
Abstract
Mesenchymal stromal cells (MSCs) are multipotent stem cells with immunosuppressive and trophic support functions. While MSCs from different sources frequently display a similar appearance in culture, they often show differences in their surface marker and gene expression profiles. Although bone marrow is considered the “gold standard” tissue to isolate classical MSCs (BM-MSC), MSC-like cells are currently also derived from more easily accessible extra-embryonic tissues such as the umbilical cord. In this study, we defined the best way to isolate MSCs from the Wharton's jelly of the human umbilical cord (WJ-MSC) and assessed the mesenchymal and immunological phenotype of BM-MSC and WJ-MSC. Moreover, the gene expression profile of established WJ-MSC cultures was compared to two different bone marrow-derived stem cell populations (BM-MSC and multipotent adult progenitor cells or MAPC®). We observed that explant culturing of Wharton's jelly matrix is superior to collagenase tissue digestion for obtaining mesenchymal-like cells, with explant isolated cells displaying increased expansion potential. While being phenotypically similar to adult MSCs, WJ-MSC show a different gene expression profile. Gene ontology analysis revealed that genes associated with cell adhesion, proliferation, and immune system functioning are enriched in WJ-MSC. In vivo transplantation confirms their immune modulatory effect on T cells, similar to BM-MSC and MAPC. Furthermore, WJ-MSC intrinsically overexpress genes involved in neurotrophic support and their secretome induces neuronal maturation of SH-SY5Y neuroblastoma cells to a greater extent than BM-MSC. This signature makes WJ-MSC an attractive candidate for cell-based therapy in neurodegenerative and immune-mediated central nervous system disorders such as multiple sclerosis, Parkinson's disease, or amyotrophic lateral sclerosis.
Introduction
M
Extra-embryonic tissues are of great interest for stem cell isolation, as they are an abundant source for cells without ethical concerns. Several studies indicate that stromal cells derived from the umbilical cord matrix or Wharton's jelly (WJ-MSC) show MSC-like features, including adherence to plastic substrates, and a similar surface phenotype and differentiation capacity [19 –21]. Furthermore, WJ-MSC display immune regulatory properties [22 –24] and improve tissue regeneration in several disease models [25,26]. Interestingly, previous studies defined that cells from extra-embryonic/fetal origin are superior to adult MSCs, showing broader differentiation potential, enhanced expansion potential, and higher cell isolation yields [10,27,28]. Accordingly, they are presented as an intermediate between embryonic stem cells and adult MSC-like stem cells [29], being derived from a more primitive tissue source than adult MSCs. In contrast, other studies suggest that MSC-like cells derived from different tissues are similar, because they originate from a common progenitor cell [30 –32]. While MSC-like cells from different tissue sources share a substantial degree of similarity in their phenotype, variation in marker expression profile and lineage-specific commitment has been reported [33,34]. As such, it is questionable whether cells from different tissues are truly similar stem cells. More likely, they show a gene expression profile related to their functions in the tissue of origin.
In this study, our aim was to define the relationship between Wharton's jelly and adult bone marrow-derived stem cell populations and describe the unique gene expression signatures of the perinatal WJ-MSC. We first compared two isolation methods for obtaining Wharton's jelly stromal cells, being the enzymatic digestion of cord matrix and microdissected tissue fragment cultures (explants). Next, our established WJ-MSC cultures were morphologically and phenotypically compared to BM-MSC. Both cell types were assessed for their growth characteristics, mesenchymal differentiation capacity, and surface marker expression. Next, we performed gene expression profiling of WJ-MSC compared to bone marrow-derived MSCs and multipotent adult progenitor cells (MAPC). Ingenuity pathway analysis was used to determine the molecular signature of the differentially expressed genes in WJ-MSC.
Collectively our data show that WJ cells resemble BM-derived MSCs in culture, but differ at the transcriptional level, with WJ cells overexpressing immune regulatory molecules and neurotrophic factors. In vivo, WJ-MSC, but also BM-MSC and MAPC exert local immune modulatory effects on T cells after subcutaneous transplantation in acute rat experimental autoimmune encephalomyelitis (EAE). Furthermore, a conditioned medium of WJ-MSC more potently induced neurite outgrowth of differentiated SH-SY5Y compared to BM-MSC. These findings indicate that stem cells derived from the Wharton's jelly are a different type of MSC displaying intrinsic immune modulatory and trophic support functions. These characteristics make WJ-MSC a promising candidate for therapeutic application in inflammation-driven neurodegenerative diseases.
Materials and Methods
Ethical approval
The collection and experimental use of human cells and tissues were approved by the Medical Ethical Committees of Hasselt University and Ziekenhuis Oost-Limburg (Genk). Umbilical cord tissues were obtained from cesarean sections after informed consent. Animal experiments were performed in accordance with institutional guidelines and are approved by the Hasselt University Ethics Committee for Animal Experiments.
Isolation and culture of WJ-MSC
Human umbilical cords (n = 10) were obtained aseptically from full-term uncomplicated pregnancies with planned cesarean section and processed for stem cell isolation within 24 h. Cells were isolated using the explant isolation method, as previously described [33], with minor modifications.
Cord blood was removed and the remaining tissue was aseptically stored in sterile phosphate-buffered saline (PBS; Lonza, Verviers, Belgium) supplemented with 1% penicillin–streptomycin (P/S; 10.000:10.000; Gibco™; ThermoFisher Scientific, Waltham, MA) and 0.2% Fungizone® (250 μg/mL; Gibco). Tissues were kept in PBS during processing. First, the umbilical arteries and veins were removed and the remaining matrix was cut into ±2 mm3 fragments (explants). Next, the explants were cultured in Knock Out™ Dulbecco's modified Eagle's medium with F-12 (KO-DMEM/F12; Gibco) supplemented with 1% P/S, 1% GlutaMAX™ (L-glut; 200 mM; Gibco), and 10% fetal bovine serum (FBS; Biochrom AG, Berlin, Germany). Cultures were kept at 37°C in a humidified atmosphere containing 5% CO2 and were left undisturbed for 10 days to allow migration of cells from attached explant tissue. Nonattached explants were discarded and the medium was renewed every 3 days. At 80%–90% confluence, cells were harvested using Stempro® accutase (Gibco). WJ-MSC intended for functional assays were harvested between passage 2 and passage 8.
For microarray analysis, WJ-MSC RNA (n = 5) was collected at the third passage in TRIzol™ Reagent (ThermoFisher Scientific) and frozen on dry ice. To compare growth of explant-derived cells in different media, WJ-MSC were harvested from the same cord and cultured in parallel in KO-DMEM/F12 or DMEM with low glucose (1 g/L; DMEM-LG; Gibco) containing the supplements described above. Enzymatic isolation was performed as described previously [35], with the difference that blood vessels were removed before digestion. To compare isolation methods, both explant and enzymatically isolated cells from the same donor were cultured in DMEM-LG with supplements.
Culture of bone marrow-derived stem cells
Human bone marrow-derived MSCs (n = 7) were purchased from Lonza and cultured according to manufacturer's instructions. For microarray analysis, BM-MSC RNA (n = 5) was collected at the third passage using TRIzol. Human MAPC pellets (n = 5; research grade MAPC®) were obtained from ReGenesys (ReGenesys bvba, Leuven, Belgium). For this, MAPC were grown in culture stacks, then harvested, and centrifuged, after which the pellet was collected in TRIzol and frozen on dry ice.
MAPC for transplantation purposes were provided as frozen stocks (n = 2). Cells were immersed in water bath until partially thawed and then inverted in DMEM low glucose (1 g/L) (Gibco) supplemented with 10% FBS (Gibco) and centrifuged at 600 g for 8 min. Supernatant was removed and cells were dissolved in fresh medium for counting. Cells were washed twice and then suspended in sterile saline solution (0.9% sodium chloride; Baxter i.v. fluids, Val d'Hony-Verdifarm, Beringen, Belgium) for animal injection (vide infra).
Culture of NTERA-2 cl.D1 and SH-SY5Y cell lines
NTERA-2 cl.D1 (NT2/D1) was purchased from LGC standards (Molsheim, France; ATCC® CRL-1973™) and cultured according to the recommended conditions. The SH-SY5Y neuroblastoma cell line (Sigma-Aldrich, St. Louis, MO) was grown in DMEM/F12 (Gibco) supplemented with 2 mM
Preparation of conditioned medium of MSC subtypes
To prepare the conditioned (secretome containing) medium of WJ-MSC (CM WJ-MSC) and BM-MSC (CM BM-MSC) for the neurite outgrowth assay, these cell types were seeded at a density of 2 × 104 cells/cm2 in their standard culture medium (vide supra). After 24 h, the medium was changed to 1 mL/5 cm2 SH-SY5Y medium containing 0.1% FBS. Forty eight hours later, the medium was collected, centrifuged at 300 g, aliquoted, and stored at −80°C for later use.
Culture kinetics
The proliferative capacity of WJ-MSC and BM-MSC was assessed by calculating cell doubling kinetics for each passage. Cell numbers were determined by means of Fuchs-Rosenthal hemocytometer counts after trypan blue exclusion (Biochrom AG). Population doublings (PD) were calculated using the formula PD = 3.32 (log (nH)–log (nS)), where nS is the number of seeded cells and nH the number of harvested cells. To yield cumulative population doubling levels (CPD), the PD value was added to the sum of population doublings of the previous passages: PDx = 3.32 (log (nHx)–log (nSx)) + PDx-1, with x being the passage number. CPDs were calculated over a period of 70 ± 5 days. For WJ-MSC, CPDs were calculated after the first passage, since the initial seeding number was unknown and the number of plastic adherent cells could only be determined after this time point.
Analysis of colony formation
Colony formation was assessed by seeding WJ-MSC in six-well plates (Greiner Bio-One, Frickenhausen, Germany) at 50 cells/cm2. Cells were cultured for 10 days, with a medium change every 3 days. Then, the cultures were fixed with 4% paraformaldehyde (PFA; Sigma-Aldrich) and stained with 0.5% crystal violet (Sigma-Aldrich) in methanol (VWR, Heverlee, Belgium). After washing twice with PBS and air-drying, colony formation was examined with a Primo Vert phase-contrast microscope (Carl Zeiss, Jena, Germany).
Multilineage differentiation
Adipogenic, osteogenic, and chondrogenic differentiation of both BM-MSC and WJ-MSC were performed using the corresponding hMSC differentiation bullet kits® from Lonza, according to the manufacturer's instructions. Subsequent histochemical analysis was performed as previously described [33].
Flow cytometry
WJ-MSC and BM-MSC were harvested, washed with FACS buffer (PBS supplemented with 2% FBS), and incubated in the dark for 30 min at 4°C with fluorescently labeled antibodies (Supplementary Table S1; Supplementary Data are available online at
Microarray analysis
For transcriptome analysis, total RNA from five different donors of WJ-MSC, BM-MSC, and MAPC was isolated using the MagMAX™-96 for Microarrays Total RNA Isolation Kit (Ambion®; ThermoFisher Scientific). Sample quality and concentration were measured using the Agilent 2100 bioanalyzer (Agilent Technologies) and NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies) respectively. Next, RNA was converted to cDNA, fragmented and labeled, and subsequently hybridized to Affymetrix GeneChip Human Gene 1.0 ST arrays using the Ambion WT Expression Kit and GeneChip WT Terminal labeling and hybridization kits, carried out in a GeneChip hybridization oven 645 (all Affymetrix UK Ltd.). Hybridized chips were stained and washed in a GeneChip fluidics station 450 and scanned using a GeneChip Scanner 3000 7G (both Affymetrix). All steps were carried out according to standard Affymetrix protocols. Quality control was performed using GeneChip Operating software (GCOS 1.4; Affymetrix). Scanned images were visually inspected and raw intensity CEL files were generated.
Subsequently, Bioconductor packages running under the R platform were used to analyze the gene expression data [36,37]. Using the oligo package [38], raw intensity files were preprocessed to obtain robust multichip average (RMA) expression values. Next, differences in gene content between two cell types were identified using an unpaired two-sided t-test. Genes were considered differentially expressed with a fold change (FC) cutoff value ≥2 and a Benjamini-Hochberg (BH) corrected P value <0.05. Data were visualized in R as volcano plots and venn diagrams using the limma package [39]. A heatmap was generated using the heatplus package [40] and principal component analysis (PCA) was plotted using the prcomp function.
For data visualization, variance-based nonspecific filtering was performed using the genefilter package to remove probe sets exhibiting the smallest variations in expression across the samples [41]. For pathway analysis, the full unfiltered data sets were used. Gene ontology (GO) analysis was performed using QIAGEN's Ingenuity® Pathway Analysis (IPA®, QIAGEN Redwood City,
Brain-derived neurotrophic factor ELISA
Supernatant from WJ- and BM-MSC cultures was collected after 7 days and snap frozen. Brain derived neurotrophic factor (BDNF) was detected using the ChemiKine™ Brain Derived Neurotrophic Factor Sandwich ELISA kit (Chemicon®, Millipore, Overijse, Belgium) following the manufacturer's protocols. Data are presented as mean concentration (pg/mL) from three independent experiments.
Neurite outgrowth assay and immunocytochemistry
To evaluate CM-induced neurite outgrowth, a two-step protocol based on retinoic acid (RA) and BDNF exposure, previously described by Encinas et al. [46] and implemented for CM-induced neurite outgrowth by Gervois et al. [47], was used. SH-SY5Y cells were seeded at a density of 2.5 × 104 cells/cm2 in standard SH-SY5Y medium on 50 μg/mL collagen type I-coated glass coverslips. After 24 h, the medium was changed to standard SH-SY5Y medium supplemented with 10 μM RA (Sigma-Aldrich) and 1% FBS instead of 10% to induce neuronal differentiation. These cells were then kept in culture for 5 days with one medium change after 3 days. Next, the cells were washed with PBS and CM WJ-MSC, CM BM-MSC, or DMEM/F12 supplemented with 50 ng/mL BDNF or 10 μM RA (Immunotools, Friesoythe, Germany), and 0.1% FBS was added to the RA-induced SH-SY5Y cells to stimulate neuronal maturation.
The cells were kept in culture for 7 days and the culture medium or CM was changed every 2–3 days. Samples of each differentiation step were fixed with 4% PFA and immunostainings were performed according to a standardized protocol for immunofluorescence as described previously [48]. The fully BDNF-differentiated SH-SY5Y cells will be used as a reference for neurite outgrowth of maturated neuroblastoma cells. The influence of RA, BDNF, CM BM-MSC, and CM WJ-MSC on neurite outgrowth of SH-SY5Y cells was evaluated by measuring the mean length of the longest neurite on neurite-bearing cells that were stained for 1 h with mouse anti-human β-III tubulin (1/2000; clone 2G10; Sigma-Aldrich) using Fiji software [49] with the NeuronJ plugin [50]. At least 100 neurites were counted in each sample. The primary antibody was detected with an Alexa fluor 555-conjugated donkey anti-mouse secondary antibody (30 min incubation, 1/500; clone A31570; Invitrogen™; ThermoFisher Scientific). The appropriate isotype control (IgG2a; clone MG2a-53; Biolegend, London, United Kingdom) was used instead of the primary antibody in an equal concentration (500 ng/mL) to verify the staining specificity.
Induction of acute experimental autoimmune encephalomyelitis and subcutaneous stem cell transplantations
Lewis rats (Janvier Labs, Le Genest-Saint-Isle, France) were housed four per cage, in a controlled environment at 22°C–24°C with 55% humidity, on a 12-h light/12-h dark cycle, with ample cage enrichment and access to rodent's chow and water ad libitum. Acute EAE induction was performed as previously described [51]. Briefly, 8-week-old female Lewis rats were injected subcutaneously in both foot pads with 100 μL immunization fluid, consisting of 250 μg/mL guinea pig myelin basic protein (MBP; generated in-house), 2.5 mg/mL H37RA heat-killed mycobacterium tuberculosis (Difco, Detroit, MI), and 60 μL Complete Freund's adjuvant (Sigma-Aldrich). Stem cell transplantations were performed on −1 dpi, 3 dpi, and 6 dpi, through subcutaneous injection in both hind limbs (100 μL each side, lateral to femur) of 1 × 106 human WJ-MSC, BM-MSC, or MAPC (n = 8 animals each) in 200 μL of sterile saline solution, or with saline only (control, n = 8). Nine days postimmunization, rats (n = 3 per group) were sacrificed by perfusion with Ringer's solution (generated in-house) for lymph node (LN) collection.
Restimulation of lymphocytes
MBP-specific lymphocytes were obtained 9 days postimmunization by isolation of the popliteal LN. LN (n = 3 rats per group) were segregated into single-cell suspensions using a 70 μm strainer and 5 mL syringe plunger (both Greiner Bio-One). Cell suspensions were separated using ficoll density gradient centrifugation (Histopaque®-1077; Sigma-Aldrich) and the mononuclear layer was aspirated for subsequent enrichment of antigen-specific lymphocytes. For this, 2 × 105 cells were cultured for 48 h in round-bottom 96-well plates (Greiner Bio-One) in a restimulation medium, consisting of RPMI 1640 (Gibco) supplemented with 0.5% P/S, 1% sodium pyruvate (Gibco), 1% nonessential amino acids (Gibco), 20 μM 2-mercaptoethanol (Sigma-Aldrich), 2% inactivated autologous serum (Lewis rat serum, generated in-house), and 10 μg/mL MBP. For control conditions, MBP was omitted.
[3H]-thymidine incorporation
The proliferation of LN cells was assayed ex vivo by 3H-thymidine incorporation. Following 48 h of restimulation, 1 μCi [3H]thymidine (PerkinElmer, Waltham, MA) was added to the lymphocyte cultures for an additional 18 h. Next, cells were collected with an automated harvester (PerkinElmer) and incorporation of radioactivity was quantified using a β-plate liquid scintillation counter (PerkinElmer). Relative proliferation is given as stimulation index, calculated as the radioactivity of the stimulated sample divided by the average radioactivity of unstimulated samples.
Statistical analyses
Statistical analyses were performed using Graphpad Prism 5 (GraphPad Software, Inc., La Jolla, CA). Data sets were tested for normal distribution using D'Agostino and Pearson omnibus normality test. A one-way analysis of variances (ANOVA, Tukey post hoc) and two-tailed unpaired t-test were used to test for significant differences. Results are reported as mean with standard deviation (SD). The Kruskal–Wallis (Dunns post hoc) and Mann–Whitney U analysis were used for data sets, which did not pass normality. Results are reported as median with interquartile range (IQR). Differences were considered statistically significant at P values <0.05.
Results
Explant-derived WJ-MSC show a mesenchymal phenotype
MSC-like cells were isolated from the Wharton's jelly of the umbilical cord using explant culturing. Approximately 7 days after isolation, fragments of cord tissue attached to the culture surface and WJ-MSC started migrating out of the tissue (Fig. 1A). After an additional 7 days of culture, cells were harvested and seeded for further expansion and characterization. By then, cultures were explant free and cells were grown to 80% confluence before harvesting. WJ-derived cells were plastic adherent and consisted of a mixed population of smaller triangular shaped cells and larger cells with fibroblast-like morphology (Fig. 1B). When cultures were grown to full confluence, WJ-MSC became spindle shaped and formed colonies (Fig. 1C). In addition, cells were able to form colonies when cultured at low density (Fig. 1C, inset). WJ-MSC cultures kept growing for 23.4 ± 1.8 CPD and 81.0 ± 7.3 days until replicative senescence occurred. Of note, culturing WJ-MSC in KO-DMEM/F12 resulted in higher growth rates compared to parallel cultures in DMEM-LG (Supplementary Fig. S1A).
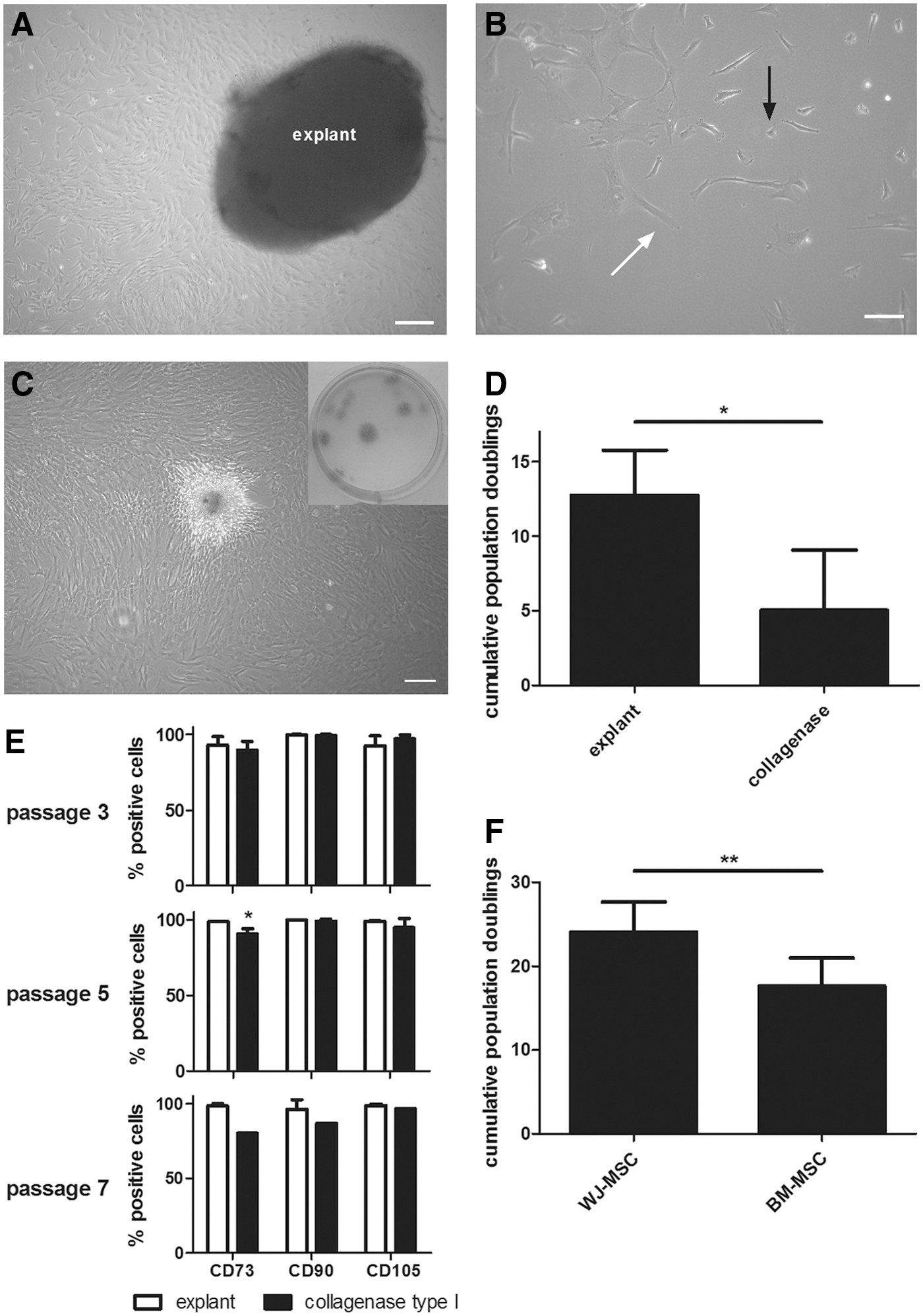
Culture characteristics of explant-derived WJ-MSC.
An alternative way to isolate cells from the Wharton's jelly comprises the enzymatic digestion of the cord matrix to release the cells [35,52]. When comparing both isolation methods, we observed that WJ-MSC obtained using the explant approach had a significantly higher growth rate compared to enzymatically isolated cells (parallel cultures; Fig. 1D). The latter displayed an aberrant morphology compared to standard cultures, having a large myofibroblast-like appearance (Supplementary Fig. S1B). This was reflected in their MSC marker profile (Fig. 1E), showing a decreased CD73 and CD90 expression upon prolonged culture (increasing passage). In addition, replicative senescence occurred more early in enzymatically isolated WJ cells, reflected by a lower growth rate and earlier growth arrest.
Next, we compared the growth characteristics, phenotype, and differentiation capacity of our established Wharton's jelly explant cultures with those of classical bone marrow-derived MSCs. The cells used for phenotypical characterization were harvested between passage 2 and passage 8. For culture kinetics analysis, cultures were grown until replicative senescence. To compare cell yield between the two cell types, CPDs were calculated over a period of 70 ± 5 days. In this time frame, all cells were still growing without any signs of replicative decline. Compared to BM-MSC (n = 7), our established WJ-MSC cultures (explant isolated + KO-DMEM/F12; n = 10) showed significantly higher CPD after 70 days of culturing (Fig. 1F).
Flow cytometry analysis demonstrated that WJ-MSC display a specific set of surface antigens (Table 1). WJ cells expressed the three main mesenchymal markers CD73, CD90, and CD105. All cultures were negative for HLA-DR and the hematopoietic markers CD14, CD19, CD34, and CD45. No expression of the pluripotency factors OCT4, SOX2, and NANOG was detected by flow cytometry or PCR (Supplementary Fig. S1C, D respectively; PCR materials and methods available in online Supplementary Data). In addition, WJ-MSC expressed several surface adhesion molecules and immunological markers, being either costimulatory or inhibitory in nature. Compared to BM-MSC, they expressed significantly higher levels of CD10, CD146, Integrin-α4 (CD49d), ICAM-1 (CD54), CD200, and PD-L2. Furthermore, both cell types expressed HLA class I molecules and costimulatory and inhibitory molecules (eg, HLA-ABC, HLA-E, PD-L and TLR-4) involved in immune cell communication. In addition, WJ-MSC expressed low levels of IFN-γR1 (CD119) and CXCR3 (CD183) receptors. Stem cell subpopulation-related markers CD146, CD117, CD271, and CD133 could be found in several samples, yet donor variability was observed for both MSC types, as shown by the high standard errors. Furthermore, WJ-MSC differentiate into the three classical mesenchymal cell types [33]. End-stage differentiation analysis by immunohistochemistry is shown in Supplementary Fig. S2 for adipocyte, osteoblast, and chondrocyte formation.
Bold values are significantly different.
P < 0.05.
P < 0.01.
P < 0.001 (unpaired t-test).
BM-MSC, bone marrow-derived mesenchymal stromal cell; WJ-MSC, Wharton's jelly-derived mesenchymal stromal cell.
Transcriptional analysis reveals WJ-MSC as a distinct type of stem cell
Differential gene expression between extra-embryonic WJ-MSC, adult BM-MSC, and MAPC was determined using Affymetrix Genechip microarray analysis. Global differences in gene expression profiles were visualized by unsupervised hierarchical clustering and PCA. As shown in Fig. 2A, B, the biological samples of each cell type cluster together and form separate groups, indicating that functionally distinct stem cell populations were obtained. Based on hierarchical clustering and Pearson's Correlation, WJ-MSC are unique stem cells, more closely related to BM-MSC than MAPC. Furthermore, clustering shows that MAPC are also distinct from BM-MSC. Volcano plots providing an indication for these differences are given in Supplementary Fig. S3. The number of differentially expressed genes in WJ-MSC (cutoff: false discovery rate 0.05 and Log FC 1) is depicted in Fig. 2A inset. Comparing WJ-MSC to BM-MSC, the expression of 795 genes was altered, of which 382 genes were upregulated and 413 genes were downregulated. Compared with MAPC, a total of 1,625 genes were altered, of which 797 were upregulated and 828 were downregulated in WJ-MSC. The top 30 most differentially expressed genes within the WJ cells are listed in Tables 2 and 3. Of note, WJ-MSC highly express multiple cell adhesion molecules and cytokines, such as ITGA2, DSG2, DSC3, IL1A, IL1B, IL8, and CD200, compared to BM-MSC and MAPC. A selection of these differentially expressed gene categories is given in Table 4.
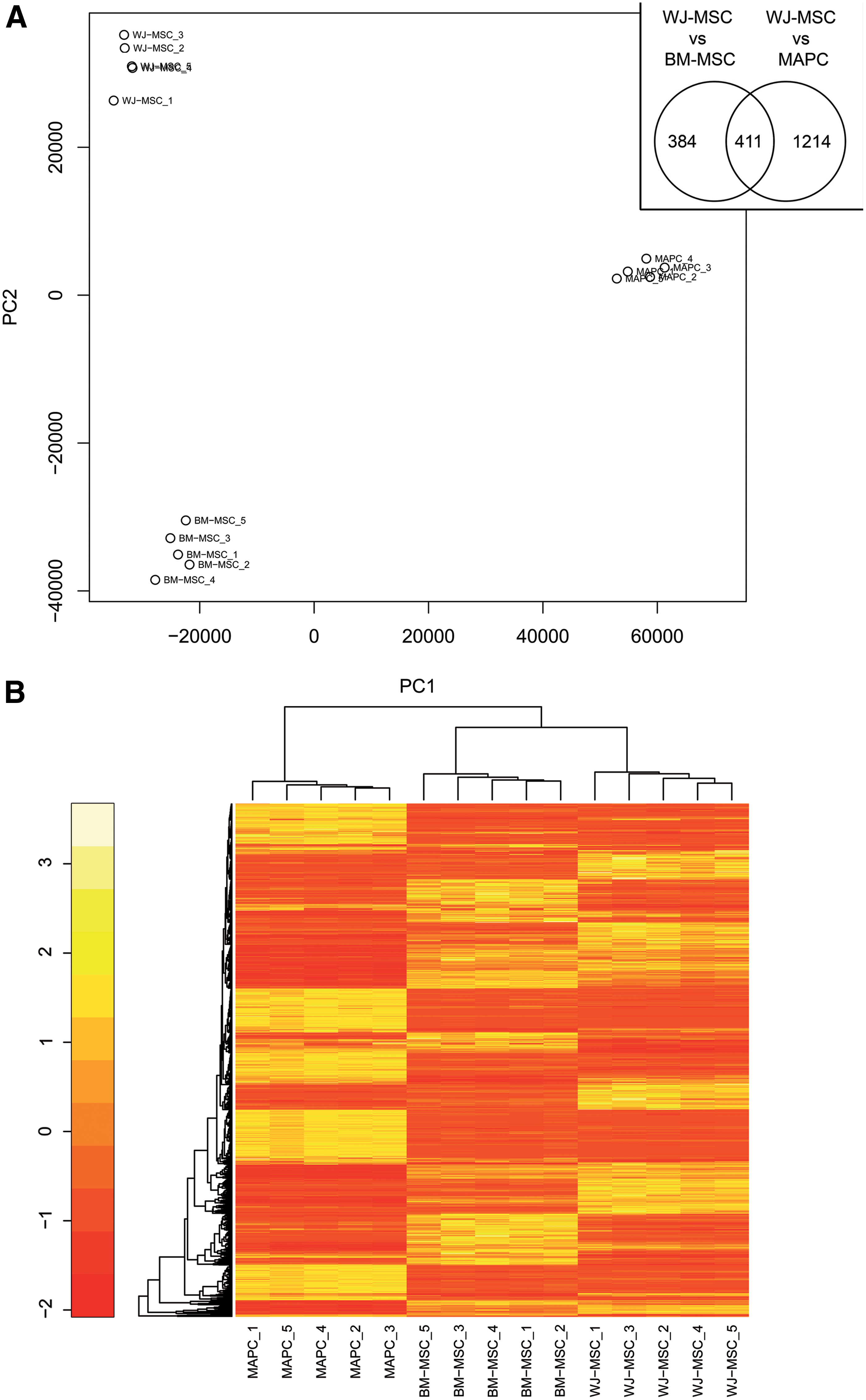
Hierarchical clustering based on differential gene expression profile of WJ-MSC, BM-MSC, and MAPC.
FC, fold change; n.d., not detectable.
Interestingly, WJ-MSC displayed 411 transcripts that were differentially regulated in comparison to both BM-MSC and MAPC (Fig. 2A inset), of which are 272 genes upregulated and 106 genes downregulated. To further elucidate the distinctive gene expression profiles, differential gene pools were screened for their related molecular signature using GO and pathway analysis software.
GO analysis identifies specialized gene expression within WJ-MSC
To determine the differences between the three cell types, we analyzed their gene expression patterns in more detail. GO analysis was performed to link the differentially regulated transcripts with their biological and molecular functions. For the analysis, we focused on the WJ group (n = 411 transcripts, differentially expressed versus both BM-MSC and MAPC). Enriched canonical pathways and functional groups are summarized in Table 5 and Supplementary Table S2.
P value* = for molecular and cellular functions and physiological system development and function, a range is given as the categories comprised multiple subfunctions
The most predominant of the overrepresented processes and functions of WJ-MSC include those pertaining to cell movement, cell–cell interactions and adhesion, cell death and survival, as well as cardiovascular system development and hepatic cell functioning. In addition, molecules related to embryonic development were enriched. In contrast, KLF4 was significantly higher expressed in BM-MSC compared to the other cell types. MAPC on the other hand showed enrichment of genes related to the cell cycle regulatory machinery, including cyclin-dependent kinases, such as CDK1 (Table 3).
The gene expression signature of WJ-MSC indicates an active cell modulatory potential through cell–cell contact and secretion of molecules
When focusing on the differentially expressed gene pools within WJ-MSC, a variety of cytokines and trophic factors such as IL8, IL6, IL1, CXCL8, BDNF, NTF3, TGFB2, VEGFA, FGF2, and LIF, as well as adhesion and immune regulatory molecules, including ICAM1, VCAM1, CD200, COX-2, PD-L1, CD49B, HLA-ABC, HLA-G,HLA-E, and HLA-F, were intrinsically overrepresented (Table 4). Moreover, BDNF protein levels were significantly increased in the supernatant of naive WJ cultures compared to bone marrow MSC (Fig. 3). In contrast, BM-MSC and MAPC overexpressed TLR4 mRNA. PTGES and HGF were found to be specifically increased in BM-MSC, and COX-1 was upregulated in MAPC. Expression of IDO-1, a T cell proliferation inhibitor [53], was not detected.

BDNF secretion by WJ-MSC and BM-MSC. BDNF concentration was measured in supernatant of WJ-MSC (n = 10) and BM-MSC (n = 7) using sandwich ELISA. Data are shown as mean absolute concentration (pg/mL) with SD. ***P < 0.001. BDNF, brain-derived neurotrophic factor.
In addition, when searching GO databases for increased biological processes and molecular functions within this differentially regulated gene pool of 411 transcripts, pathways involved in cell movement, cell–cell signaling, as well as cellular adhesion were enriched (Table 5 and Supplementary Table S2).
MSC secretome promotes neurite outgrowth in SH-SY5Y cells
To verify the neurotrophic activity of WJ- and BM-MSC, the conditioned medium was collected and used to mature differentiating neuroblastoma cells (SH-SY5Y). Neurite outgrowth gradually increased during the differentiation process (Fig. 4A–F) as was also demonstrated quantitatively (Fig. 4G). RA treatment (45.2 ± 0.4 μm) significantly increased the mean neurite length compared to controls (36.38 ± 4.36 μm; P < 0.05) (Fig. 4A, B), but prolonged RA exposure (Fig. 4C) did not additionally improved neurite outgrowth (45.14 ± 3.6 μm). Quantification of neurite outgrowth after β-III tubulin staining showed a significantly longer mean neurite length (P < 0.001) for cells treated with CM BM-MSC (66.79 ± 3.24 μm), CM WJ-MSC (90.71 ± 5.58 μm), and BDNF (82.32 ± 5.11 μm) (Fig. 4D–F respectively) compared to control, RA-induced, and RA-maturated SH-SY5Y cells (Fig. 4A–C respectively). BDNF- and CM WJ-MSC-maturated SH-SY5Y cells have significantly longer neurites than CM BM-MSC-maturated cells (P < 0.01 and P < 0.001, respectively). No significant difference in mean neurite length could be observed between CM WJ-MSC- and BDNF-maturated SH-SY5Y cells.
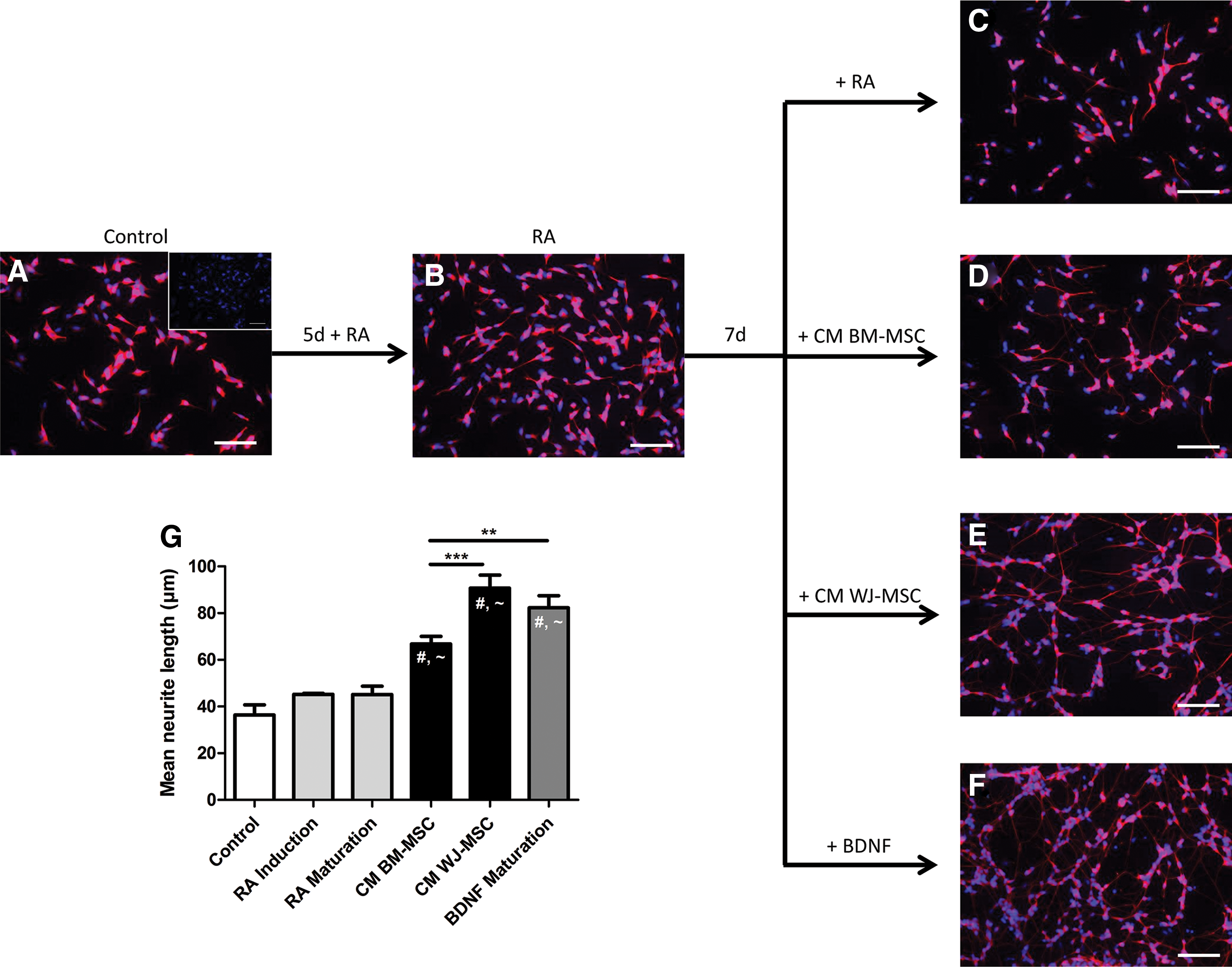
Neurite outgrowth of SH-SY5Y cells following exposure to CM BM-MSC and CM WJ-MSC. Control SH-SY5Y cells
Subcutaneous stem cell transplantation in acute EAE exerts local immune modulatory effects on T cells
To verify the immune suppressive phenotype of the stem cells, in vivo transplantation in acute EAE was performed. To assess potential effects on peripheral immune cell activation, stem cells were repeatedly transplanted subcutaneously near the immunization site 1 day before disease induction, and on day 3 and day 6 during EAE immune system priming. On day 9, lymphocytes were harvested from local LN and assessed for sensitivity to the immunizing antigen. MBP-specific proliferation of LN cells (mainly T cells) was measured and compared between treated and control animals (Fig. 5). LN-derived T cells from all treatment groups showed a significantly reduced reactivity toward MBP. These data show that WJ-MSC-, BM-MSC-, and MAPC-treated EAE animals exhibited a reduced lymphocyte proliferation upon restimulation with MBP, indicating that local application of stem cells interferes to some extent with their activation.

Subcutaneous stem cell transplantations exert local immune modulatory effects in Lewis rat EAE. Stem cells were subcutaneously transplanted 1 day before disease induction, and on day 3 & 6 after EAE induction. Leukocyte cultures derived 9 days postimmunization from the popliteal lymph nodes (LN) of WJ-MSC, BM-MSC, and MAPC treated (n = 3 each) and control animals (n = 3) were restimulated with MBP. Cultures without MBP served as baseline proliferation controls (dotted red line). The stimulation index is presented as mean ± SD. *P < 0.05 and **P < 0.01 versus restimulated cultures of saline-treated animals. Color images available online at
Discussion
MSCs are widely investigated as candidates for tissue engineering and cell transplantation in inflammatory degenerative diseases due to their differentiation capacity, their extensive immune modulatory properties, as well as trophic support functions [54]. The human umbilical cord has gained much attention over the years as an alternative source of multipotent MSCs. Furthermore, many reports attribute a broader expansion and differentiation potential to these cells than adult tissue-derived stem cells [10,29]. To investigate these extensive biological features of umbilical cord-derived WJ-MSC in comparison to adult bone marrow-derived stem cells, we first identified the most optimal method to isolate and expand the WJ-MSC. Afterward, we assessed their phenotype and defined their whole genome transcriptional profile.
We found that the explant isolation technique was superior compared to enzymatic culturing. The explant cell morphology more closely resembled that of fibroblast-like MSCs from bone marrow and the cells exhibited better growth rates than their enzymatic counterparts. Moreover, enzymatic cultures presented with earlier growth arrest and the spontaneous appearance of large myofibroblast-like cells. We speculate that this population is induced by the applied isolation procedure itself. In fact, enzymatic isolation has been reported to potentially alter the immunophenotype and functions of the cell [25,55], as well as preselect for endothelial progenitors [56]. In concordance with other studies [57,58], we show that the Wharton's jelly explant method yields lower variability and better growth rates than enzymatic isolation, here performed as collagenase type I digestion. The latter was previously reported to enrich for MSCs [35]. In our hands, a heterogeneous stem cell isolate containing MSCs was initially isolated, but expression of MSC markers declined with increasing culture time (passage 5). Similar morphological and marker expression changes were previously reported for senescent BM-MSC [59] or long-term cultured BM- and amnion-derived MSC [60]. Although enzymatic digestion is the fastest way of obtaining cells from the cord matrix [52], explant or whole cord cultures are considered highly enriching for MSCs, since spontaneous migration out of the tissue needs to occur [25,33,61].
Our further experiments were performed with explant-derived Wharton's jelly stromal cells. Similar to previous reports [26,62], the cells exhibited MSC-like properties based on their culture morphology, ability to differentiate into the classical mesenchymal cell types (bone, fat, and cartilage), and their surface marker expression profile. However, we observed subtle differences in expression of immune-related and adhesion molecules. In summary, these findings indicate that explant-derived WJ-MSC express the previously defined mesenchymal phenotype [4], but differ from BM-MSC regarding their cell surface molecules and growth characteristics.
Extra-embryonic tissue-derived stem cells are attributed extended therapeutic properties over adult stem cells [63 –65]. To further find evidence for that, we performed a whole genome transcriptional comparison of WJ-MSC to both lineage-restricted (classical BM-MSC) and lineage-nonrestricted MAPC. In this way WJ-MSC were compared to adult cells derived from the same tissue, but with different stem cell potency (multipotent BM vs. pluripotent MAPC). MAPC, which are derived from bone marrow under defined culture conditions, yield a unique population of stem cells, which have the ability to differentiate into cells from all three germ layers, express markers similar to embryonic stem cells, and show higher replicative potential compared to standard MSCs [66 –69].
In this study, microarray analysis revealed clear transcriptional differences between stem cells derived from Wharton's jelly and bone marrow. Our data indicate that WJ cells overexpress a multitude of factors with proregenerative functions related to cell–cell adhesion, immune modulation, and neurotrophic support. It does not imply that in this comparison, the bone marrow-derived stem cell populations do not express these factors. Ample in vitro and preclinical data are available on their phenotype and regenerative properties [67,70 –72]. Moreover, both types of bone marrow-derived cell populations showed a distinct transcriptional profile. Specifically for WJ-MSC, an upregulated expression of anti-inflammatory molecules, such as, for example, CD200, PD-L1, and nonclassical HLAs, along with adhesion molecules, such as ICAM1 and VCAM1, was observed. Such molecules have been implemented in the immunosuppressive actions of stem cells both in vitro and in vivo [73,74].
In this study, we confirm the immune modulatory activity in vivo, after stem cell administration near the immunization site in Lewis rat EAE. This setup allows for direct assessment of T cell modulation, and although the intrinsic immune modulatory profile of the three cell types was different, we found no differences in their ability to reduce T cell proliferation. In this regard, the reduction in autoantigen-induced T cell proliferation for WJ-MSC, BM-MSC, and MAPC is indicative of an active interplay between these cells and the inflammatory milieu (licensing on the spot), and an effector mechanism involving paracrine signaling, activation of immune modulatory processes, and stem cell homing to LN after subcutaneous implantation. Multiple molecules might be involved in suppression of T cells and they are not necessarily the same for each cell type. We have previously shown that IDO-1 expression in WJ-MSC is involved in suppression of T cell proliferation, but only appears after licensing [53]. In this study, IDO was not detected in the naive (unlicensed) stem cell populations. Moreover, other immune cells in the inflammatory cascade might be affected. It was previously shown for neural precursor cells that their secretome affected antigen-presenting cell functioning after subcutaneous injection in EAE [75]. Regarding LN homing, we and others have previously shown that WJ-MSC possess the proper trafficking potential for homing to the peripheral lymphoid tissues [53,76]. Furthermore, the majority of intravenously transplanted cells are trapped in the pulmonary vasculature and spleen. By subcutaneous administration, cell trapping was avoided.
In addition to the expression of immune signaling molecules, we show constitutive higher levels of BDNF secretion in normal WJ-MSC cultures compared to BM-MSC. Previously, we demonstrated a similar gene activity pattern in WJ-MSC, together with a beneficial effect on neuroinflammation and neurodegeneration after transplantation in an animal model of multiple sclerosis [53]. Expression of immune suppressive and neurotrophic factors is a crucial feature for triggering endogenous repair pathways [25,74]. Hence, the paracrine potential of WJ- and BM-MSC to induce neurite outgrowth in human SH-SY5Y neuroblastoma cells was investigated. In this study, an adapted protocol based on sequential RA and BDNF signaling was used [47]. In the experimental setup, RA-treated SH-SY5Y cells were exposed to CM of WJ-MSC or BM-MSC. In parallel, BDNF-induced maturation was included as a positive control. This RA stimulation before the addition of CM or BDNF is preferable as it increases the expression of Tyrosine Kinase (TK) A and B receptor, thereby improving the responsiveness for neurotrophins such as BDNF, which was shown to be secreted by the stem cell populations that were used in this study [46,77]. The results described in this study are in line with Pires et al. [78], who reported that the secretome of BM-MSC and human umbilical cord perivascular cells (HUCPVC) boosts neurite outgrowth of SH-SY5Y cells.
Due to the absence of direct contacts between BM-MSC, WJ-MSC, and SH-SY5Y cells, the observed effects are solely attributed to the paracrine factors produced by these stem cell populations. BDNF was one of the neurotrophins identified as increased in the secretome of our WJ-MSC cultures and is a well-described neurite outgrowth inducer [79], thereby likely responsible for the observed increase in neurite outgrowth. Martins et al. recently showed that BDNF derived from HUCPVC is a significant determinant for axonal outgrowth in rat embryonic cortical neurons [80]. They observed enhanced axonal growth upon local CM application, which was lost when BDNF was depleted from the CM by TK receptor blocking. Nevertheless, the neurite outgrowth observed in our setup can also be attributed to other factors than the CM-derived BDNF. In our cultures, the concentration of BDNF secreted by WJ-MSC and BM-MSC is less than 400 pg/mL, compared to the positive control containing 50 ng/mL BDNF, thereby hinting at the involvement of other effector molecules. This hypothesis is in line with Crigler et al. who demonstrated that the effect of BM-MSC-produced BDNF is only partially responsible for the observed effects on SH-SY5Y cells [81]. The involvement of several distinct molecular categories, including neurotrophic and angiogenic growth factors, cytokines, and microvesicles, have been reported [64,74,78,82,83]. Likewise, a number of in vivo studies showed that the expression of such cytokines and neuroprotective growth factors by WJ-MSC correlates with a beneficial outcome in rat models of Parkinson's disease [84], ischemic stroke [85,86], and spinal cord injury [87].
Finally, WJ-MSC display a plethora of other molecules for immunological interaction, cell adhesion, and homing (reviewed elsewhere [62,88]). Importantly, homing capacity together with a reduced costimulatory surface profile and immunomodulatory phenotype are essential for effective cell communication and migration toward sites of injury in both autologous or allogeneic application. Similar to the study by Fong et al., we observed comparable results for the expression of HLA class I genes, cytokines, and adhesion molecules, but we found no expression of pluripotency markers (eg, OCT4, SOX2, and NANOG) in WJ-MSC [63].
Although many tissue-derived stem cells are designated as MSCs, most comparative in vitro studies of stem cells derived from different tissue sources have indicated functional differences in gene and protein expression, postulating the novel stem cell subtype to be better than the other. Plausible reasons for such differences could be as follows: (1) no uniform marker set identifies MSCs from all these sources, (2) variations in isolation technique and culture medium components [89], or (3) MSCs acquire culture-induced differences in cell potency [89,90]. In addition, the aseptic collection and processing of tissues is critical in not only preventing microbial contamination but also stem cell activation through, for example, TLRs [91]. Roobrouck et al. indicated that both phenotype and functional properties of human mesangioblasts, BM-MSC and MAPC, were partially influenced by the culture conditions [90]. Gatta et al. reported genotypical changes after extended in vitro culturing of WJ-MSC (12 passages) [92]. In this study, both BM-MSC and WJ-MSC were harvested at early passage (passage 3), minimizing the extent of prolonged culturing effects. In addition, all three cell types were cultured in different media inherent to their isolation method. We found a similar picture for umbilical cord MSC as was previously reported by Klingemann et al., who compared the overall transcriptional profiles of umbilical cord- and bone marrow-derived MSC cultured under the same conditions [93].
Nevertheless, to date, it remains to be determined whether the isolation method used or additional growth factors in culture have a potential impact on the observed expression profile of the WJ cells. Based on our data and others (described above & [25,94]), we suggest that the isolation method already selects for a certain cell phenotype, which depending on the amount of physical stress during the handling (eg, enzymatic cleaving of membrane receptors, mechanical stress of forceps, or centrifugation) predestines for the specific gene expression patterns observed later on in cultured cells.
In summary, this study reports the isolation of mesenchymal-like cells from the Wharton's jelly that express a distinct transcriptional signature compared to lineage-restricted and lineage-nonrestricted bone marrow-derived stem cells. Overall, our data support the previous notion that WJ-MSC are equipped with potent trophic and immunomodulatory properties, related to their functions in vivo at the fetus–maternal interface [24,27,73]. Furthermore, we specifically observed an overrepresentation of immune modulatory and neurotrophic growth factors in WJ-MSC, indicating a potential beneficial role for transplantation in neurodegenerative diseases like spinal cord injury, stroke, multiple sclerosis, Parkinson's disease, or amyotrophic lateral sclerosis, for which there is currently no cure.
Footnotes
Acknowledgments
We thank Goele Willekens, Katrien Wauterickx and Christel Bocken (Hasselt University, Biomedical Research institute), Kristel Gijbels (ReGenesys), Sabina Vanherle, and Dr. Patrick Lindsey (University Maastricht Genome Center) for their assistance with the stem cell cultures or microarray analysis. Especially, we thank Stephane Plaisance (VIB-BITS) for his advice and support with the pathway analysis software.
This research was promoted by grants from the IWT (agentschap voor Innovatie door Wetenschap en Technology), Alma-in-Silico (EMR INT4.-1.3.-2008-03/003), the VIB (Vlaams Instituut voor Biotechnologie), the transnational University Limburg, Hasselt University, and Limburg Sterk Merk. Pascal Gervois is supported by grant number 12 U7718 N of the Fonds Wetenschappelijk Onderzoek Vlaanderen. The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the article.
Author Disclosure Statement
The authors declare that no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.