
Abstract
Mesenchymal stem cells derived from bone marrow (BM-MSCs) have multifunctional properties that have made them a promising therapeutic agent for many regenerative, anti-inflammatory, and autoimmune applications. Under chronic pathological conditions, however, BM-MSCs can become functionally compromised due to long-term exposure to changes in the systemic and localized stem cell niche microenvironments. In addition to the fact that functionally compromised BM-MSCs may be therapeutically ineffective, impairment of BM-MSCs is potentially a contributing factor to disease progression and development of comorbidities. For the purpose of this review, MSC-based therapies for treatment of nonhealing wounds in diabetic patients will be used as an example to demonstrate the effect that the diabetic host environment has on the regenerative capacity of endogenous BM-MSCs. This review will also discuss the mechanism by which the pathogenesis of diabetes mellitus leads to intrinsic dysfunction of the bone marrow stem cell niche that ultimately results in MSC failure and will highlight potential strategies for counteracting the functional decline of BM-MSCs.
Introduction
S
Bone marrow mesenchymal stem cells (BM-MSCs), however, are multifunctional and in addition to their ability to differentiate into multiple mesodermal lineages (adipocytes, osteoblasts, chondrocytes, myocytes, and keratinocytes) [9], they have immunomodulatory and growth-promoting functions [10,11]. These multifunctional properties of BM-MSCs are mediated through cell-to-cell interactions, cytokine/chemokine secretion, and extracellular vesicles [9 –11]. The functional capacity of BM-MSCs, together with their responsiveness to the microenvironment, has made them an attractive potential agent for many regenerative, anti-inflammatory, and autoimmune applications [10]. This is particularly true for diseases and disorders such as diabetes (type 1 and type 2) and its secondary complications, for which no or only partially effective treatments are available.
MSC-based therapies for type 1 diabetes (T1D) (an autoimmune disease characterized by the complete loss of insulin secretion leading to hyperglycemia) are mostly focused on alleviating hyperglycemia by stimulating pancreatic β cell regeneration while taking advantage of the immune-suppressive properties of BM-MSCs [12]. In contrast to this, MSC-based therapies for type 2 diabetes (T2D) (multifactorial disease that is associated with insulin resistance-induced hyperglycemia) are more focused on the treatment of comorbidities such as erectile dysfunction [13], retinopathy [14], and nonhealing wounds [15]. Autologous BM-MSCs are derived from the same individual who requires therapy, thereby reducing the risk of the patient developing graft-versus-host disease, in which an immune response can potentially reject the transplanted cells. A shortcoming in current autologous MSC-based therapeutic strategies, however, is that success is unpredictable, particularly in the presence of underlying disease. The advancement of autologous MSC therapies is therefore dependent on an improved understanding of the effect that the extrinsic host environment has on stem cell function.
Diabetes Mellitus Compromises MSC Function
Refer to Figs. 1 and 2 for detailed illustrations of how the pathogenesis of diabetes leads to stem cell dysfunction as will be discussed throughout this review.
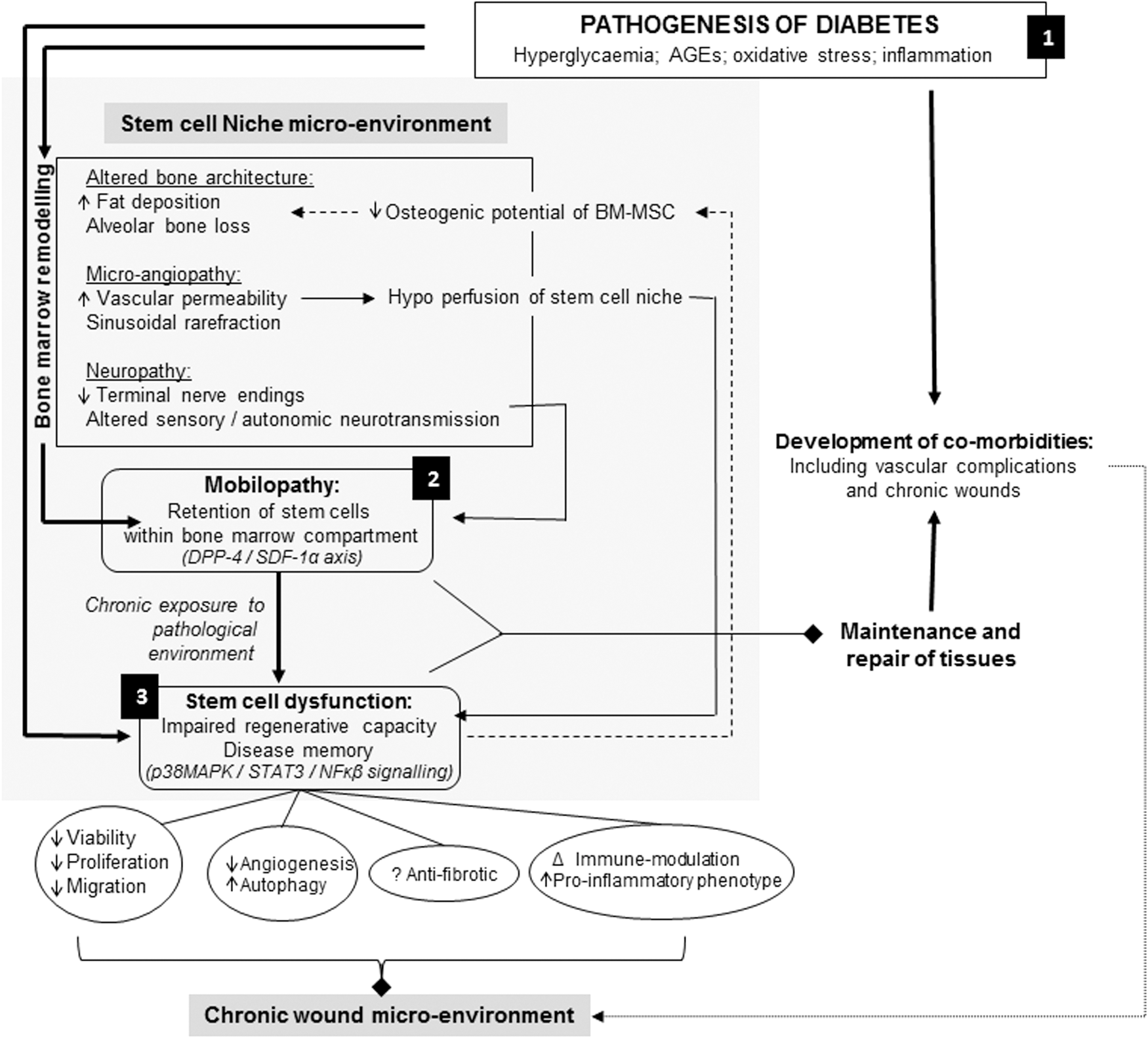
Overview of the effect that the pathogenesis of diabetes has on bone marrow stem cell function. The combined effect of hyperglycemia, AGEs, oxidative stress, and inflammation causes remodeling of the bone marrow stem cell niche microenvironment. This contributes to retention of stem cells within the bone marrow compartment, where long-term exposure to the pathological microenvironment impairs the regenerative capacity of stem cells. Bone marrow remodeling is characterized by alterations in bone architecture, microangiopathy, and neuropathy. Alterations in bone architecture are due to increased fat deposition and alveolar bone loss that are a direct consequence of the impaired osteogenic potential of BM-MSCs. Hypoperfusion of the stem cell niche within the bone marrow as result of microangiopathy in turn contributes to the functional decline of stem cells within their niche. Dysregulation of the p38 MAPK/STAT3 and downstream NFκB signaling pathways has been implicated in the dysfunction of stem cells, whereas dysregulation of the DPP-4/SDF-1α axis together with neuropathy has been identified as a contributing factor to mobilopathy. Mobilopathy together with stem cell dysfunction contributes to the development of comorbidities since the vascular integrity of tissues cannot be maintained and repair of subsequent tissue damage is impaired. The overall reduction in stem cell viability, proliferation, migration capacity, together with limited proangiogenic potential, and changes in the immune modulation capacity of BM-MSCs significantly impair the ability of these stem cells to respond to environmental cues. Upon transplantation, dysfunctional BM-MSCs can therefore not restore the localized wound microenvironment to promote healing.
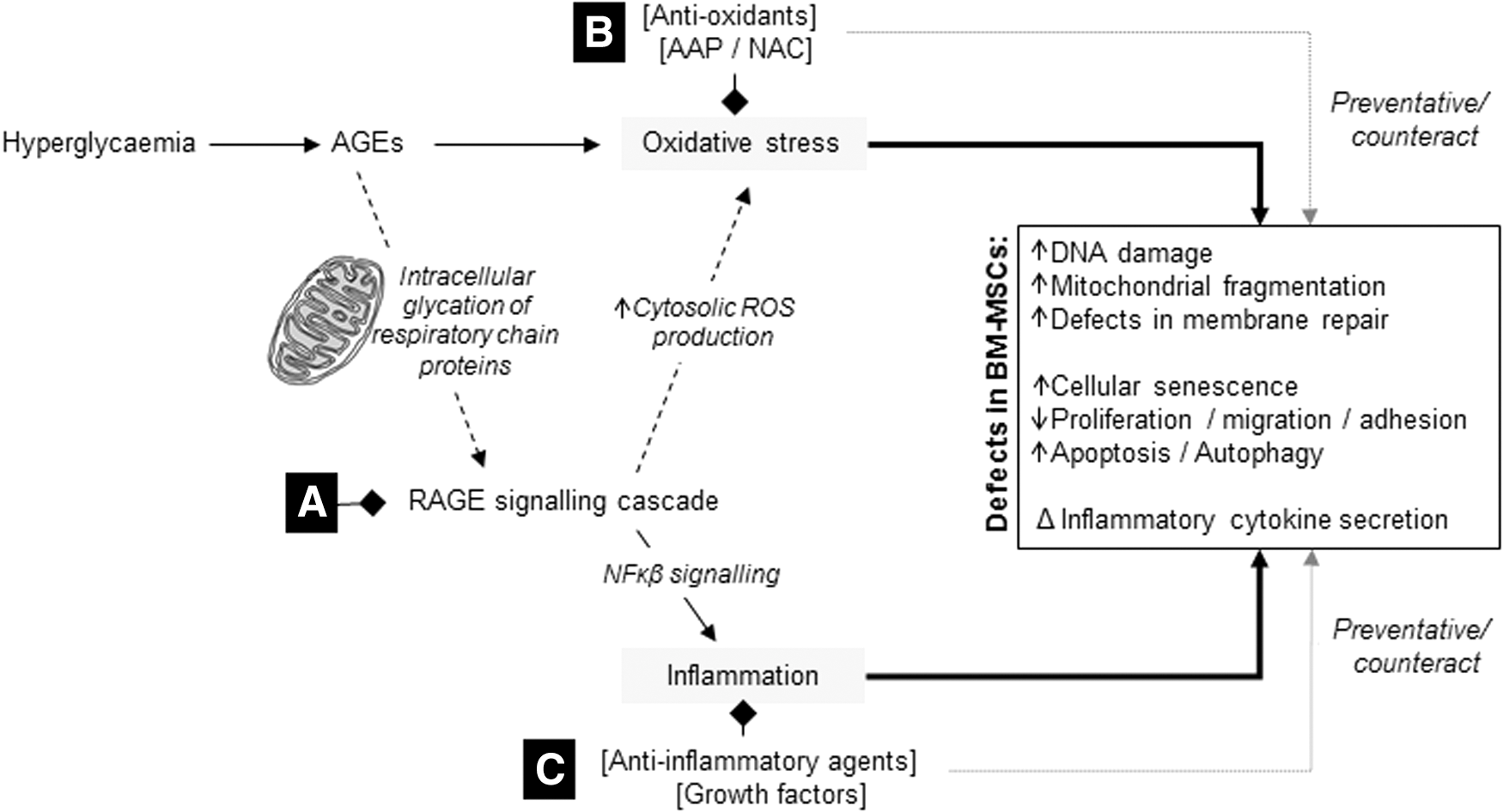
Molecular mechanisms underlying stem cell dysfunction in diabetes. Hyperglycemia-induced formation of AGEs leads to excessive production of ROS by mediating the intracellular glycation of mitochondrial respiratory chain proteins and triggers a cascade of events through activation of RAGE. In turn, chronic RAGE signaling can cause defects in cellular membrane repair; trigger intracellular damage by inducing oxidative and endoplasmic reticulum stress through elevations in cytosolic ROS production; and amplify inflammation through NFκB-mediated signaling. The negative effect of AGEs on BM-MSC function is thus thought to result from increased oxidative stress and an amplified inflammatory response. Potential therapeutic strategies include
Implications for autologous MSC-based therapies
For the purpose of this review, MSC-based therapies used for the treatment of nonhealing wounds in diabetic patients will be used as an example to demonstrate the effect that the diabetic host environment has on the regenerative capacity of BM-MSCs. Although autologous BM-MSC therapy has shown some promise in animal models [16 –20] and preclinical case reports [16,17,21 –32], the outcomes of these studies were variable and not as effective as initially hoped. The clinical outcomes and wound etiology of 14 studies with a total number of 123 chronic wound patients who had undergone autologous BM-MSC therapy between the years 2003 and 2015 are summarized in Table 1. From the table, it is clear that while most chronic wound studies show an overall reduction in wound size post autologous BM-MSC application, some patients were unresponsive to this treatment. In most cases, complete wound closure only occurred when BM-MSC therapy was combined with skin grafting. The unpredictability of autologous MSC-based therapies for the treatment of nonhealing diabetic foot ulcers in particular can, at least in part, be explained by recent advances demonstrating that (1) stem cell impairment is a major complication of diabetes [33,34] and (2) that impaired MSCs cannot respond adequately to the hostile chronic wound microenvironment post-transplantation (Refer to [35 –37] for detailed reviews on the pathophysiology of a chronic wound microenvironment).
The time frame that the chronic wounds from the patients have failed to heal before inclusion in the respective studies is indicated as (years) in the wound etiology column.
BM-MSCs, bone marrow-derived mesenchymal stem cells; DFU, diabetic foot ulcer; G-CSF, granulocyte colony-stimulating factor; HIV, human immunodeficiency virus; T2DM, type 2 diabetes mellitus; ↓, reduction.
Shin and Peterson [33] investigated the influence of the pathogenesis of T2D on the therapeutic potential of endogenous BM-MSCs and demonstrated that the diabetic mice in their study possessed fewer BM-MSCs and that these cells exhibited impaired proliferation and survival in vitro. Using an excisional splint wound model, the study also indicated that in addition to a reduced number of endogenous BM-MSCs homing to wounded areas in diabetic mice, engraftment of diabetic BM-MSCs produced limited improvement and could not produce the same therapeutic outcomes as BM-MSCs derived from healthy donors [33].
In a clinical study, Badiavas et al. [24] confirmed these observations and highlighted that BM-MSCs derived from chronic wound patients fail to reach optimal growth in vitro and that BM-MSC expansion to reach sufficient numbers for autologous transplantation purposes proved to be challenging. Despite attempts to optimize the isolation technique for these proliferative impaired BM-MSCs, it was noted that BM-MSCs derived from chronic wound patients had an altered secretome and that production of growth factors and cytokines was deficient [24,34]. Alterations in gene expression and the secretome of BM-MSCs can have detrimental effects on proregenerative functions of BM-MSCs that include antifibrotic, anti-inflammatory/immunomodulatory, and proangiogenic properties.
Despite the lack of studies investigating the effect of the diabetic host environment on the intrinsic antifibrotic capacity of BM-MSCs, it is known that immunomodulatory properties of diabetic BM-MSCs are skewed to favor a proinflammatory environment [34]. Our previous study indicated that inflammatory mediators [ccl-2, ccl-3, ccl-4, interleukin (IL)-6, IκBα, macrophage chemotactic protein (MCP)-1/5, macrophage inflammatory protein (MIP)-1α/β, MIP-2, and tumor necrosis factor alpha (TNF-α)] are overexpressed in diabetic compared with control BM-MSCs [34]. In vivo, the overexpression of MIP-1α/β and MCP-5 would result in the attraction of proinflammatory (M1) macrophages [38,39] to the wounded area. This macrophage phenotype promotes tissue destruction and prevents wound healing [40]. Other genes associated with proinflammatory pathways that were upregulated included IκBα [upstream of nuclear factor kappa B (NFκB) signaling] and IL-23 (known to promote chronic inflammation through induction of IL-1β and TNF-α production) [41]. Diabetic BM-MSCs with a skewed inflammatory phenotype would thus be unable to perform the imperative function of restoring the proregenerative conditions within the wound microenvironment needed for healing. T2D has furthermore been shown to affect the proangiogenic properties of BM-MSCs.
In a recent animal study, Ribot et al. [42] indicated that BM-MSCs derived from diabetic Zucker rats secrete soluble bioactive and chemotactic mediators that promote endothelial cell (HUVEC) migration and the formation of tubular structures. An increased expression of proangiogenic genes [ANPEP, MCP-1, MIP-2, HIF-2, insulin-like growth factor (IGF-1), IL-6, PLAU, TIE1, and TNF-α] and a decreased expression of antiangiogenic genes [COL18A1, COL4A3, F2, interferon-γ (IFN-γ), and TGF-β1/3] were evident in diabetic versus control BM-MSCs [42]. There is, however, very little evidence that supports the promotion of healing by diabetic BM-MSCs through angiogenesis in an in vivo hostile wound microenvironment.
In contrast to Ribot et al. [42] who investigated the ability of diabetic BM-MSC-derived conditioned media to promote angiogenesis in healthy endothelial cells, Rezabakhsh et al. [43] investigated the impact of human T2D serum samples on the angiogenic differentiation capacity of primary (healthy) BM-MSCs. The authors demonstrated that a 7-day incubation of healthy BM-MSCs with T2D serum samples completely abolished angiogenesis tube formation, decreased chemotaxis, and inhibited endothelial differentiation rate in response to subsequent in vitro stimuli compared with incubation with control serum samples. A crosstalk between the abnormal regulation of P62 during angiogenesis and excessive autophagy signaling was identified as the underlying cause of BM-MSC dysfunction [43]. Taken together, these studies indicate that long-term exposure to the chronic inflammatory and hyperglycemic environments characteristic of T2D affects the intrinsic function of endogenous BM-MSCs [33,34,44].
Functional comparisons of BM-MSCs derived from T1D patients with those derived from healthy controls have, however, demonstrated that T1D and healthy BM-MSCs exhibit no differences in terms of morphology, immunosuppressive activity, and migration capacity [45 –47]. Nonetheless, these studies revealed differential expression of genes related to cytokines, immunomodulation, and wound healing potential between healthy and T1D BM-MSCs [45,46]. Unlike T1D, T2D is a multifactorial disease that is associated with insulin resistance, hyperglycemia, persistent systemic inflammation, increased oxidative stress, and the accumulation of advanced glycation end products (AGEs) (proteins and lipids that become glycated due to hyperglycemia) [48]. The pathophysiology of T2D and the associated changes in the bone marrow microenvironment can therefore affect multiple aspects of BM-MSC function and/or morphology and seem to exacerbate stem cell failure to a greater extent than T1D. It is, however, still largely unknown whether distinct mechanisms underlie BM-MSC dysfunction in T1D compared with T2D.
In addition to the fact that functionally compromised BM-MSCs may be therapeutically ineffective, the impairment of BM-MSCs may be a contributing factor to disease progression and the development of comorbidities [49]. In a recent review of the literature, Fadini et al. [50] highlight the clinical implications of stem cell defects in diabetes and suggest that a better understanding of the underlying molecular and cellular mechanisms that regulate the bone marrow stem cell niche and mobilization of BM-MSCs should be the focus of new therapeutic strategies for patients with diabetes. We are in agreement with Fadini et al. [49,50], especially since the exact nature of stem cell impairment is not yet clear and it is also not clear whether in vitro treatments to optimize function would last when cells are reintroduced to the pathological microenvironment upon transplantation.
Bone marrow stem cell niche dysfunction and mobilopathy
The complex nature of diabetic pathophysiology is known to induce significant end-organ failure that includes remodeling of the bone marrow compartment within the trabecular structure of long bones [49,51]. The bone marrow compartment has an extensive microvascular network and is thought to contain two types of stem cell niches: the endosteal or osteoblastic niche, where primitive hematopoietic stem cells (HSCs), BM-MSCs, and macrophages reside, and the vascular niche that is located close to sinusoidal vessels where committed HSCs, endothelial progenitors, and perivascular cells reside [51]. Pathology-induced alterations of the bone marrow microenvironment can thus have detrimental effects on these stem cell niches and their function.
In diabetic patients, bone marrow remodeling is associated with (1) altered bone architecture due to increased fat deposition and alveolar bone loss as a direct consequence of the impaired osteogenic potential of BM-MSCs and depletion of the osteoblast progenitor cell pool [52 –54]; (2) microangiopathy: altered vascular permeability and sinusoidal rarefaction (vascular leakage of proteins and leukocytes) resulting in hypoperfusion of stem cell niches [51,55]; and (3) neuropathy: a reduction in the number of terminal nerve endings (due to fusion of BM-MSCs to nerves, causing premature apoptosis) and altered sensory and autonomic neurotransmission [56 –58]. This remodeling of the bone marrow together with hyperglycemic conditions has been shown to affect the maturation of endothelial progenitor cells and BM-MSCs, to promote a proinflammatory/profibrotic macrophage (M1) phenotype, and to lead to the potential retention of stem/progenitor cells in the bone marrow compartment [59 –62].
The retention of stem/progenitor cells in the bone marrow is referred to as mobilopathy and results in an altered ratio in terms of stem/progenitor cells present in the bone marrow compared with peripheral blood. The chronic exposure of stem cells to the pathological bone marrow environment due to mobilopathy exacerbates the functional decline of BM-MSCs. Diabetic mobilopathy is furthermore considered a contributing factor in the development of secondary complications since the mobilization of stem/progenitor cells into peripheral circulation plays a crucial role in the maintenance and repair of tissues and their vascular integrity [63]. The molecular mechanisms underlying this maladapted compartmentalization are complex, but thought to involve altered stromal-derived factor 1 alpha (SDF-1α) signaling [63 –66].
SDF-1α, also known as C-X-C motif ligand 12 (CXCL12), is an active chemokine that acts through the C-X-C receptor 4 (CXCR4) and serves as a retention signal for stem/progenitor cells [67]. Mobilization is achieved when a chemotactic SDF-1α gradient is established toward the peripheral blood. This is accomplished by degradation of SDF-1α in the bone marrow through the protease activity of dipeptidyl peptidase-4 (DPP-4, also known as CD26); suppression of BM-MSC activity; release of chemotactic SDF-1α from ischemic/injured tissues into circulation; and suppression of plasma DPP-4 activity [63 –66]. Current strategies for improving stem/progenitor cell mobilization are thus mostly focused on targeting the molecular mechanisms that regulate the DPP-4/SDF-1α axis. In a comprehensive review of the literature, Albiero et al. [63] describe the pharmacological targeting of the DPP-4/SDF-1α axis using granulocyte colony-stimulating factor (G-CSF), CXCR4 blockers (AMD3100), statins, DPP-4 inhibitors, erythropoietin, and/or angiotensin-converting enzyme inhibitors to counteract vascular complications. Pharmacological-mediated mobilization by targeting the DPP-4/SDF-1α axis is, however, dependent on nervous system stimulation and the responsiveness of DPP-4 [65,68,69]. Fadini et al. [66] demonstrated that diabetic patients are unresponsive to G-CSF stimulation due to dysregulation of DPP-4 activity and neuropathy is furthermore known to be a complication of diabetes. Consequently, solo targeting of the DPP-4/SDF-1α axis is unlikely to successfully mobilize stem/progenitor cells in patients suffering from these diabetic complications.
The dysregulation of bone marrow function in diabetes is also associated with an imbalance in macrophage polarization and it has been hypothesized that excessive bone marrow macrophages with an inflammatory phenotype (M1) contribute to the retention of stem/progenitor cells in the bone marrow [64]. Albiero et al. [64] confirmed that diabetic patients have an increased number of M1 macrophages and that depletion of bone marrow macrophages in a diabetic mouse model restored stem cell mobilization. It was established that M1 macrophages (but not anti-inflammatory M2 macrophages) release oncostatin M (OSM) that in turn induces SDF-1α expression by BM-MSCs through a mitogen-activated protein kinase (MAPK) p38–signal transducer and activator of transcription-3 (STAT3)-dependent pathway [64]. In support of this observation, our group recently demonstrated that the STAT3 signaling pathway is dysregulated in BM-MSCs derived from obese prediabetic mice and that the gene expression of diabetic BM-MSCs was clearly skewed to favor a proinflammatory environment [34]. Albiero et al. [64] confirmed their observations by indicating that higher plasma OSM levels in diabetic patients correlated with the ratio of stem/progenitor cells in peripheral blood versus bone marrow. Accordingly, OSM antagonism was suggested as a strategy to restore bone marrow function in diabetes.
Strategies focused on restoring stem/progenitor cell mobilization are, however, limited, in that stem cell damage can occur at the tissue level (within the bone marrow niche) before mobilization into peripheral blood. This was confirmed by an observation by Januszyk et al. [70]: the pathogenesis of both T1D and T2D may deplete specific subpopulations of BM-MSCs and this defect cannot be corrected by restoring glucose homeostasis. In addition to affecting BM-MSC viability and functional capacity, long-term exposure to the pathological bone marrow niche environment can induce a certain degree of disease memory in BM-MSCs [71]. Madhira et al. [71] demonstrated that in primary BM-MSC cultures derived from a T2D rodent model (WNIN/GR-Ob mutant rats), in which the animals suffer from several secondary complications (retinopathy, nephropathy, infertility, vascular complication), BM-MSCs retained a memory of the disease even after removal into optimal culture conditions. The in vitro culture of BM-MSCs is thus a feasible model, in which the disease mechanisms underlying stem cell impairment can be studied and which can be used to assess the efficacy of potential treatments to counteract functional impairment of BM-MSCs.
Molecular mechanisms underlying stem cell dysfunction
The accumulation of AGEs is one of the most pronounced mechanisms that underlies MSC dysfunction in diabetes. In short, formation of AGEs leads to excessive production of reactive oxygen species (ROS) by mediating the intracellular glycation of mitochondrial respiratory chain proteins and triggers a cascade of events through activation of the receptor for AGEs (RAGE) [72 –74]. In turn, chronic RAGE signaling can cause defects in cellular membrane repair [75]; trigger intracellular damage by inducing oxidative and endoplasmic reticulum stress through elevations in cytosolic ROS production; and amplify inflammation through NFκB-mediated TNF-α signaling [72 –75]. The negative effect of AGEs on BM-MSC function is thus thought to result from increased oxidative stress and an amplified inflammatory response [76,77].
In vitro, exogenous AGE stimulation-induced ROS production in healthy BM-MSCs impairs cellular proliferation, migration, and adhesion; alters inflammatory cytokine secretion; and induces cellular apoptosis [52,77 –80]. BM-MSCs derived from T1D RAGE-knockout mice, on the other hand, are protected from apoptosis and express high levels of the stemness genes, Nanog and Oct 4 [81]. Aikawa et al. [81] therefore suggested that targeting the RAGE signaling pathway may be a viable approach to preserving BM-MSC function in diabetes. In agreement, Denu and Hematti [82] recently reviewed the effects of oxidative stress on MSC biology and highlighted the need to identify novel strategies to optimize ROS levels in BM-MSCs to enhance their regenerative capabilities and longevity during the ex vivo expansion process before transplantation.
Potential Preventative Measures Against Stem Cell Impairment
Antioxidants: N-acetylcysteine and ascorbic acid 2-phosphate
Ascorbic acid 2-phosphate (AAP) can preserve the stemness of healthy BM-MSCs during prolonged in vitro culture by promoting cellular proliferation, telomerase activity, and intracellular levels of antioxidant enzymes, such as superoxide dismutase (SOD), catalase, and phosphorylated forkhead box 01 (FOXO1) [83 –85]. AAP can also stimulate the secretion of extracellular matrix components (collagen and glycosaminoglycan) without affecting the phenotype or differentiation potency of healthy BM-MSCs [86]. Even though AAP can partially rescue healthy BM-MSCs from oxidative stress, the antioxidant N-acetylcysteine (NAC) has shown greater promise as a protective agent against oxidative injury [80,87,88] and has been shown to improve postamputation stump healing in an animal model of T1D [89].
In vitro, Ali et al. [87] demonstrated that preconditioning of diabetic mouse-derived BM-MSCs with NAC ameliorated hydrogen peroxide-induced apoptosis and oxidative stress by upregulating the prosurvival genes (Akt and Bcl-2) and downregulating the expression of proapoptotic and stress genes (Caspase-3, Bax, Bak, p53, p38, and NFκB). This is supported by findings reported by Li et al. [88], which indicate that in addition to downregulation of proapoptotic genes, the combined antioxidant treatment of BM-MSCs with NAC and AAP reduces ROS generation, stabilizes mitochondrial membrane potential, and decreases mitochondrial fragmentation following hydrogen peroxide-induced oxidative stress. NAC has also been shown to prevent cellular senescence [90], reduce DNA damage and chromosomal abnormalities [91], and block ROS-induced adipogenic differentiation [92 –94] in cultured BM-MSCs. Despite the ability of NAC to scavenge ROS and block its production, Li et al. [95] showed that NAC was unable to protect BM-MSCs against membrane damage induced by oxidized low-density lipoprotein, known to be elevated in the serum of diabetic patients [96,97]. In a recent in vivo study, Zayed et al. [89] did, however, demonstrate that daily intraperitoneal NAC injections were able to improve postamputation stump healing and muscle neovascularization over a period of 7 days in a streptozotocin-induced T1D mouse model. Although these studies highlight the therapeutic potential of NAC, its benefit in preventing stem cell impairment in T2D patients has not yet been demonstrated.
Anti-inflammatory agents and growth factors
The chronic production of excessive inflammatory cytokines can attenuate the paracrine responsiveness of stem cells to their microenvironment. While investigating the impact of diabetes on BM-MSC function during bone fracture healing, Ko et al. [98] concluded that reducing the activity of proinflammatory TNF-α in vivo may preserve the regenerative potential of BM-MSCs in diabetic patients. This is supported by other studies implicating proinflammatory cytokines such as TNF-α and IFN-γ in the impairment of healthy BM-MSCs through the downstream activity of excessive NFκB signaling [98,99]. Upstream of NFκB signaling, in vitro inhibition of p38 MAPK has been shown to attenuate the effects of acute exogenous AGE stimulation on healthy BM-MSC proliferation by preventing the production of proinflammatory ccl2, ccl3, ccl4, and IL-1β [78]. The p38 MAPK-mediated STAT3 signaling pathway has furthermore been implicated in the retention of stem/progenitor cells in the bone marrow [64], and we recently demonstrated for the first time that BM-MSCs derived from prediabetic obese and healthy control mice differed in the expression of genes associated with the STAT3 signaling pathway [34]. Our study confirmed that normal gene expression can be restored by targeting STAT3 signaling with either recombinant IL-6 treatment or with a conventional diabetes drug (pioglitazone) with anti-inflammatory properties. Despite restoration of mRNA expression of intracellular signaling proteins and cell surface receptors, acute anti-inflammatory treatment could not restore the migration capacity of severely impaired diabetic BM-MSCs [34].
Preconditioning of functional BM-MSCs with growth factors, on the other hand, can promote the regenerative function of these cells in hostile environments by enhancing paracrine/autocrine functions [100,101]. Khan et al. [100] demonstrated that preconditioning of T1D BM-MSCs derived from streptozotocin-induced diabetic mice with IGF-1 and fibroblast growth factor (FGF)-2 upregulated the expression of growth-promoting genes (IGF-1, FGF-2, Akt, GATA-4, and Nkx 2.5) and downregulated proapoptotic genes (p66, p53, Bax, and Bak). Preconditioned T1D BM-MSCs furthermore showed increased antioxidant enzyme activity (SOD) under oxidative stress conditions and had a higher chemotactic ability in response to SDF-1α stimulation compared with control (not preconditioned) BM-MSCs in vitro. It is, however, important to note that T1D BM-MSCs are not severely impaired and retain their ability to proliferate and migrate, unlike BM-MSCs derived from obese T2D animal models that have refractory mobility.
In support of the notion that growth factors can promote the regenerative function of BM-MSCs, Peng et al. [101] established that BM-MSCs can be reprogrammed to have enhanced angiogenic effects in vitro and tissue regeneration functions in vivo. The authors describe the use of platelet-rich clot releasate (PRCR) as a driver of gene programming through stimulation of the platelet-derived growth factor receptor/PI3K/Akt/NFκB signaling pathways. In a mouse model, engraftment of PRCR-preconditioned BM-MSCs into surgical wounds was furthermore shown to attenuate apoptosis, improve epithelialization, and promote regeneration of blood vessels. There was, however, no underlying disease present in the study performed by Peng et al. [101], and it is not clear whether in vitro preconditioning would restore the function of severely impaired diabetic BM-MSCs before transplantation. It is furthermore unclear whether the beneficial effects of in vitro preconditioning will be sustained upon transplantation back into a hostile environment, especially since diabetic BM-MSCs can retain memory of the disease. Together, these studies do, however, make it clear that the use of anti-inflammatory agents and antioxidants should be emphasized in patients with diabetes undergoing MSC therapy.
Future Perspective for Clinical Implications
The advancement of autologous BM-MSC therapy is dependent on an improved understanding of the alterations that occur in BM-MSC function when their niche is chronically exposed to pathological environments. Future studies should thus investigate the exact nature of stem cell impairment in chronic diseases and focus on strategies to either correct the impairment before transplantation or prevent it from occurring. Given the unpredictability of autologous BM-MSC therapies in T2D patients, commercially available allogeneic MSC preparations have been developed as an alternative therapeutic option. Several ongoing clinical trials are currently investigating the efficacy of allogeneic MSC therapy for the treatment of chronic wounds in diabetic patients [102 –104]. Nonetheless, preventative measures against stem cell dysfunction should remain a priority in high-risk T2D patients in an attempt to restrict the development of comorbidities such as nonhealing wounds.
The potential beneficial effects of NAC in the prevention of oxidative stress [105] and the treatment of diabetic complications, such as contrast-induced nephropathy [106 –109], ocular complications [110], endothelial dysfunction [111], and hypertension-linked cardiac complications [111,112], have been demonstrated in T2D patients. There is, however, still a paucity of studies investigating the efficacy of antioxidants and anti-inflammatory agents in reversing the functional decline of BM-MSCs in patients.
Footnotes
Acknowledgments
This work is based on the research supported, in part, by the National Research Foundation of South Africa (grant no. 105921) and the Faculty of Medicine and Health Sciences at Stellenbosch University.
Author Disclosure Statement
No competing financial interests exist.