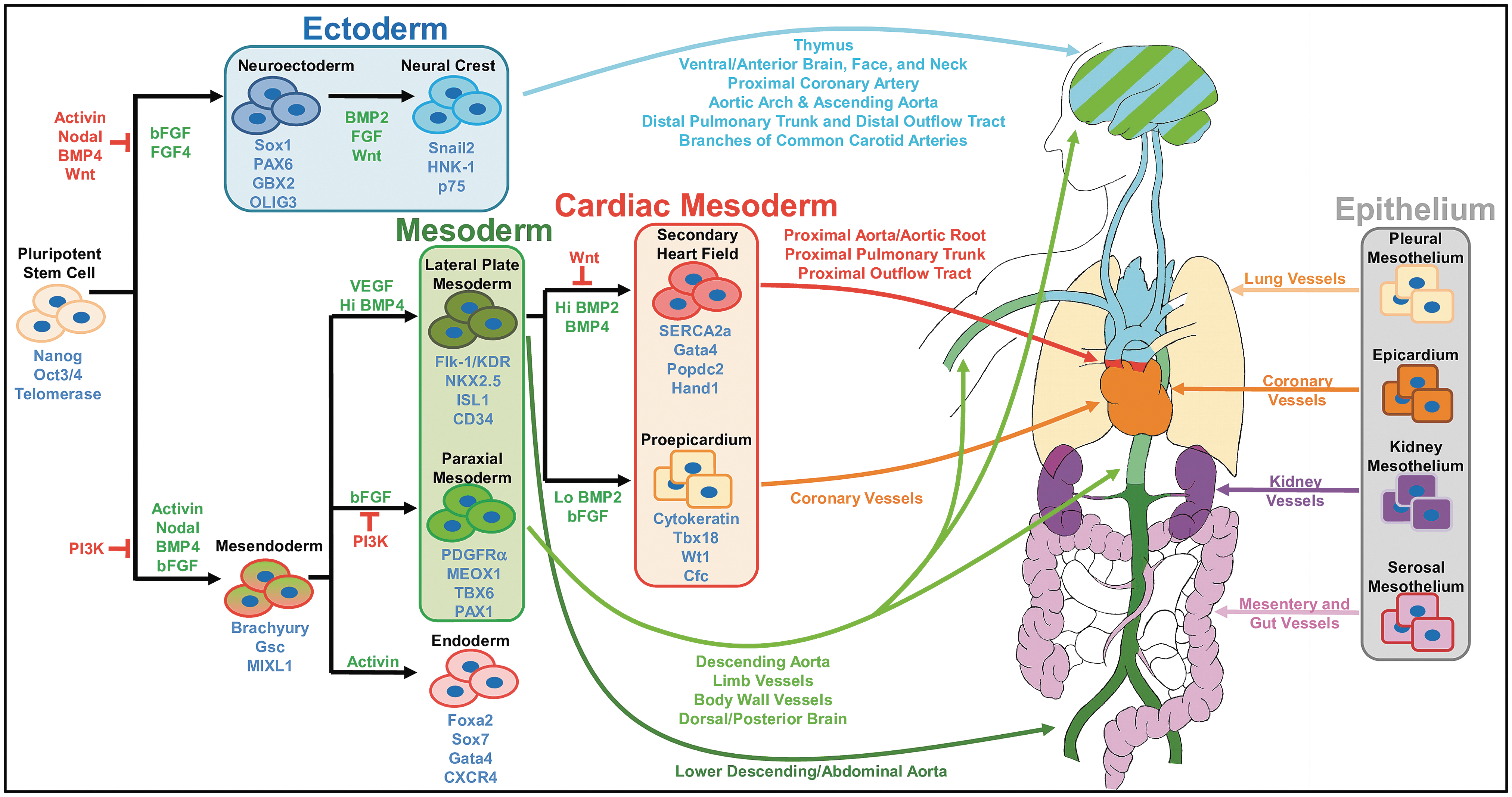
Heterogeneity of mural cell development. Stem and progenitor cells cycle through a number of developmental stages before ultimately committing to a mural cell fate. Moreover, the specific path a cell undertakes will determine its final anatomic location and specific function. The signaling pathways depicted in this study are a culmination of information from various chemically defined in vitro differentiation schemes and where data were lacking, from in vivo and ex vivo studies. However, not all steps in embryonic development have been recapitulated in vivo and in vitro, nor have all known mural cell subphenotypes been generated using in vitro differentiation methods.
Many embryonic populations are actually heterogeneous sets of precursors. For example, the LPM includes a variety of cardiac and hematopoietic progenitors with different multipotential [21]. As a result, some protocols derive embryonic progenitors into these more specific subtypes. For example, PSC differentiation protocols have produced more specialized subregions of germ layers, such as the well-studied neuroectoderm [22]. Furthermore, in vivo progenitors do not arise directly from PSC, but rather mature through many branches of a progenitor tree. As such, some protocols aim for faithful recapitulation, as exampled by the stepwise generation of the mesendoderm, the in vitro analog to the PS [20]. Thus far, chemically defined derivation protocols exist for the generation of not only all three germ layers but also a variety of more specific embryonic intermediates.
As previously reviewed by Majesky [13], VSMC arise from a number of embryonic sites (Fig. 1), including the following: the NC of the ectoderm, the LPM, and the somites of the PM. In general, mural cells located above the heart originate from the NC. NC-originating mural cells have been found in the perivascular regions starting from the distal pulmonary trunk and outflow tracts up to the neck, face, and ventral/anterior brain [23 –26]. Similarly, the aorta, which partially loops above the heart, is invested with NC-originating VSMC at the aortic arch, ascending aorta, and branching arteries, including the carotid arteries and right subclavian artery [23,26]. However, mural cells of the dorsal/posterior brain derive from the PM [26]. VSMC also arise from the LPM-derived secondary heart field and proepicardium. They are situated at the heart level and include the coronary arteries and veins, the proximal pulmonary trunk and outflow tract, aortic root [24,25,27 –30], and abdominal aorta [30,31], although some NC-derived VSMC have also been found in the proximal coronary artery [23]. Last, PM-derived VSMC typically constitute regions most distal from the heart, such as the vessels in the limbs, body walls, and upper and middle descending aortas [30,32,33].
In addition to these distinct developmental regions, VSMC can also arise from the mesothelial lining of their respective organs (Fig. 1). So far, it has been shown that the serosal mesothelium develops into VSMC of the mesentery and gut, the pleural mesothelium into lung VSMC, the kidney mesothelium into kidney VSMC [4], and the LPM-derived proepicardium into coronary artery VSMC [27,29,34,35]. Moreover, locations in the developing embryo may be transiently invested with VSMC of one origin before quickly being replaced by another. Specifically, LPM-derived VSMC of the developing mouse and chick constitute the descending aorta before being displaced by PM-derived VSMC, which then remain throughout maturity [30,32].
Distinct from the VSMC, PC are mural cells often found lining microvessels, but like VSMC, PC possess origin-specific heterogeneity. Although PC ontogeny is less clear [36], evidence implies that PC and VSMC arise from the same embryonic origins. Both aortic VSMC and PC develop from the somites of the PM [33], while both mural cell types of the brain and face have been found to originate from the NC [26,37]. Further implicating common origins, immunohistochemistry of murine retinas suggest that VSMC and PC may share a common progenitor that coexpresses VSMC and PC markers [38,39]. In fact, it is highly probable that PC themselves are progenitors to VSMC [4,40].
Developmental diversity not only contributes to different functions at maturity but also determines the pathway for differentiation and recruitment for mural cells. For example, the NC, but not the LPM or PM, requires the transcriptional coactivator MKL2 for differentiation into VSMC [19]. Also, the NC mural progenitors’ TGF-β differentiation pathway relies primarily on the Smad2- and myocardin (MYOCD)-related transcription factor-B as opposed to PM progenitors, which use Smad3 and MYOCD for mural differentiation [41,42]. Furthermore, while the platelet-derived growth factor (PDGF) pathway is known to be crucial for PC recruitment, it is not required for PC of the liver and thymus [43,44].
Mural cell sources in adult organisms
Mural cell progenitors are found not only in the developing embryo but also within adult tissues (summarized in Table 1). Approximately, one tenth of the postdevelopment perivasculature consists of cells that express progenitor markers such as stem cell antigen-1 (Sca-1), c-kit, CD34, and CD309 [45,46]. In vivo, these cells are held undifferentiated within progenitor niches, while mature cells reside around them. These progenitor reservoirs become active and serve as replenishable cell sources during tissue disease and wound healing and, as such, are studied for their potential in cellular therapies. It is speculated that adult progenitors are embryonic precursors held back in progenitor niches [40] and presumably lie in between an embryonic progenitor and a mural cell along the developmental timeline. Therefore, in vitro analogs of mural progenitors, which generate mural cells through PSC-derived analogs of adult progenitor intermediates, may serve as an additional checkpoint for further purification and verification of the developmental path.
Mural progenitor cells can arise from a number of different sources in the adult body and possess a wide variety of properties regarding surface marker expression, cell function, and differentiation potential. This table summarizes the source, characterization, and multipotency of the various adult mural and mural progenitor cells.
αSMA, alpha smooth muscle actin; CIV, collagen type IV; EC, endothelial cell; FGF, fibroblast growth factor; FN, fibronectins; MSC, mesenchymal stem cells; MVSC, multipotent vascular stem cell; PC, pericytes; PDGF, platelet-derived growth factor; Sca-1, stem cell antigen-1; SHH, sonic hedgehog; VSMCs, vascular smooth muscle cells; vWF, Von Willebrand factor.
Among the most well-studied SMC progenitors are mesenchymal stem cells (MSC, sometimes referred to as mesenchymal stromal cells). MSC are found in many tissues in the body and exhibit multipotential for many different cell types, including nonmesenchymal lineages. The MSC is strictly defined by a set of protein markers and multipotential ability [48]. Although SMC potential is not in the MSC's minimal criteria, several studies have shown the ability of MSC to derive SMC [60]. In addition, MSC are known to secrete angiogenic signals that promote vessel formation [61]. So far, there have been several published protocols for generating MSC from PSC [62] and contractile SMC have been generated from a PSC-MSC intermediate [63]. Moreover, it has been shown that MSC derived from PSC are less senescent and more effective at promoting regeneration than their in vivo counterparts [9].
While VSMC and PC are both classified as mural cells that stabilize blood vessels, there is strong evidence that PC can differentiate into VSMC [4,40,50]. It has been known for some time that PC share many markers and multipotential ability with MSC, a known progenitor of SMC [50]. PC also share some markers, possess similar developmental origins, and show similar developmental dynamics compared with VSMC [38], hinting that plasticity among mural cells is likely. Only recently has Volz et al., demonstrated that PC are an intermediate population in the in vivo epithelial–mesenchymal transition of heart epicardium to VSMC [4]. While Özen et al., agree that PC are multipotent in vivo [64], Guimarães-Camboa et al., demonstrate that PC do not act as stem cells [49] even though they may be multipotent in vitro [50]. The discrepancy between these studies hints at a heterogeneous nature of PC that can further complicate mural cell identity and their in vivo versus in vitro behavior. So far, PC have been generated from PSC as a minority population in mesoderm EC protocols where PC are purified by negative sorts of early EC CD31+ cells [65 –67].
A third well-studied mural progenitor is the Sca-1+ progenitor. The Sca-1+ label actually represents a heterogeneous population of progenitor cells that are scattered throughout body and possess multipotential for a variety of tissue [51,68]. Sca-1+ progenitors are known progenitors for VSMC [52,69] and contribute to atherosclerotic lesions during vascular disease [46]. Sca-1+ cells have been generated from PSC and further differentiated into SMC [69]. EC have also been implicated as a potential source of VSMC by endothelial-to-mesenchymal transitions (EndMT).
In vivo, EndMT is the process where EC migrate into the surrounding mesenchyme and acquire fibroblastic characteristics, a process particularly prevalent in fibrosis [70]. In vitro, EndMT can be induced in CD31+ ECs with factors such as TGF-β1, where the resulting transdifferentiated EC will lose EC functions and gain VSMC contractile markers such as alpha smooth muscle actin (αSMA) and smooth muscle protein 22-alpha (SM22α) [71,72]. Similarly, the coexpression of endothelial markers and αSMA was detected in a subpopulation of cells in frozen sections of aortic valves, suggesting the transdifferentiation may occur in vivo. However, no study yet has induced the contractile VSMC marker smooth muscle myosin heavy chain (SMMHC) from EndMT EC.
Aside from these well-studied mural progenitors, there are a many more cells likely to have, but are yet without, PSC-derived analogs. Indeed, generation of progenitors from PSC is a difficult task given that progenitor cells usually do not have a single, distinct protein marker. The markers that do identify these cells are often nonspecific and immature (Table 1). Furthermore, many presumably distinct progenitor marker sets overlap with the profiles of others and cells can change their profiles between the different in vivo and in vitro environments. As such, multiple markers are needed to identify these cells, and it is not an uncommon strategy to screen for a large panel of markers when characterizing or searching for new progenitors.
The generation of mural progenitors is further complicated by the cells’ spontaneous differentiation in unoptimized, especially serum-containing, culture media. Therefore, it is difficult to capture these cells in their progenitor form. In vivo, these cells exist within progenitor niches that prevent cell differentiation. For example, some Sca-1+ progenitors lie within the blood vessel adventitia where an abundance of sonic hedgehog (SHH) signaling presumably silences transcription factors of VSMC differentiation [51]. Similarly, multipotent vascular stem cells (MVSC) experience a drastic change in marker expression when exposed to conditions of vascular disease or in vitro culture and are further differentiated when exposed to serum culture [55]. Specialized stem cell media may, at least, partially recapitulate the factors within a progenitor niche, and commercially available progenitor media have been developed for maintenance of these fickle cells. For instance, MVSC can remain undifferentiated in neural stem cell media [55], saphenous vein progenitors in EC media [45], and vascular wall-resident multipotent stem cells in MSC media [54].
Function is related to origin
In normal vasculature, the core functions of VSMC of different origins are still the same, providing stability and contractile function. However, there are nuances in their signaling and response that can differ between VSMC of different origin. In vivo, angiotensin II (AngII) induces hyperplasia in the ascending aorta (NC origin), but induces hypertrophy in the descending aorta (PM and LPM origin) [73]. In vitro, TGF-β1 is able to induce proliferation, a hallmark function of dedifferentiated VSMC in diseased vasculature, in NC-VSMC, but inhibits said response in LPM or PM-VSMC [31,74]. Cheung et al. confirmed these origin-specific responses by demonstrating that PSC-derived NC-VSMC are more proliferative in response to TGF-β1 and AngII compared to PSC-VSMC of PM or LPM origin [19]. Furthermore, they showed that LPM-VSMC are more migratory, PM-VSMC were more likely to contract in response to vasoactive agonists, and PM, LPM, and NC-VSMC had differential expressions of MMPs and their TIMPs in response to the inflammatory cytokine interleukin 1 (IL-1). This demonstrates that the mural cell's origin-based heterogeneity is expressed in their PSC-derived analogs.
The downstream functions of VSMC from different origins also play an especially impactful role in vascular disease. During the onset of atherosclerosis, VSMC make significant contributions to atherosclerotic lesions during vessel injury, where they lose contractility and gain fibroblastic properties in response to signals such as IL-1 [75]. Indeed, sections of vessels containing VSMC of different origins are variably susceptible to atherosclerosis and experience different rates of disease progression and recurrence [76]. For example, studies on mice explants under hyperphosphatemic conditions and live mutant mice lacking a calcification inhibitor showed that the aortic arch, which contains NC-VSMC, is more prone to calcification than the mesoderm-derived descending aorta [77]. Furthermore, in canine studies where atherogenesis-prone vessels were grafted into normally resistant areas, the grafted region remained disease prone [78]. This indicates that diversity in disease response is a result of inherent differences in the vessels itself and not the environment of a different anatomic location.
While PC are primarily known for stabilizing microvasculature, they also possess specialized functions specific to their residing anatomy [36]. Brain PC play a role in the formation of the blood–brain barrier (BBB) and continue to stabilize it through interactions with astrocytes and regulation of BBB-specific genes [79]. Brain PC also exhibit macrophage-like behavior and even possess macrophage markers [80]. In the kidney, a specialized form of microvascular PC, known as the mesangial cells, exhibit specialized contractile function for controlling capillary flow and glomerular filtration in concert with podocytes [81]. Finally, specialized liver PC, hepatic stellate cells, hold a large reservoir of vitamin A, and like VSMC, dedifferentiate into a fibroblastic phenotype in pathology [82].
Considering origin heterogeneity in vitro
Taken together, these studies demonstrate that embryonic origins significantly shape mural cell behaviors such as organ-specific functions, response to pathology, as well as, signaling and recruitment requirements. While in vitro models have only generated PC from the LPM, VSMC have been generated from the NC, LPM, PM, and CM, and display a diversity of properties that match their in vivo counterparts [19,83].
Like their embryonic counterparts, adult mural progenitors may also convey their origin-specific heterogeneity to their mural derivatives. Furthermore, adult progenitors serve as good examples of intermediates present in the later stages of in vitro development. As such, these intermediates can be used as additional checkpoints for characterization and purification of PSC-derived mural cells.
To obtain functions that most accurately represent the desired organ-specific VSMC, it is suggested that the in vitro differentiation strategies used for generating mural cells should recapitulate, as closely as possible, their embryonic progenitor origins. This is especially true with clinical applications given the connections between VSMC origins and disease susceptibility. However, this is complicated by the fact that each mural cell may possess cells from multiple origins. Indeed, there are many subtle paths, intermediate steps, and signaling requirements in the developmental tree that have yet been thoroughly characterized and recapitulated.
Signaling Pathways Directing Mural Fate
Commitment to a mural precursor only brings the cell halfway toward its ultimate identity. Before mural cells can be found in the developing embryo, mural precursors can be identified as PDGF receptor-β (PDGFR-β)-positive cells scattered around the developing vessel mesenchyme [43,84]. These cells are recruited into the first layer of mural cells by a number of soluble, cell–cell, and cell–matrix signals [85]. Likewise, adult mural precursors will differentiate when their precursor niche is altered by disease states.
The many in vivo signals known to be critical to mural recruitment can be used to direct mural fate in vitro (Fig. 2). Briefly, the soluble signals PDGF-ββ and TGF-β1 are the most well known for in vivo and in vitro mural development. However, less studied factors such as sphingolipids, cell–cell contact signals, retinoids, mechanical forces, and ECM have also been shown to be necessary in mural fate.
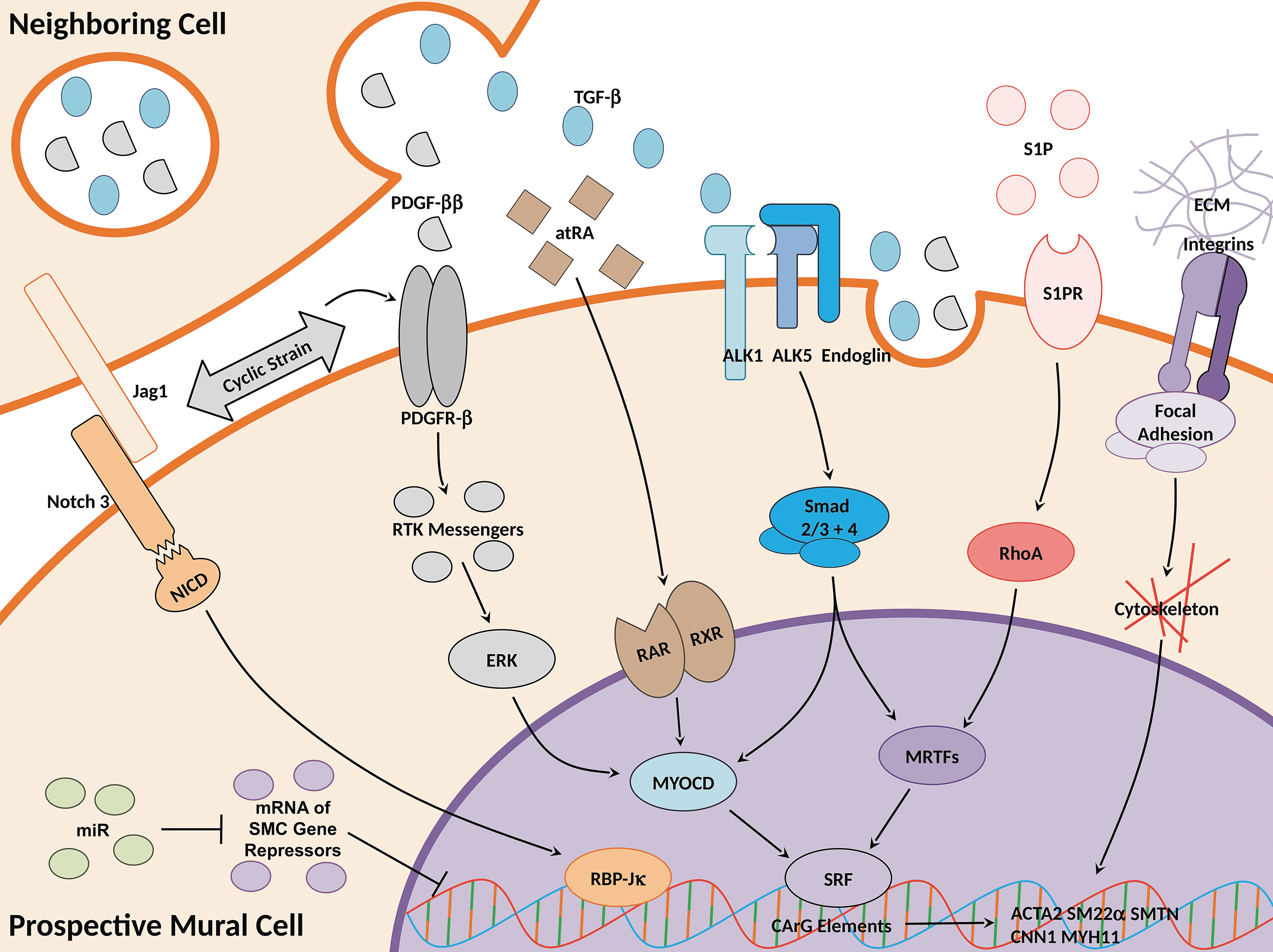
Signaling pathways for mural differentiation. Mural development occurs through the combinatorial effects of multiple signaling pathways. Shown in this study is a simplified depiction of the major pathways and their key ligands, receptors, intermediates, and transcriptional regulators necessary for mural differentiation.
Mural cell signaling pathways have been shown to direct mural differentiation from many progenitor types of different species and origins. Therefore, many mural differentiation signaling pathways may be universal across different mural progenitors. Furthermore, many of these developmental pathways are retained in mature VSMC and continue to upregulate many mural-specific contractile markers in adult cells (a notable exception is PDGF-ββ, which negatively regulates mature VSMC markers [5]). As such, signaling studies for VSMC development may extrapolate toward signaling studies for VSMC phenotype modulation and vice versa.
Transcriptional regulation
Transcription factor serum response factor (SRF) is critical for epigenetic and transcriptional regulation of SMC genes. SRF cofactors MYOCD and the two isoforms of MYOCD-related transcription factor (MRTF-A/B) have been found downstream of many major mural differentiation pathways such as PDGF-β [86], TGF-β [41,87], and sphingosine-1-phosphotase (S1P) [86,88]. When activated by its cofactors, SRF binds to CArG elements within many of the genes encoding SMC contractile markers [89]. As such, many studies use SRF, MYOCD, and MRTF as markers of SMC differentiation and regard them as transcriptional switches of SMC fate. To note, SRF, MYOCD, and MRTF are not SMC specific, but regulate the expression of cardiac and neuronal genes [90,91]. Furthermore, these transcriptional elements do not regulate certain SMC genes such as smoothelin-B (SMTNB) [92]. To the best of our knowledge, SRF, MYOCD, and MRTF have not yet been directly implicated in PC development.
SMC development is significantly reduced without these transcriptional elements as demonstrated in vivo by MYOCD knockout mutant mice [93], as well as in vitro by dominant-negative MYOCD and SRF SMC precursors [92,94]. However, mutant mice showed that VSMC may still form, although fewer in number, in the absence of MYOCD [95]. In vitro, overexpression of MYOCD in PSC induces the expression of multiple SMC genes, although cardiac genes were also expressed [92]. Similarly, knockout of MRTF-B in vivo [96,97] and MRTF-A attenuation of an in vitro MSC/S1P model inhibited VSMC development [98].
Furthermore, SMC transcriptional elements are regulated, in part, by epigenetic regulators such as histone deacetylase (HDAC) (reviewed in detail by Alexander and Owens [99]). These epigenetic regulators modulate the binding of transcriptional activators such as SRF [100] and MYOCD [101]. Although most evidence for the significant role of epigenetics in SMC gene expression comes from studies of SMC phenotypic modulation, there is also evidence that epigenetics play an important role in differentiation. Mutant mouse vessels lacking HDAC3 had decreased SMC marker expression, while HDAC3-negative explants of NC cells could not differentiate into SMC [102]. In vitro, spliced HDAC7 binds to SRF and upregulates SMC markers during the differentiation of mESC [103]. Also, multipotent A404 progenitors were dependent on the acetylation of histones H3 and H4 to commit to SMC fate [100]. Therefore, the presence of transcriptional regulation alone is not enough to drive SMC fate and epigenetic modifications are required to expose VSMC DNA sequences.
Platelet-derived growth factor-ββ
EC secrete PDGF-ββ during the early stages of vessel development where the ligand finds its way to surrounding mural precursors and recruits them to the developing mural layers [43]. PDGF-ββ’s important role in VSMC and PC development is highlighted by murine mutant models where inhibition of the ligand, receptor, or downstream events usually result in poor mural recruitment as indicated by sparse mural coverage, leaky vessels, and embryonic lethality [2,36,43,104].
While PDGF-ββ and its receptor PDGFR-β are the most implicated in mural development, there exists several isoforms of both the ligand and receptor. In total, there are four ligand monomers (A–D), which form five dimers (PDGF-αα, αβ, ββ, CC, and DD), and two receptor monomers that form two receptor tyrosine kinase (RTK) dimers (PDGFR-α, αβ, β) [105]. Our isoforms of interest, PDGF-ββ and PDGFR-β, have especially high binding affinity to each other, although the ββ ligand can also activate the other two receptor isoforms and the β receptor can also be activated by PDGF-DD [106].
On presumptive SMC, PDGF-ββ, binds to the RTK PDGFR-β and proceeds to activate a multitude of RTK pathways [107]. In this study, Src, Grb2, PI3K, Ras, SHP-2, PLCγ, and other unstudied pathways act coordinately to induce SMC differentiation where ablation of a single pathway does not completely abolish PDGF-ββ’s effects. One target of these pathways is ERK, which is known to be upstream of MYOCD, a well-studied transcriptional cofactor of SMC genes [86].
The PDGF-ββ pathway is often associated with mural proliferation as it induces the proliferative/synthetic phenotype in mature VSMC [5]. Similarly, PDGFR-β+ cells in the pericapillary of the developing embryo proliferate in response to PDGF-ββ. Knockout of the ligand inhibits this proliferation and causes lethal vascular defects [43], while overexpression exacerbates proliferation resulting in an abnormally thick medial layer [108]. Although proliferation is often viewed as mutually exclusive to differentiation, PDGF-ββ has been shown to facilitate VSMC differentiation in many in vitro models ranging from PSC-derived LPM, PM, and NC cells [19,109,110] to a wide variety of adult progenitors [52,53,55,57,69].
Transforming growth factor-β
TGF-β is another growth factor that induces mural differentiation in vivo and in vitro. This ligand comes in three isoforms, TGF-β1/2/3, and targets several receptors on mural cells—Alk-5 (also known as TGF-βRI), Alk-1 (TGF-βRII), and endoglin (CD105) [111,112]. The three receptors coordinately direct the phosphorylation of Smad1/5/7 or Smad2/3, both of which associates with Smad4 and localizes to the nucleus to activate transcription factors.
In mouse models, impairment of the TGF-β pathway through SMC-specific deletion of Alk-5 [113] or endoglin [114] leads to poor SMC recruitment, while epicardium-specific deletion of Alk-1 prevents the epithelial-to-mesenchymal transition of the epicardium into VSMC [115]. Similarly, in vitro, TGF-β1 has been used extensively for VSMC differentiation as demonstrated with multipotent adult stem cells [52,55,116] and in vitro analogs of embryonic precursors [19,110,117,118]. Although less utilized, TGF-β3 has been demonstrated to induce SMC differentiation in MSC [87,116].
Although it is unclear how the TGF-β receptors activate the different Smad pathways [119], Smad2/3 has been demonstrated to be critical for mural development. During mural differentiation, the Smad2/3/4 complex translocates to the nucleus where it activates transcriptional activators of SMC genes such as MYOCD of MSC in vitro [87] and MRTF-B of NC cells in mice and in vitro [41]. Furthermore, siRNA inhibition of Smad2/3 impairs the differentiation of SMC from embryoid bodies (EB) [120].
Sphingolipids
Sphingolipids are a class of plasma membrane lipids that also serve as important signaling ligands for vascular development [121]. One of the most studied phospholipids is S1P, which is accompanied by five different S1P receptors (S1PR1–5, also known as endothelial differentiation gene, Edg), of which S1PR1–3 are expressed in VSMC [86].
In the S1P-mediated differentiation of mural precursors, S1PR2 activates RhoA, which initiates actin polymerization for the nuclear translocation of SRF cofactor MRTF-A [86,88]. In vivo, universal knockout of S1PR1 inhibits the maturation of vessels by impairing the recruitment of VSMC [121]. Although S1PR2 or S1PR3 knockouts alone yield milder phenotypes, double or triple knockouts of S1PR1 with S1PR2–3 result in more severe embryonic vascular defects [122]. In vitro, S1P was able to induce MSC and 10T1/2 mesenchymal progenitors to express SMC characteristics [88,123]. Sphingosylphosphorylcholine (SPC), another sphingolipid, has also been used to differentiate MSC into contractile SMC where this differentiation pathway depends on RhoA/MRTF-A like S1P-induced differentiation [98,116]. Interestingly, SPC did not induce mural fate in PSC-derived NC, PM, or LPM [19].
Cell–cell contact signaling
The Notch contact signaling pathway encompasses two transmembrane Jagged (Jag1–2) and three Delta-like ligands (Dll1, 2, and 4) along with four Notch receptors (Notch1–4) [124]. In the developing vasculature, Jag1 on EC binds to Notch3 on prospective VSMC, enabling the recruitment of the first VSMC layer [125,126]. The first VSMC then produce their own Jag1 in a positive feedback loop, allowing subsequent medial layers to recruit through Notch signaling. Upon binding of Jag1, Notch intracellular domain (NICD) is cleaved from the receptor and translocates to the nucleus to form a transcription complex for mural differentiation. RBP-Jκ/CBF-1 is a necessary component in this transcription complex, as demonstrated with the in vitro differentiation of 10T1/2 precursors into SMC [127]. Similarly, NC-specific in vivo and ex vivo ablation of mastermind-like (MAML), another member of the Notch-mediated transcription complex, interferes with SMC development [124]. Although it has not been explicitly shown in mural differentiation, RBP-Jκ and MAML likely associate with other factors such as Ski-interacting protein and p300 [86,128]. This complex transcribes SMC genes independent of SRF, although the two complexes can coordinately upregulate SMC genes in parallel [127].
In vivo, disruption of the Notch pathway in developing SMC or the Jag1-expressing EC results in poor recruitment of both VSMC [4,124,129,130] and PC [131]. In vitro, activation of the Notch pathway and subsequent differentiation of SMC precursors can be achieved with exogenous introduction or overexpression of Jag1 or NICD [126,129]. VSMC differentiation studies have also activated the Notch pathway with coculture of EC or other cells that express Notch ligands [127,132].
Notch has also been reported to activate other pathways for SMC differentiation. In the embryonic mouse, deletion of RBP-Jκ in epicardial cells ablates expression of PDGFR-β and Smad2/3 [133]. Furthermore, in mouse embryonic epicardial explants, TGF-β inhibitor attenuates the expression of NICD-induced SMC gene expression, while NICD upregulates the expression of TGF-β ligands. Cooperation of Notch with the TGF-β pathway is bridged through interactions of RBP-Jκ and Smad2/3 [129]. Taken together, the collective evidence shows that the Notch pathway is critical to mural cell differentiation through its own unique transcriptional elements and by mediating other mural differentiation pathways.
Although less studied, connexin-mediated (Cx) contact signaling has also been shown to be necessary for VSMC differentiation. VSMC express Cx37, Cx40, Cx43, and Cx45, which are required for gap junctions that allow direct exchange of signaling molecules and ions between cells [120,134,135]. In vivo, knockout of Cx45 resulted in the impaired recruitment of VSMC and attenuation of TGF-β1 signaling during which early VSMC appear [136]. Similarly, Cx43 is required for EC coculture-induced differentiation of VSMC where knockout of Cx43 attenuated TGF-β1 concentration in the coculture media [137]. Therefore, Cx-mediate contact signaling regulates VSMC differentiation, at least, partially through the TGF-β pathway.
Retinoic acid
Retinoic acid (RA), also categorized under vitamin A, is responsible for the development of many tissues. RA can bind to three nuclear membrane RA receptors (α, β, or γ RARs), which then form dimers with RXRs (α, β, or γ) [138,139]. After recruiting coactivators, the dimer may induce epigenetic changes or bind to cofactors to activate the transcriptional activity of development-related genes [140].
The earliest models of PSC to SMC differentiation used RA in an EB model [141]. Since then, RA has been used to induce SMC in a variety of cell types of different species in EB or monolayer induction [120,135,142,143]. Interestingly, SMC origin need not be specified as RA and serum can direct PSC toward SMC in one-step differentiation protocols, making RA protocols among the simplest to execute. Origin specification is still an option as RA can also guide multipotent SMC precursors toward SMC fate [19,142]. Furthermore, it has been shown that RA upregulates MYOCD during in vitro SMC differentiation [142].
Although RA mediates vascular development in vivo [144], its effects are not limited to vascular cells, but contribute to the organization of many embryonic organs [145]. Likewise, RA-mediated differentiation of PSC yields not only SMC but also cardiomyocytes, skeletal muscle, and neuronal cells depending on the concentration of the ligand [146,147].
MicroRNAs
MicroRNAs regulate SMC gene expression and differentiation by inhibiting translation or degrading mRNA [148]. miR-145/143 are derived from the same primary miR and are simultaneously expressed to promote VSMC differentiation [149]. miR-145 promotes the activity of SMC gene transcription factor MYOCD and antagonizes pluripotency factor Klf4, while miR-143 represses MYOCD's competitor Elk-1 [150]. In vivo, miR-145/143 are expressed in mural progenitors around the time when early SMC are recruited. Indeed, deletion of miR-145/143 yields a nonlethal phenotype with deficient VSMC recruitment indicated by a reduced medial layer [149]. In vitro, exogenous miR-145, but not miR-143, induced NC cells into contractile VSMC [150], and Notch-mediated signaling in MSC/EC coculture results in increased miR-145/143 [151]. MiR-1 and miR-10a are also critical to SMC differentiation as demonstrated in the RA-mediated differentiation of mESC to SMC. Moreover, miR-10a represses HDAC4, an epigenetic inhibitor of SMC gene expression [152], while miR-1 suppresses the expression of pluripotency transcriptional regulator KLF4 [153].
Biochemical signaling in the ECM
Biochemical regulation of mural fate is not limited to soluble signals as mural precursors are also stimulated by ECM ligands through integrin receptors. There are a total of 18 α and 8 β integrin subunits that combine to form at least 24 integrin dimers [154]. These integrins mechanically couple the cell to the ECM and activate focal adhesions and downstream pathways that play direct roles in regulating cytoskeletal components and therefore cell migration, cell shape, and cell differentiation [155].
Typically, VSMC-specific knockout of integrin receptor subunits such as β1 [156], α4 [157], α7 [158], α5, and αv [159], but not α5 alone [160], yields similar phenotypes with disturbed mural recruitment and vessel organization. However, these defects are not the result of ablated mural differentiation, but rather impaired migration, proliferation, and association with maturing vessels. Knockout of the downstream focal adhesion protein α-parvin yields similar effects where mural cells recruit improperly and respond abnormally to PDGF-ββ-mediated chemotaxis [161]. Similarly, the deletion of ECM proteins themselves can cause deficient mural recruitment as was the case with brain vessels and astrocyte-specific laminin (LN)-γ1 knockout [162].
It is clear that ECM are critical components for mural development, but which ECM proteins constitute the correct microenvironment? This is difficult to answer as the vessel's ECM changes as the vessel matures, and there are multiple distinct vessel layers each with a unique ECM composition. Furthermore, the vessel layers form sequentially during embryonic development, starting with the intima and building toward the adventitia. Therefore, vascular precursors sense different ECM compositions at different stages of vascular maturity.
In a mature blood vessel wall, the bulk of the main structure and mechanical strength is constituted by fibronectins (FN), LN, elastin, and collagens. Further structural support and function are provided by fibrillins and fibulins, which form microfibrils, nidogen, and perlecan, which tie together other ECM, and heparin sulfate proteoglycans (HSPG), which provide additional binding sites for integrins and soluble proteins. Note this is a general overview, as detailed descriptions of each ECM protein will indicate overlapping roles (reviewed in detail by Rhodes and Simons [163] and Kelleher et al. [164]).
The blood vessel architecture is formed from multiple distinct layers with unique ECM compositions [164]. At the lumen face, the tunica media contains EC held primarily within FN. This layer rests on the basement membrane, which consists mainly of collagens, LN, enactins, Von Willebrand factor (vWF), HSPG, and in microvasculature, PC [165]. Around the basement membrane lies the internal elastic laminae, a cell-free tissue of primarily elastin and microfibrils. Between the internal and a similarly composed external elastic laminae lies the tunica media. The tunica media is constituted by alternating layers of VSMC in collagens and more cell-free lamellae composed of elastin and microfibrils [155,164]. Circumferenced around the other layers, the adventitia comprised fibroblasts in collagen-rich ECM. These specific ECM compositions play key roles in vascular function and transduce biochemical and mechanical signals necessary for proper recruitment of mural cells.
For in vitro cell culture, the ECM substrates typically used are FN, LN, vitronectin, collagen, or its denatured form, gelatin. Of these ECM, collagen-type IV (CIV) and gelatin have been used extensively to derive VSMC [15,19,143,166] and PC [67,110]. Indeed, blocking antibodies against integrin subunits α1, αv, or β1 repressed the CIV-mediated mural differentiation of mESC-derived Sca-1+ progenitors [69]. Although few studies directly compare the in vitro effects of ECM composition on mural differentiation, it was shown that cardiac progenitor cells in SMC maintenance media with PDGF-ββ most preferentially differentiated into SMC in FN compared to LN, CIV, vitronectin, and gelatin [167]. In contrast, MSC in MSC maintenance media have the highest expression of SMC markers on CIV over LN and FN [168]. However, with a simple media of DMEM and serum, MSC prefer SMC differentiation under collagens, elastin, and FN over LN [169]. These studies highlight the fact that ECM effects on SMC differentiation are coupled with the progenitor type and other microenvironment conditions.
Growth factors in the ECM
A major role of the ECM is to bind soluble ligands that would otherwise have limited exposure to their target cells. Able to bind both cell integrins and soluble growth factors, the ECM is able to concentrate soluble signals to cell surface receptors [170]. This localization is primarily attributed to HSPGs [171], but other proteins, such as LN and FN [170], also have soluble signal binding domains. Many growth factors and cytokines bind to ECM, including the PDGF and TGF-β families that are necessary for mural development [170,171].
Currently, there are no in vitro studies that demonstrate ECM-bound soluble signals for the differentiation of mural cells. However, the in vitro implications are demonstrated through in vivo studies where matrix-bound PDGF-ββ has been shown to be necessary to maintain the microenvironment for mural development.
In vivo, mutant mice with the deleted ECM sequence that binds PDGF-ββ (known as the retention motif) resulted in impaired PC recruitment to the retinal and glomerular microvessels [2]. In addition to decreased PC density, it was found that PC migration and dendritic extensions were likely guided by the concentration gradient of ECM-bound PDGF-ββ. Similarly, interference with the N-sulfation of heparin sulfates prevents proper recruitment of PC to mice hindbrain microvessels [172]. In mice tumor microvessels supplemented with exogenous retention motif, PC demonstrated improper recruitment with enlarged microvasculature compared to wild-type controls [173]. Overexpression of PDGF-ββ by the tumor cells did not restore tight PC-EC attachment, demonstrating that ECM-bound PDGF-ββ has unique mural recruitment mechanisms distinct from that of its soluble counterpart.
Mechanical signaling and the ECM
The alternating elastin and collagen layers in the elastic laminae and tunica media, respectively, provide the blood vessel with the bulk of its elasticity and mechanical strength. These physical properties create the inherent recoil necessary to transduce the pulsatile nature of blood flow into cyclic, radial expansions that mold the mechanical microenvironment of prospective VSMC. Cells in the vessel wall sense three key elements of this force: (1) the type of strain, which is always cyclic and uniaxial in normal physiology, (2) the frequency of the cycle, which is ∼1 Hz in the resting adult human and faster in fetal and murine circulatory systems [174,175], (3) the magnitude of circumferential strain, which is <10% in aging human vessels and, in theory, higher in more elastic younger and fetal vessels [175,176].
During development, VSMC first appear shortly after initial blood flow and its accompanying cyclic strain [4,177]. As development progresses, the vessel wall accumulates ECM, thickens, and matures its mechanical properties in response to the increasing blood pressure [178]. The changing hemodynamics also serve to recruit VSMC that reciprocally regulate the vessel wall's ECM content and mechanical properties. The significance of the mechanical environment is highlighted in elastin knockdown models where the vessel wall experiences abnormal wall strains, and as a result, abnormal vessel wall thickens [179,180]. Furthermore, the vessel wall becomes thick to the point of obstruction, and VSMC are no longer circumferentially aligned, but longitudinally aligned [181]. As blood pressure increases with elastin deficiency, it has been proposed that maintaining a constant tension per lamellar unit (VSMC layer+elastin layer) is a regulatory driving force for building the vessel wall and recruiting VSMC layers [178]. When the mechanical effects of blood pressure is completely nullified in Isl-1 knockout mutants with vessels disconnected from the main circuit, developing PC failed to express their normal markers Jag1 and Notch3 [4]. Indeed, the Notch pathway is coupled with the circulation as hemodynamic forces are sensed by EC cilia and then transmitted to VSMC through Notch [182].
In vitro, mechanical strain has been shown to induce VSMC differentiation in primary and PSC-derived progenitors. This has been demonstrated with a variety of cells, including mouse C3H/10T1/2 mesenchymal progenitors [183], mesoderm progenitors from mESC [166], NC stem cells from human induced PSC [184], human MSC [185], and rat bone marrow progenitor cells [186]. Typically, the uniaxial mechanical strain, with physiological parameters, is coupled with soluble signals for enhanced VSMC differentiation. Mechanical strain may also activate mural differentiation pathways independent of soluble ligands as demonstrated with the PDGFR-β pathway on mESC-derived mesoderm progenitors [166].
The stiffness (elastic modulus) of the vessel wall may additionally regulate the commitment of mural progenitors. One study implicated the potential role of low stiffness (2 kPa) in directing MSC toward an EC versus an SMC fate, although it is unclear whether it is the nanofibrous architecture compared with polystyrene culture dishes or the signaling from lower stiffness of the nanofibers that might be directing EC fate [187]. Although there is little direct evidence to date that stiffness guides mural fate, it is well known that stiffness can modulate a progenitor's shape, cytoskeletal organization, and consequently its differentiation (reviewed in depth by Reilly et al. [188]).
In sum, in vivo and in vitro evidence paints a complex picture where mechanical forces are processed by the ECM and sensed by mural precursors directly through multiple pathways and indirectly through neighboring cells. Indeed, it would be sensible that the multilayered blood vessel, its varied ECM compositions, and consequent mechanical environments [189] would provide a mechanical environment unique to developing mural cells.
Considerations for combinatorial signaling in vitro
By far, the most popular in vitro differentiation signals for mural differentiation are PDGF-ββ and TGF-β1. However, there are many more microenvironment factors and pathways that are required for mural fate. These necessary signals are implied to be endogenously expressed, but few studies explore their exogenous potential. Ideally, although impractically, these factors would be tightly controlled in the perfect in vitro differentiation model. Likewise, the parameters of each signal, such as ligand concentration in soluble signals, would be considered. For now, differentiation protocols typically focus on one or two of these signals and, from that, another question arises. If two mural progenitors develop through different microenvironments with different signals, would this dissimilarity impart heterogeneity in the resulting mural cells?
Another issue is apparent when comparing the in vivo signaling for VSMC and PC recruitment. Both mural cells appear to share commonalities in developmental origins and developmental microenvironments. Yet, why do the few in vitro PC differentiation studies yield PC only as a subpopulation in EC differentiation protocols? In contrast, common mural recruitment signals such as PDGF-ββ have guided many types of mural progenitors into VSMC, but not PC. Part of the answer may lie within the incompletely defined and difficult to characterize nature of PC. Perhaps the multipotential PC population is too transient to observe without a niche microenvironment to capture its undifferentiated state. Maybe PC require close cell–cell communications with nascent EC, hence why PC derivation protocols are all adapted from EC protocols. Or are there unknown, but significant, factors that drive progenitor commitment to PC and not VSMC? More studies in vivo and in vitro are required to elucidate these concerns.
Developmental Timescale and Mural Cell Function
Thus far, this review has covered the source and means of mural development. During in vitro differentiation, the proceeding final step is the assessment of the product cell and determination of protocol success. Typically, this process involves the characterization of markers and functions of the product cell. There are two key properties to cell markers and function, specificity and timescale. Together, the information on the specificity and kinetics of differentiating cells can be used to determine a cell's degree of commitment to a specific lineage and resemblance to its in vivo counterpart. In the following section, we will review mural cells and their properties as they transition from an embryonic progenitor to a mature, fully committed mural cell.
In vivo, initiation of heartbeat, blood flow, and consequent hemodynamic forces occur right before the onset of VSMC marker expression [4,177]. It is thought that these hemodynamic forces, particularly circumferential cyclic strain, play a significant role in the recruitment of mural cells. The earliest indicator of mural lineage is the expression of PDGFR-β in the developing vascular mesenchyme [84]. These cells migrate toward the endothelium where they form an initial perivascular layer in direct contact with EC [30,84]. Around this time, these cells will express the first contractile marker, αSMA [33,84,94,190]. VSMC progenitors may also express the SMC gene regulator, SRF, which will catalyze the eventual synthesis of intermediate to late stage VSMC contractile markers [94]. At this time in development, there is no clear distinction between the future PC-populated basal lamina and VSMC-populated tunica media [84]. Potentially, the early αSMA+ cells may be αSMA+/neural/glial antigen 2+ (NG2) (some of which are also desmin+ [43]) ancestors of both VSMC and PC [38,39]. This progenitor's lineage choice may be distinguished by its αSMA expression, the persistence of which may indicate VSMC fate and the discontinuation of which may indicate PC fate [38].
If a mural progenitor chooses VSMC fate, it will then begin building SMC-specific contractile machinery. Chronologically (Fig. 3), the presumptive VSMC will express SM22α [190], calponin-1, h-caldesmon [38,190], and then the SM1 isoform of SMMHC [191]. Sometime after caldesmon expression, the VSMC-ensheathed vessel will gain stability and resistance to hyperoxia [38]. Vascular cells will begin to upregulate production of collagens and elastin, ECM proteins that constitute the majority of the tunica media [164]. VSMC will also begin forming additional perivascular layers, while aligning circumferentially [84]. As the contractile complex develops, VSMC will acquire intermediate filaments, followed by dense bodies, and then thick filaments [192]. Finally, the VSMC will complete its toolset with the expression of SMTNB [193], and then close to birth, the expression of the SM2 isoform of SMMHC [194 –196].
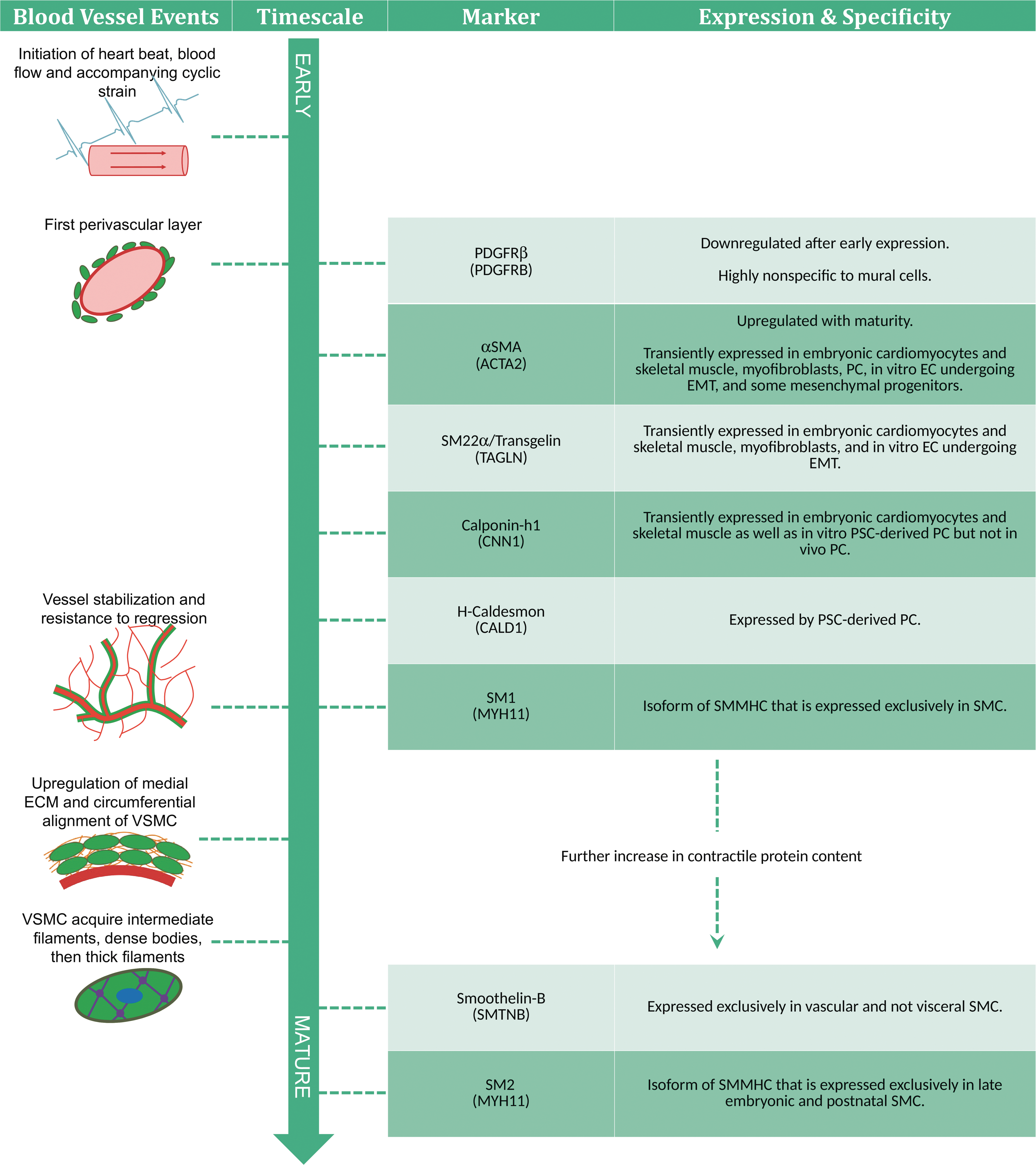
Developmental timeline of the vascular smooth muscle cell. VSMC express their multitude of characteristics with varying lineage specificity and time points in development. Therefore, the presence or absence of certain traits can indicate the cell's degree of VSMC commitment and maturation level. Note that in embryonic development, timescales will vary across species and vessel location. As such, this diagram estimates the relative occurrence of events and marker acquisition. VSMCs, vascular smooth muscle cells.
Generally, SMC markers later in the developmental timeline are more lineage specific. One of the earliest markers PDGFR-β is the most nonspecific VSMC marker mentioned in this review. While it is expressed in both mural cells, it is also expressed in other vascular cells, including vascular progenitors [53], ECs [197], fibroblasts [198], and a number of nonvascular embryonic and adult cell types [49]. αSMA is the next earliest marker and is also found in vascular progenitors [55,59], while αSMA and SM22α can both be found in myofibroblasts [199]. Furthermore, αSMA, SM22α, and calponin-h1 are all transiently expressed in early embryonic cardiomyocytes and skeletal muscles [190,200 –202]. The markers that are exclusive to SMC are h-Caldesmon, SMTNB, and the SM1 and SM2 isoforms of SMMHC [203].
Functionally, mature VSMC regulate vessel tone and by contracting in response to agonists such as AngII, endothelin, and carbachol [135,204,205]. Also, VSMC would exhibit phenotypic modulation (downregulation of contractile markers and increase of proliferation, migration, and ECM remodeling proteins) under dedifferentiation factors such as PDGF-ββ and serum (reviewed in detail by Owens [5]).
An important consideration: vascular SMC are not easily distinguishable from visceral SMC. Aside from anatomic location, the most apparent distinction is in contraction, where SMC of different organs will display varying degrees of tonic or phasic activity [206]. However, this distinction is lost in vitro as the molecular makeup of the SMC types is not so different and cellular contractions are not signaled by their native microenvironment. Indeed, all VSMC contractile markers are also expressed in visceral SMC with the exception of the vascular-specific SMTNB [19,193,207]. Other differences between vascular and visceral SMC are relatively minor, including the vascular SMC's higher vimentin–desmin ratio and αSMA content [208], and the visceral SMC's additional 7 amino acid sequence on SMMHC heads [209]. Furthermore, large-scale gene analysis reveals that vascular SMC, compared to visceral SMC, preferentially express genes related to the activation of the TGF-β pathway, activation of the immune system, and communication with EC [210]. This hints at many potential distinctions between the two SMC and further studies are needed to distinguish specific properties for in vitro identification.
The PC developmental chronology has not been mapped as extensively as VSMC, and lineage-specific characteristics of PC are not known [36]. In vivo, PC are typically defined by their unique position within the perivascular basement membrane [79]. Lacking this essential definition, in vitro studies necessitate multimarker panels coupled with tests of cell function, such as ECM production, multipotential, and migration as exampled by Wanjare et al. [110]. PC markers include, but are not limited to NG2 [39], PDGFR-α [44], PDGFR-β [43], and CD146 [50] (reviewed in detail by Armulik et al. [79] and Bergers and Song [3]). Like VSMC, PC may also exhibit contractile function in response to carbachol [110], endothelin, and AngII [211]. Although PC express the early VSMC contractile markers αSMA and desmin [38], the PC contractile machinery is vastly different as it lacks intermediate and late VSMC markers such as calponin and SMMHC [212]. Furthermore, PC are nonterminally differentiated and can possess multipotential for skeletal muscle [50], smooth muscle [4], neuronal cells [213], adipocytes, and osteoblasts [110]. As previously mentioned, there is a large degree of heterogeneity across PC of different origins and anatomic locations resulting in a wide range of potential functions [3]. Therefore, a single PC may not concurrently express all known PC characteristics, adding additional complexity to the identification of these cells.
To note, mural cells developed or generated in vitro may express unusual characteristics without an in vivo analog. To list some examples, αSMA, SM22α, and calponin-h1 can be expressed by in vitro EC undergoing EndMT [214,215]. PSC-derived PC may express calponin-h1 and h-caldesmon where PC in vivo do not [38,53,66,110,212]. Furthermore, h-caldesmon is found on BC3H1 skeletal myoblasts that exhibit dual SMC and skeletal muscle character [216].
A key function of mural cells is the integration and stabilization of vasculature. This behavior can be observed by simply seeding mural cells with EC undergoing angiogenesis. Typical techniques include in vivo implantations with Matrigel plugs [19,110], in vitro Matrigel vasculogenesis assays [52], and microfluidic devices for generation of in vitro perfusable vessel formation [217]. With the inclusion of mural cells, these nascent vessels may display attributes associated with stability and tone regulation such as the narrowing of vessel diameter, increased longevity, and decreased permeability and contractile function [217 –219].
Furthermore, mural cells are significant contributors to vessel wall remodeling and express a variety of ECM, MMP, and TIMP [110,220,221]. However, most cell types contribute to the surrounding ECM to some degree, and ECM proteins are not specific to any one tissue. Therefore, there is a large degree of nonspecificity regarding the expression of any ECM and ECM-related protein.
Considerations for generation of pure populations in vitro
A major challenge in stem cell engineering is the generation of pure cell populations. Heterogeneous populations can exhibit unpredictable and undesirable behaviors when used in research or clinical applications. Specifically, for the clinical application of PSC-derived populations, any remaining pluripotent cells risk the formation of cancerous teratomas. In fact, the ability to form teratomas is a hallmark of PSC [222]. So how can we achieve the generation of pure mural populations? Typically, cells can be purified by flow cytometry where the cell in question must express a highly lineage-specific surface marker set. Failing that, as in the case with both mural cells, the process of generating pure populations becomes a significant challenge.
However, unlike MSC, PC have no standardization for marker characterization; so they are usually characterized and purified by a set of nonspecific markers. In addition, the widely accepted definition of a PC, a perivascular cell in the basal lamina [36], relies on the anatomic location, which is absent for in vitro studies. Furthermore, many PC functions, such as multipotency, can be found in other mesenchymal progenitors [223]. The lack of specific properties complicates the identification and purification of PC as extensive characterization is required for a convincing proof of identity [110].
VSMC do not express lineage-specific surface receptors or receptor combinations. All specific proteins are located within an internal contractile complex, making purification by flow cytometry impossible without modifications to the cell. Because VSMC uniquely express SMMHC, which has not been found in any other cell type thus far [191,224], the expression of SMMHC alone is near definitive proof of VSMC lineage. Transgenic cells that express fluorescent labels or puromycin resistance have been developed with SMMHC [204,205,225]. While these transgenic cells can be purified to produce high-purity VSMC populations, the additional gene modification step may not be practical in a clinical application setting. However, VSMC progenitors are varied and numerous with many potential surface markers (Table 1). A viable alternative would be the purification of an intermediate population combined with a highly efficient differentiation protocol.
Conclusion
The generation of specialized mural cells from PSC is desirable for vascular therapies as well as understanding vascular development and disease states. Although many studies have demonstrated mural development in vitro, several challenges remain. First, mural cells are functionally heterogeneous as determined by their many embryonic and adult origins. Second, numerous factors of the microenvironment can guide a progenitor to mural fate, yet many are seldom explored in vitro. Finally, the purification of VSMC and the characterization of PC remain difficult. These issues hinder in vitro models’ ability to generate pure, physiologically representative mural cells. However, hints at resolving these issues can be found in vivo, where the development of mural cells is extensively characterized. By understanding in vivo development, we can better work toward the in vitro generation of mural cells.
Footnotes
Acknowledgments
The authors thank funding sources from the California Institute for Regenerative Medicine (CIRM) Basic Biology Award (no. RB5-07414), NSF Integrative Graduate Education and Research Traineeship (IGERT) Award no. 0965918, and NSF-Science and Technology Center (STC) for the Emergent Behavior of Integrated Biological Systems (EBICS) Award no. 0939511.
Author Disclosure Statement
No competing financial interests exist.