
Abstract
The physiological root resorption of deciduous teeth is a normal phenomenon, but the mechanisms underlying this process are still unclear. In this study, deciduous dental pulp stem cells (DDPSCs) and permanent dental pulp stem cells (DPSCs) were derived from deciduous teeth and normal permanent teeth at different stages of resorption. In the middle stage of root resorption, DDPSCs exhibited an increase in the ability to induce osteoclast differentiation. Activation of the alpha 7 nicotinic acetylcholine receptor (α7 nAChR) by secretory mammalian Ly-6 urokinase-type plasminogen activator receptor-associated protein 1 (SLURP-1) caused a significant increase in the expression levels of NF-κB, receptor activator of nuclear factor-kappa B ligand (RANKL), and the ratio of RANKL/osteoprotegerin (OPG). These effects were inhibited by alpha-bungarotoxin (α-BTX). Furthermore, the expression levels of RANKL/OPG were significantly reduced following inhibition of NF-κB. High-strength, dynamic positive pressure increased the expression of SLURP-1 and α7 nAChR in DDPSCs in the stable stage. These data indicated that mechanical stress stimulated the expression of SLURP-1 and α7 nAChR in DDPSCs. Additionally, SLURP-1 activated α7 nAChR, thereby upregulating the expression of NF-κB and enhancing its activity, thus regulating RANKL/OPG expression and affecting the ability of DDPSCs to influence osteoclastogenesis, which likely enhances root resorption and leads to the physiological loss of deciduous teeth.
Introduction
P
Previous studies identified stem cell niches in dental pulp and deciduous dental pulp, from which cells can proliferate and differentiate rapidly and participate in tissue repair with external stimuli [5 –7]. The expression of RANKL in deciduous dental pulp stem cells (DDPSCs) is significantly higher than in dental pulp stem cells (DPSCs), suggesting that DDPSCs may be involved in the regulation of root resorption [8]. However, the specific mechanism underlying the actions of DDPSCs in the regulation of osteoclast differentiation in physiological root resorption is unclear.
The alpha 7 nicotinic acetylcholine receptor (α7 nAChR) is a ligand-gated ion channel receptor with unique ion conduction characteristics that can exert control over the extracellular Ca2+ concentration [9,10]. Since Grando et al. [11] discovered nAChR in non-nervous system tissues, α7 nAChR has been shown to mediate the process of bone resorption in oral periodontal tissues through a series of actions [12]. Recent studies have shown that α7 nAChR mediates osteoclastogenesis [13]. In vivo, RANKL-mediated osteoclastogenesis has been shown to be reduced in mice lacking α7 homomeric nAChR or β2-containing heteromeric nAChRs, and bone histomorphometry revealed an increased bone volume and impaired osteoclastogenesis in male mice lacking the α7 nAChR. However, the ligand of α7 nAChR in an environment without nicotine and the downstream signaling pathway involved in the regulation of osteoclast differentiation after its activation have yet to be identified.
Secretory mammalian Ly-6 urokinase-type plasminogen activator receptor-associated protein 1 (SLURP-1) is a member of the Ly-6 protein superfamily that can act as an endogenous ligand to specifically activate α7 nAChR and is involved in transmembrane signal transduction, cell activation, and cell adhesion processes [14,15]. Some researchers have reported that SLURP-1 can mediate the influx of Ca2+ through α7 nAChR and activate a variety of downstream signaling pathways, which causes upregulation of NF-κB in Het-1A human oral keratinocytes [16].
Physiological root resorption of deciduous teeth is a complex biological process, and studying the mechanisms underlying this process is difficult due to the nature of the physical environment, where one of the most significant factors is occlusive force. Some researchers believe that primary root resorption of deciduous teeth may also be related to changes in occlusive force [17,18]. In the replacement period of dentition, the occlusive force exerted on the deciduous teeth gradually increases with the development of the jaw, facial growth, and increased masticatory muscle function. When the force applied to the deciduous teeth exceeds the tolerance thresholds of the periodontal ligament, it may cause the local tissue to produce a variety of cytokines to promote odontoclast activation and initiate root resorption [18]. During masticatory movements, pulp is also stimulated by force [19,20], and force can alter the intracellular signaling, proliferation, osteogenic differentiation, and gene expression of DPSCs [19].
In this study, we hypothesized that the expression of SLURP-1 and α7 nAChR in DDPSCs would change under mechanical stress. Following the activation of α7 nAChR by SLURP-1, the ratio of RANKL/OPG expression was altered through upregulation of NF-κB, thereby regulating osteoclastogenesis ability. The aim of this study was to investigate the possible role of DDPSCs in osteoclast differentiation through α7 nAChR during physiological root resorption.
Materials and Methods
Tooth samples
Deciduous incisors and permanent premolars were extracted because of retained deciduous teeth or orthodontic treatment needs. All the teeth are healthy and in good periodontal condition. According to the stage of root resorption, the teeth were subdivided into four groups (Fig. 1A): stable stage (S group, incisor teeth showing lingual root resorption <1/3 of the root length, n = 4); middle stage (M group, resorption between 1/3 and 2/3, n = 7); final stage (F group, resorption >2/3, n = 6); and premolar teeth (P group, n = 6). All of the subjects came from the School of Stomatology, Fourth Military Medical University and donors were free from any recent clinical acute or chronic infections. Before the investigation, the participants and their guardians were informed of the objectives of the study. Written informed consents were obtained from all subjects before the initiation of the study. Ethical approval was obtained from the Ethics Committee of the School of Stomatology, Fourth Military Medical University.

Isolation and identification of DDPSCs and DPSCs.
Cell culture
DDPSCs and DPSCs were isolated and cultured as previously described [8,21,22]. All of the teeth were washed in sterile phosphate-buffered saline (PBS) and were cut around the cementum–enamel junction using sterilized dental fissure burs to expose the pulp chambers. The pulp tissue was gently separated from the root and then cut into small pieces (1 mm3). Pulp tissues were digested with type I collagenase (Sigma, Santa Clara, CA) for 40 min at 37°C. Single cell suspensions were generated through filtration through a 70-μm strainer, followed by washing with PBS and resuspension in Alpha Minimum Essential Medium (a-MEM; HyClone, Logan, UT) supplemented with 10% fetal bovine serum (FBS; HyClone), 0.292 mg/mL glutamine (Invitrogen, Carlsbad, CA), 100 U/mL penicillin (Invitrogen), and 100 mg/mL streptomycin (Invitrogen). The cell suspension was then incubated at 37°C in a humidified atmosphere of 95% air and 5% CO2. The culture medium was changed every 3 days. The explants were maintained in six-well plates (Costar, Cambridge, MA) for 2 weeks until the proliferating fibroblasts became subconfluent.
Osteogenic/adipogenic differentiation of DDPSCs and DPSCs
Osteogenic/adipogenic differentiation of DDPSCs and DPSCs was performed according to previously reported methods [18 –23]. DDPSCs and DPSCs (fourth passage) were placed in six-well plates (1 × 104 cells/well) and cultured in α-MEM supplemented with 5% FBS, 100 U/mL penicillin, 100 mg/mL streptomycin, and 50 mg/mL anti-ascorbic acid for 24 h. Then, the medium was replaced with osteogenic induction medium (100 nM dexamethasone, 50 pg/mL ascorbic acid, and 5 mM beta-glycerophosphate; Sigma) or adipogenic induction medium (0.5 mM methylisobutylxanthine, 0.5 mM hydrocortisone, and 60 mM indomethacin; Sigma) and the cells were incubated for 21 or 28 days, respectively, with the medium being changed every 3 days. Finally, induced cells were fixed with 75% ethanol and stained with 2% Alizarin Red (Sigma) and Oil Red O solution, respectively. After washing with PBS, the cells were examined and imaged using an inverted microscope (Olympus, Japan).
Immunophenotype analysis
DDPSCs were stained with antibodies for stem cell surface markers and analyzed through flow cytometry as previously described [18 –22]. To identify the phenotypes of the DDPSCs, 5 × 105 cells (fourth passage) were incubated with phycoerythrin-conjugated monoclonal antibodies for human CD146 (BioLegend, San Diego, CA), CD105 (eBioscience, San Diego, CA), and CD34 (BioLegend) and with fluorescein isothiocyanate-conjugated monoclonal antibodies for human CD90 (eBioscience), CD45 (eBioscience), and CD14 (eBioscience), according to the manufacturer's instructions. The incubation procedure was conducted at 4°C in the dark for 1 h. After washing with PBS, cells were subjected to flow cytometry (Beckman Coulter, Fullerton, CA) analysis.
Coculturing with RAW264.7 cells and tartrate-resistant acid phosphatase staining
Coculturing with RAW264.7 cells and tartrate-resistant acid phosphatase (TRAP) staining were performed according to previously published methods [24]. DDPSCs and DPSCs (fourth passage) were seeded into a 24-well culture plate (1 × 105 cells/mL). After 12 h, RAW264.7 cells (1 × 106 cells/mL) were directly placed in culture with DDPSCs, DPSCs, and α-MEM (containing 30 ng/mL M-CSF; Wobai, Shanghai, China). The medium was partially changed every 3 days. After 7 days, the cells were stained for TRAP analysis using the Acid Phosphatase Leukocyte Kit (Sigma-Aldrich). The cells were then observed and imaged using an inverted microscope. Five different imaging fields were acquired for each group to quantify the number of TRAP (+) cells. The average number of TRAP (+) cells was compared between the groups.
RNA interference
SLURP-1 siRNAs and the universal negative control dicer substrate duplex were purchased from RiboBio Co. Ltd. (Guangzhou, China). DDPSCs from the M group (fourth passage) were trypsinized, counted, and seeded at 1 × 105 cells/well into 24-well culture plates, such that the cells achieved 30%–50% confluence on the day of transfection. Then, siRNA transfection was conducted with riboFECT™ CP (RiboBio Co. Ltd., Guangzhou, China) according to the manufacturer's instructions. The DDPSCs were subsequently incubated at 37°C with 5% carbon dioxide, and the DDPSCs were cultured for another 24 and 72 h for quantitative reverse transcriptase–polymerase chain reaction (qRT-PCR) and western blot analysis, respectively.
SLURP-1, alpha-bungarotoxin, and pyrrolidine dithiocarbamate treatment
DDPSCs were treated with SLURP-1 (Abcam, Cambridge, United Kingdom), alpha-bungarotoxin (α-BTX; Toctis, Bristol, United Kingdom) or pyrrolidine dithiocarbamate (PDTC; Beyotime, Shanghai, China) as previously described [25 –27]. DDPSCs from the M group (fourth passage) were trypsinized, counted, and seeded into six-well culture plates at 5 × 104 cells/well. Full-length recombinant SLURP-1 (1 μg/mL), alpha-BTX (10−8 M), or PDTC (5 × 10−5 M) was added to the culture medium when cells grew at a density of 80%. The cells were subsequently cultured at 37°C in a humidified atmosphere of 95% air and 5% CO2 for 24 h.
Total RNA extraction and qRT-PCR
Total RNA from DDPSCs and DPSCs was isolated using the TRIzol reagent (Invitrogen). Reverse transcription–polymerase chain reaction (RT-PCR) was performed with 3 μg of RNA using the SuperScript First-Strand Synthesis Kit (TaKaRa, Dalian, China). The real-time PCR system was prepared using the QuantiTect SYBR Green PCR Kit (TaKaRa) and the Applied CF BiosystemsX96™ Real-Time sequence detection system (Applied Biosystems, Darmstadt, Germany). The primers used in this study are listed in Table 1.
α7 nAChR, alpha 7 nicotinic acetylcholine receptor; α-BTX, alpha-bungarotoxin; OPG, osteoprotegerin; RANKL, receptor activator of nuclear factor-kappa B ligand; SLURP-1, secretory mammalian Ly-6 urokinase-type plasminogen activator receptor-associated protein 1.
Protein isolation and western blot analysis
Protein isolation and western blotting was performed according to previously described protocols [27,28]. The Nucleoprotein Extraction Kit (Sangon Biotech, Shanghai, China) was used for nuclear protein preparation based on the manufacturer's instruction. Generally, cells at the fourth passage were harvested on ice and hypotonic buffer (Sangon Biotech) was mixed with cells. After centrifuging and collecting precipitate for three times, lysis buffer for nuclear protein isolation (Sangon Biotech) was added. After 20 min incubation on ice, nuclear protein was prepared in the supernatant. The primary antibodies used in this study included rabbit anti-human SLURP-1(dilution 1:50,000; Abcam), rabbit anti-human α7 nAChR (dilution 1:500; Abcam), rabbit anti-human NF-κB p65 (dilution 1:2,000; Abcam), rabbit anti-human RANKL (dilution 1:1,000; Abcam), rabbit anti-human OPG (dilution 1:3,000; Abcam), and mouse anti-human β-actin (dilution 1:500; Boster, Wuhan, Hubei, China). Then, the membranes were incubated with horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG secondary antibodies (Boster Biological, Wuhan, China). The blots were visualized using an Enhanced Chemiluminescence Kit (Amersham Biosciences, Shanghai, China) according to the manufacturer's instructions. Semiquantitative analysis was conducted by determining the densities of the labeled protein bands for each group, with β-actin being used as the internal reference (ImageJ, Rawak Software, Inc., Germany). The semiquantitative results were normalized to β-actin expression.
Experimental application of mechanical stress
Mechanical stress application was performed according to a previously described protocol [29 –31]. To mimic compressive stress on the dental pulp during chewing movements, a dynamic positive pressure-controlled cellular strain unit was used. Cell culture plates were placed inside a pressure chamber, which was then placed within a CO2 culture incubator (37°C). Pressure on the cells was generated by compressing the gas phase (2% CO2, 95% N2) at a specific frequency in the pressure chamber. The DDPSCs (fourth passage) from the S group were seeded in six-well plates at a density of 1 × 105 cells/mL and randomly divided into six groups. One group was a blank control, cultured for 3 days under normal condition, and the other five groups were exposed to 0–45, 0–90, 0–135, 0–180, or 0–225 KPa dynamic positive pressure, with a frequency of 0.1 Hz for 2 h/day, with 3 days of continuous mechanical stimulation.
Statistical analysis
All experiments were performed in triplicate. Data were expressed in a mean ± standard deviation form. The statistical differences between two groups were determined using two-tailed unpaired Student's t-test, whereas that for more than two groups one way analysis of variance was used followed by Tukey's post-test. SPSS 13.0 software was utilized and P value less than 0.05 was considered as statistically significant.
Results
Isolation and characterization of DDPSCs and DPSCs
The DDPSCs and DPSCs isolated from the teeth of each group exhibited a long fusiform shape, with a round nucleus that occupied most of the cell body and long processes on both sides of the cell (Fig. 1A). The cells were arranged in a radial or vortex-like distribution. Compared with that of DDPSCs, the DPSCs exhibited a longer growth period of 7–10 days, and their cell bodies were larger, but the morphology of the DPSCs and DDPSCs were similar, and the difference was not obvious. After osteogenic/adipogenic induction for 21 and 28 days, respectively, both the DDPSCs and DPSCs formed mineralized nodules (Alizarin Red staining, Fig. 1B) and lipid droplets (Oil Red O staining, Fig. 1B), indicating that the cells had multidirectional differentiation abilities. The flow cytometry assay revealed a typical pattern for MSCs: CD146 (+), CD105 (+), CD90 (+), CD34 (−), CD45 (−), and CD14 (−) (Fig. 1C).
Induction of osteoclast differentiation by DDPSCs and DPSCs
DDPSCs and DPSCs were cocultured with RAW264.7 cells for 7 days, and TRAP staining was then performed. The results showed that the DDPSCs and DPSCs from each group could induce TRAP (+) osteoclast-like cell formation (Fig. 2A). Quantitative analysis of TRAP (+) osteoclast-like cells (Fig. 2B) showed that the number of TRAP (+) osteoclast-like cells induced by DDPSCs from group M was significantly higher than that in the other groups (P < 0.05). There was no significant difference in the number of TRAP (+) osteoclast-like cells formed in the other groups. The expression of RANKL and OPG mRNA and protein in DDPSCs and DPSCs was examined through real-time PCR and western blotting (Fig. 2C–E). In the DDPSCs from group M, real-time PCR showed that the expression of RANKL increased significantly (P < 0.05) and it is higher than that in the group S, F, and P. The expression of OPG was lower (P < 0.05) and the ratio of RANKL/OPG from group M was significantly higher (P < 0.05) than that from any other group. The results of western blotting were consistent with those of the real-time quantitative PCR.

Induction of osteoclast differentiation by DDPSCs and DPSCs.
Expression of α7 nAChR in DDPSCs and DPSCs
Gene and protein expression of α7 nAChR in cells derived from each group were then examined. Compared with group S, F, and P, the expression of α7 nAChR was significantly enhanced in DDPSCs from the M group (P < 0.05), as demonstrated by qRT-PCR (Fig. 3A). The western blot assay (Fig. 3B, C) showed that the protein expression of α7 nAChR was also higher in DDPSCs from the M group than that from any other group (P < 0.05). This difference was statistically significant.

Differential expression of α7 nAChR in DDPSCs and DPSCs.
SLURP-1 regulates the ability of DDPSCs to induce osteoclast differentiation through α7 nAChR
The expression of SLURP-1 in DDPSCs and DPSCs was detected through real-time PCR and western blotting. The real-time PCR analysis (Fig. 4A) showed that the expression of SLURP-1 in the group M DDPSCs was significantly higher than that in the other groups (P < 0.05), and SLURP-1 expression was slightly higher in the S group compared with the P group (P < 0.05). The results of western blotting (Fig. 4B, C) were consistent with those of the real-time quantitative PCR.
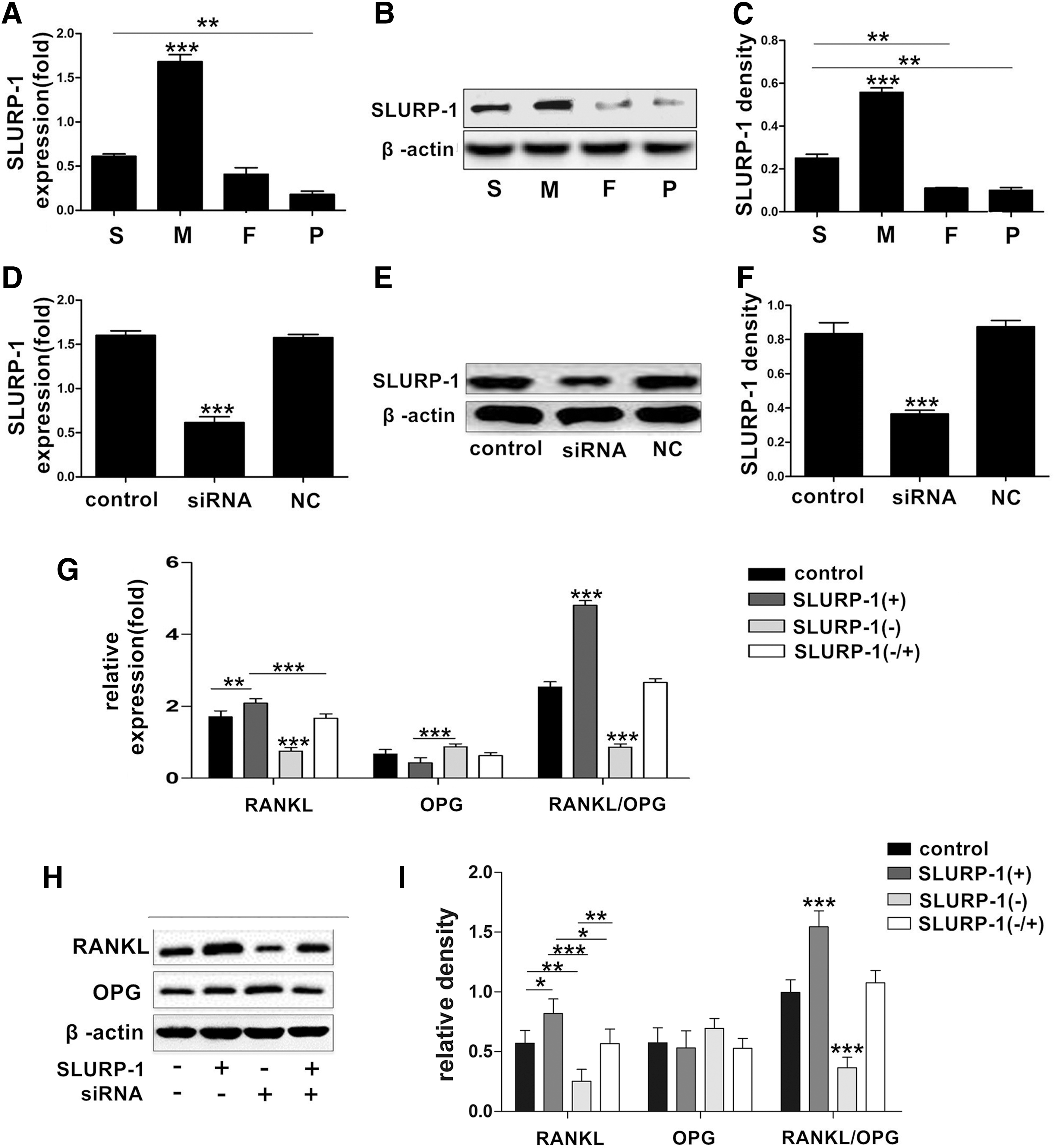
SLURP-1 regulates RANKL/OPG expression levels in DDPSCs.
siRNA was transfected into the M-group DDPSCs to downregulate the expression of SLURP-1 (Fig. 4D–F), and exogenous SLURP-1 protein was then added to achieve SLURP-1 overexpression. The expression levels of RANKL and OPG in the DDPSCs were detected through real-time quantitative PCR and western blotting. The results showed that the expression levels of RANKL and RANKL/OPG increased significantly (P < 0.05) when SLURP-1 was overexpressed (Fig. 4G–I). The expression of RANKL and RANKL/OPG decreased after SLURP-1 was silenced (P < 0.05) (Fig. 4G–I). The expression of RANKL and OPG was not significantly different from the blank control group when SLURP-1 protein was added after SLURP-1 was silenced (Fig. 4G–I).
The gene and protein expression levels of the RANKL and OPG were detected through real-time quantitative PCR and western blotting following the treatment of SLURP-1 protein and/or α-BTX to the group M DDPSCs. The results (Fig. 5A–C) showed that the expression of RANKL and RANKL/OPG increased significantly (P < 0.05) when SLURP-1 protein was given, whereas RANKL and RANKL/OPG expression decreased significantly (P < 0.05) when the α7 nAChR blocker α-BTX was added, and their expression remained decreased (P < 0.05) when SLURP-1 protein and α-BTX were given simultaneously. In addition, authors cocultured DDPSCs from group M with RAW264.7 cells. TRAP staining results (Fig. 5D, E) further indicated that α-BTX, the specific antagonist of α7 nAChR, would suppress the upregulation effect of SLURP-1 on DDPSCs to induce the precursor cells to differentiate into osteoclast cells.
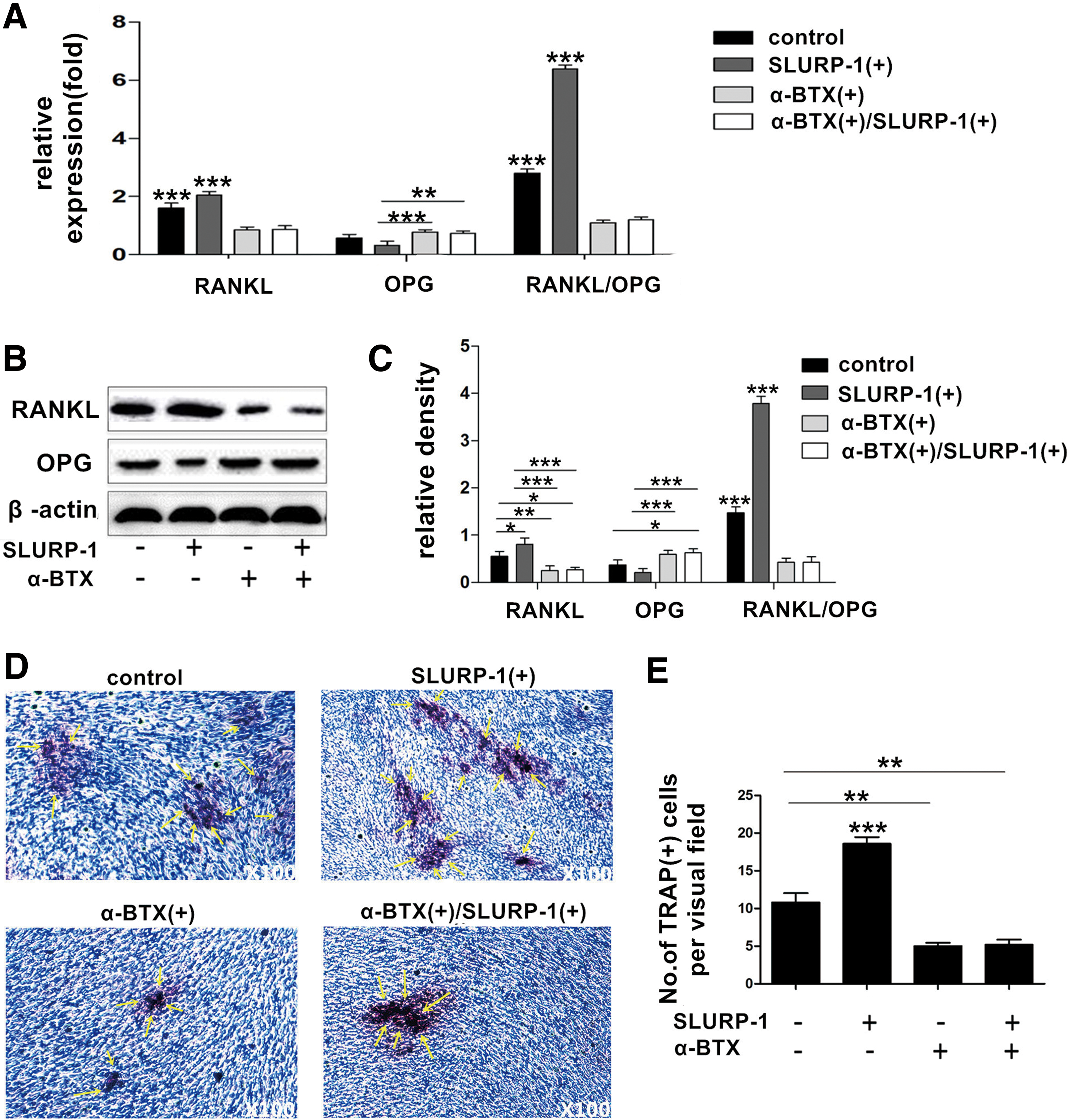
SLURP-1 regulates the ability of DDPSCs to induce osteoclast differentiation through α7 nAChR.
NF-κB regulates RANKL/OPG in DDPSCs
The expression of the NF-κB gene and nuclear p65 protein in DDPSCs and DPSCs was detected through real-time quantitative PCR and western blotting, respectively. The results (Fig. 6A–C) showed that the expression of the NF-κB gene and the nuclear p65 protein was significantly increased (P < 0.05) in the M group of DDPSCs than that in the other groups.
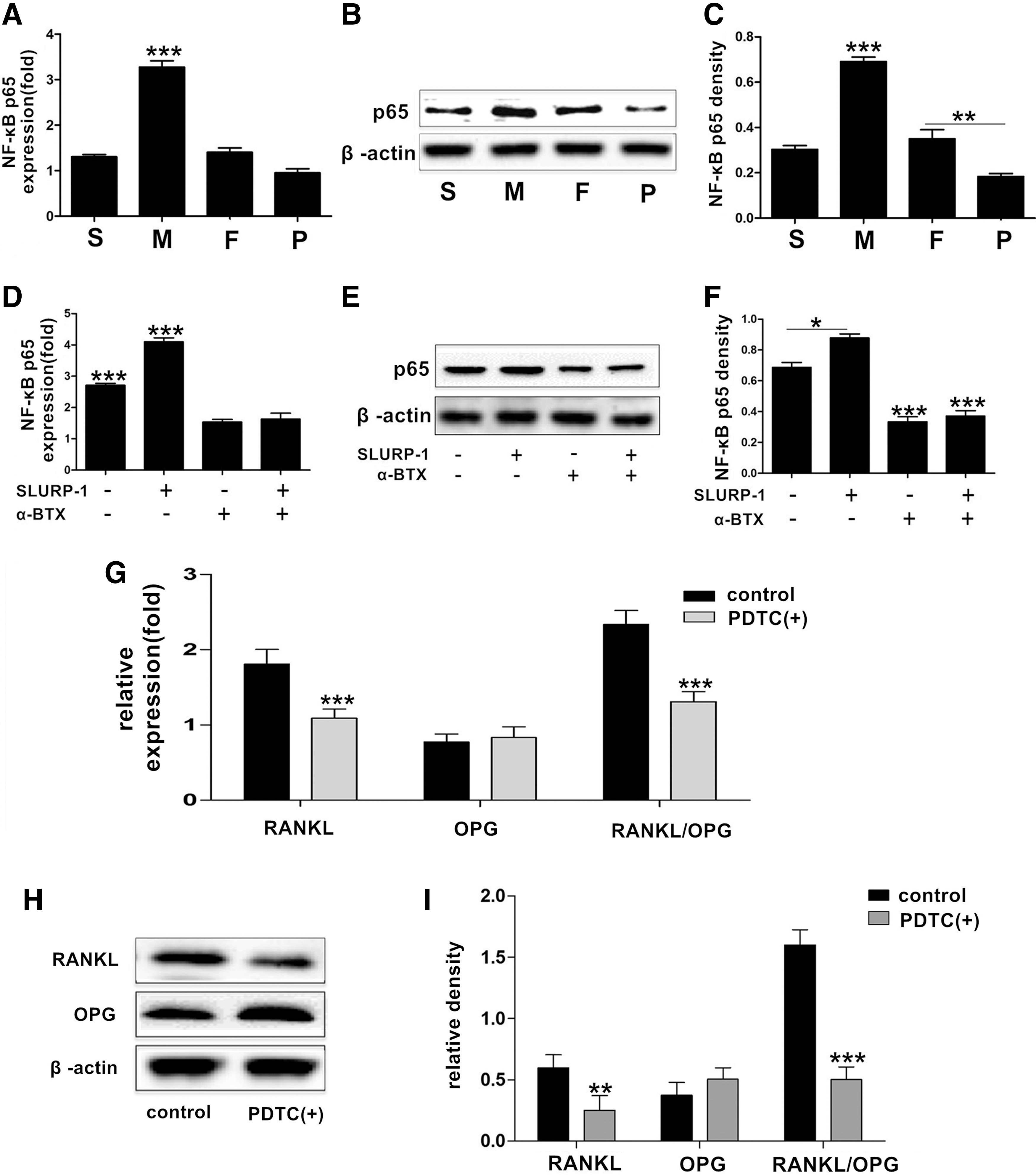
NF-κB regulates the expression of RANKL/OPG in DDPSCs.
The expression of the NF-κB gene and the nuclear p65 protein were detected through real-time quantitative PCR and western blotting, respectively, following the treatment of SLURP-1 protein and/or α-BTX to the M group DDPSCs. The expression levels of NF-κB mRNA and p65 protein in the nucleus were increased after SLURP-1 was added (P < 0.05) (Fig. 6D–F), but were significantly decreased after α-BTX was given (P < 0.05) (Fig. 6D–F), and NF-κB mRNA and p65 protein levels remained decreased when both SLURP-1 protein and α-BTX were added (P < 0.05) (Fig. 6D–F).
The expression of RANKL and OPG was detected through real-time quantitative PCR and western blotting, respectively, following the treatment of the NF-κB inhibitor PDTC to the M group DDPSCs. The expression levels of RANKL and RANKL/OPG were significantly decreased after PDTC was added (P < 0.05) (Fig. 6G–I).
Effects of mechanical compressive stress on the expression levels of SLURP-1 and α7 nAChR in DDPSCs
The expression levels of SLURP-1 and α7 nAChR in the S group of DDPSCs were detected through real-time quantitative PCR and western blotting. The results (Fig. 7A–E) showed that the expression levels of SLURP-1 and α7 nAChR increased significantly (P < 0.05) under dynamic positive pressures of 0–180 and 0–225 KPa.
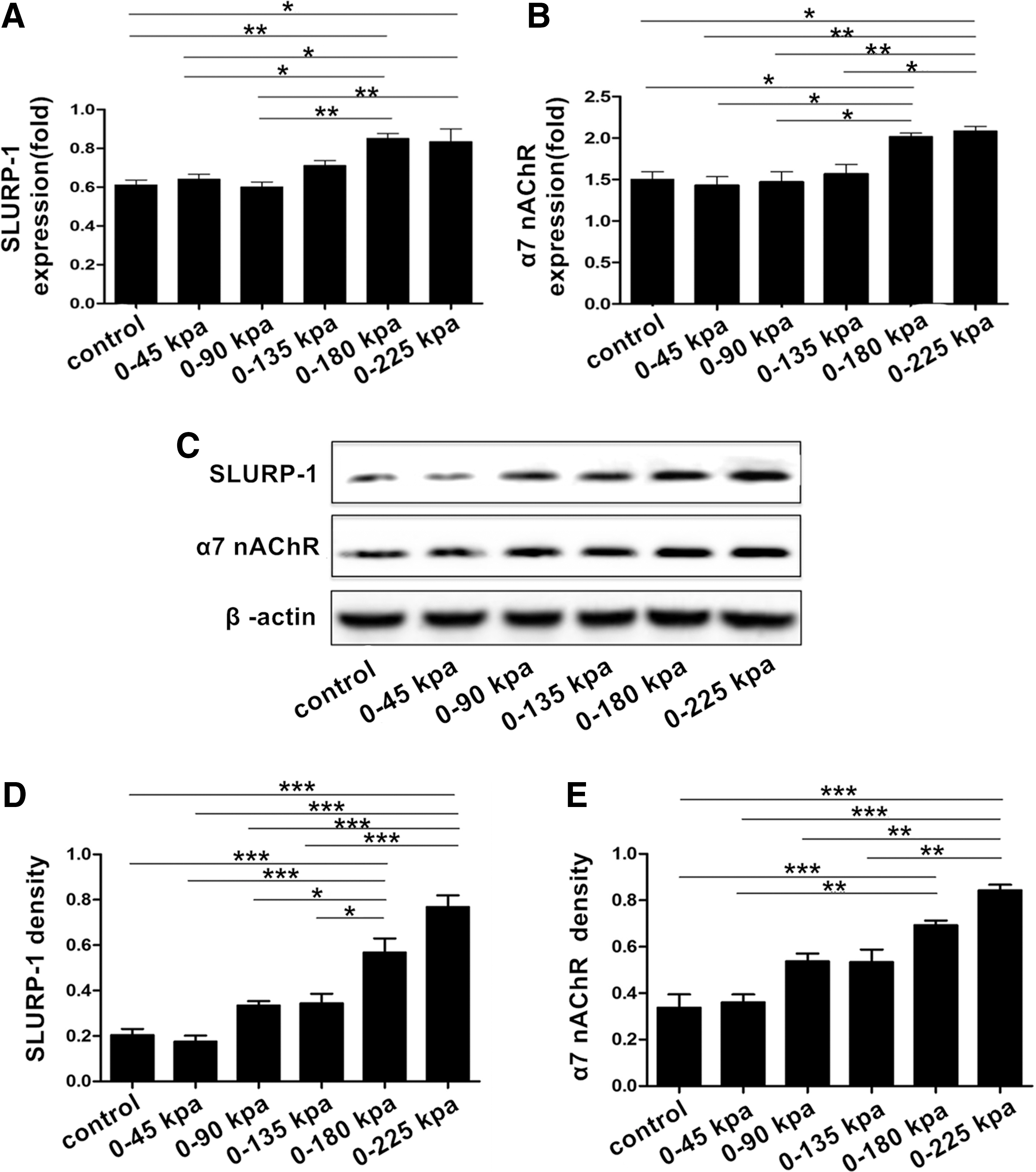
Effect of mechanical pressure on the expression of SLURP-1 and α7 nAChR in DDPSCs.
Discussion
During the process of the replacement of primary and permanent teeth, there is no unified, recognized point of view regarding the factors that lead to root resorption of deciduous teeth. Previous studies have shown that the mechanical pressure exerted by the permanent tooth germ, the dental follicle of succedaneous tooth, occlusive force, and the periodontal ligament may be related to physiological root resorption [1,32 –35]. Most studies have focused on the external resorption of the root, for example, studying the regulation of root resorption by deciduous periodontal ligament stem cells [24]. However, internal resorption of the root also occurs during physiological root resorption [8]. In addition, the root can be absorbed in the absence of the permanent tooth germ, but the speed is relatively slow, and absorption begins in the root canal cavity [36,37]. Therefore, we chose to focus on DDPSCs in this study. In recent years, some studies have found that the expression of RANKL in DDPSCs is significantly increased in the active root resorption phase, suggesting that DDPSCs may be involved in the regulation of root resorption [8]. However, the mechanism underlying the DDPSC regulation of osteoclast differentiation in physiological root resorption is unclear, and the factors that regulate RANKL and OPG secretion also need to be explored.
Because the effect of RANKL is not species restricted, we examined the ability of DDPSCs to support osteoclastogenesis in coculture with RAW264.7 cells [24,38]. In this experiment, we cocultured DDPSCs and DPSCs with RAW264.7 cells and found that the DDPSCs in the M group could induce more TRAP (+) osteoclast-like cells. RANKL and OPG have been recognized as important factors for osteogenesis/osteoclastogenesis. RANKL binds to its receptor RANK on the surface of osteoclastic precursor cells to promote the differentiation of osteoclasts. OPG can compete with RANK to bind to RANKL and inhibit osteoclast differentiation and maturation [3]. For a more detailed assessment of the expression of RANKL and OPG, we calculated the RANKL/OPG ratio and used it to specifically reflect the ability of RANKL to induce osteoclast differentiation [39]. We found that the expression level of RANKL increased, whereas OPG decreased in the group M DDPSCs. Hence, the ratio of RANKL/OPG was significantly increased in the M group compared with that in other groups. The results demonstrated that RANKL/OPG was highly expressed in the DDPSCs during the active phase of root resorption, which could promote osteoclast formation.
α7 nAChR is a ligand-gated ion channel-type receptor. Previous studies have shown that nicotine could aggravate the inflammation of periodontal tissue and the loss of bone tissue through upregulation of NF-κB, IL-1, IL-6, TNF-α, and other inflammatory factors through α7 nAChRs in periodontal ligament cells [27]. In addition, α7 nAChRs in periodontal ligament cells can regulate the expression of RANKL/OPG through CD4+ T cells following activation by nicotine [12]. A previous study demonstrated that α7 nAChR regulated osteogenesis/osteoclastogenesis by activating the Wnt signaling pathway [40]. However, whether α7 nAChR participates in the regulation of osteoclastic differentiation in physiological root resorption remains unknown. We have shown that α7 nAChR is expressed in DDPSCs and that its expression increases in the most active phase of root resorption, which is consistent with the elevated osteoclastogenesis ability of DDPSCs observed in this period. This study also revealed that α7 nAChR in DDPSCs may be involved in the regulation of osteoclast differentiation during physiological root resorption. Therefore, α7 nAChR is not only involved in the pathological processes of related diseases but also plays a critical role in normal physiological processes. α7 nAChR requires a ligand for activation and can be activated by nicotine, as observed in the periodontal ligament cells of patients who smoke [27,40]. However, there is no nicotine present in the environment when physiological root resorption occurs, and the α7 nAChR ligand and the downstream signaling pathways involved in the regulation of osteoclast differentiation are unknown.
SLURP-1 is a member of the Ly-6 protein superfamily that can act as an endogenous ligand to specifically activate α7 nAChR and is involved in signal transduction, cell activation, and cell adhesion [14,15]. Some researchers have reported SLURP-1 expression in Het-1A human oral keratinocytes. SLURP-1 plays a similar role to α7 nAChR agonists by regulating Ca2+ influx mediated by α7 nAChR and activation of the downstream NF-κB signaling pathway [16]. This study also confirmed the expression of SLURP-1 in DDPSCs. Similar to α7 nAChR, the expression of SLURP-1 in DDPSCs was significantly increased in the active phase of root resorption. Because the high ability to induce osteoclast differentiation and some related factors were expressed higher in the group M DDPSCs, we choose the group M DDPSCs as the target to explore the mechanism. By silencing and overexpressing SLURP-1 in the group M DDPSCs, SLURP-1 was shown to upregulate RANKL and RANKL/OPG in DDPSCs. In addition, the expression levels of RANKL and RANKL/OPG in the M group were significantly increased following the treatment of exogenous SLURP-1, and these effects were blocked by α-BTX, indicating that SLURP-1 might regulate the ability of DDPSCs to induce osteoclast differentiation through α7 nAChR.
NF-κB is a key cytokine that regulates innate and acquired immune responses, cell proliferation, and other biological functions [41 –43]. Studies have shown that NF-κB is involved in the regulation of osteoclast formation and activation [44]. In addition, NF-κB can be regulated by α7 nAChR in PC12 cells, and nicotine promotes neuroprotection through the α7 nAChR-JAK2 (NF-κB; STAT3)-Bcl-2 prosurvival signaling pathway [45]. However, the relationship between NF-κB and α7 nAChR in DDPSCs and its effects associated with osteoclastogenesis in physiological root resorption remain unclear. In this study, we also found that SLURP-1 acts on DDPSCs to promote NF-κB mRNA expression and nuclear transfer of the NF-κB subunit p65 protein and that this effect can be inhibited by an α7 nAChR antagonist. In addition, the expression levels of RANKL and RANKL/OPG were significantly decreased in DDPSCs treated with an NF-κB inhibitor. The results suggested that α7 nAChR in DDPSCs was activated by SLURP-1 to upregulate the expression of NF-κB and enhance its activity, resulting in the promotion of osteoclastogenesis during the physiological root resorption of deciduous teeth.
However, we have not explored the specific mechanism of NF-κB that regulates the expression of RANKL and OPG in physiological root resorption. Previous studies have shown that NF-κB plays an important role in immune inflammatory responses by regulating target gene expression, which leads to increased expression of TNF-α, IL-1β, IL-6, and other inflammatory factors [42,46,47]. These inflammatory factors can regulate the expression of RANKL and OPG; thus promoting the activation of osteoclasts [48 –50]. Therefore, the occurrence and development of the physiological root resorption of deciduous teeth may be tightly associated with the inflammatory microenvironment.
Some researchers believe that the physiological root resorption of deciduous teeth may also be related to changes in occlusive force, which can act as an exogenous factor to initiate various mechanisms involved in root absorption [34]. Previous studies have focused on changes in periodontal ligament cells with mechanical stress during physiological root resorption [33]. Some researchers have speculated that in the replacement period of dentition, the occlusive force exerted on deciduous teeth gradually increases with the development of the jaw, facial growth, and increased masticatory muscle function. When the force applied to the deciduous teeth exceeds the tolerance thresholds of the periodontal membrane, it may cause the local tissue to produce a variety of cytokines, such as IL-6 and dinoprostone, to promote odontoclast activation and initiate root resorption [18].
The root can be absorbed in the absence of the permanent tooth germ; under such conditions, absorption begins in the root canal cavity, and internal resorption dominates the process [36,37]. Therefore, the dental pulp also plays an important role in root resorption. During chewing movements, the pulp encircled by the hard tissue of the tooth can also be stimulated by mechanical force [19 –21]. Mechanical stress promotes odontoblastic differentiation through the heme oxygenase-1 pathway in a human dental pulp cell line [21], in addition to dynamic, hydrostatic pressure-enhanced DPSC survival and odontogenic differentiation [19]. However, whether mechanical stress may affect SLURP-1 and α7 nAChR expression in DDPSCs as an extrinsic factor to regulate osteoclastogenesis is unknown.
It is challenging to simulate the physiological stress applied to DPSCs in vitro. Until now, there was no mechanical device that could simulate the complex force microenvironment. In this study, we used a cell pressure loading apparatus created in our laboratory; this instrument has been used in many studies [29 –31]. To mimic the occlusive force, the physiological stress on DDPSCs during chewing movements, a dynamic positive pressure-controlled cellular strain unit was employed. Cell culture plates were placed inside a pressure chamber within a CO2 culture incubator (at 37°C). Pressure on the cells was generated by compressing the gas phase (2% CO2, 95% N2) in the pressure chamber at a specific frequency. The pressure levels at every frequency varied randomly, but were all within the range of the prior highest level. This type of dynamic pressure on the cells mimicked the mechanical environment of occlusive force experienced by DDPSCs in vivo.
In this study, the group S DDPSCs, which were at the initial stage of root resorption, were subjected to mechanical stimulation. The results showed that the expression levels of SLURP-1 and α7 nAChR were significantly elevated under positive pressures of ∼0–180 and 0–225 KPa (P < 0.05). We hypothesized that mechanical stress is closely related to physiological root resorption and may serve as an initial factor in the promotion of osteoclastogenesis in DDPSCs. However, this hypothesis has yet to be validated in an ideal animal model and the specific mechanism underlying how mechanical pressure promotes SLURP-1 and α7 nAChR expression still needs to be explored.
In this study, we selected cells from representative groups to explore the mechanisms involved in the physiological root resorption of deciduous teeth. In the future, we will conduct a more comprehensive study of the mechanisms underlying this process and establish an animal model to validate our hypothesis.
Summary
In this study, we first explored the possible role of α7 nAChR in the physiological root resorption of deciduous teeth in vitro. We confirmed that the expression levels of SLURP-1 and α7 nAChR in DDPSCs increased with mechanical force stimulation. α7 nAChRs in DDPSCs were activated by SLURP-1 to upregulate the expression of NF-κB and enhance its activity, which resulted in the promotion of osteoclastogenesis during the physiological root resorption of deciduous teeth. These findings provide a theoretical basis for studying the physiological root resorption and replacement of deciduous teeth. They also provide possible references regarding the prevention and treatment of retained deciduous teeth and the preservation of deciduous teeth in the absence of permanent tooth germ in an in vitro aspect. However, further in vivo studies are needed to consolidate the in vitro results and help researchers to understand the exact mechanisms of physiological root resorption.
Footnotes
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (81470743 and 81500805). The authors thank the patients for participating in this research.
Author Disclosure Statement
No competing financial interests exist.