
Abstract
Bone morphogenetic protein (BMP) signaling plays critical roles on the development of a large array of embryonic organs and promotes the in vitro formation of pancreatic cystoid colonies containing insulin-producing cells. However, this signaling and its underlying mechanism on in vivo development of prenatal pancreas have not been clearly understood. To address these questions, we analyzed, with a variety of techniques, the prenatal mouse pancreas after Pdx1 (the pancreas and duodenum homeobox factor 1 gene)-driving deletion of the BMP receptor type 1a gene (Bmpr1a). In this study, we report that the Pdx1-driving deletion of Bmpr1a transiently disrupted only the assembly of architectural structure of prenatal islets. The differentiation of endocrine lineage cells and the development of pancreatic acinar tissue were comparable between Bmpr1a-deleted fetuses and -undeleted Controls throughout the period examined. Molecular studies revealed that among many proteins surveyed, the key cell–cell interaction molecule E-cadherin (E-cad) only was expressed significantly less at both messenger RNA (mRNA) and protein levels in Bmpr1a-deleted than Control fetal endocrine cells. We thus conclude that BMP signaling transiently regulates the expression of E-cad and the establishment of prenatal islet architecture.
Introduction
B
BMP signaling is a critical regulator of embryonic development of a large array of tissues and organs [3]. For example, this signaling cascade regulates the early embryo development, as the deletion of Bmpr1a leads to mouse embryonic lethality at the stage of gastrulation [4]. After gastrulation, the pancreas is given rise from the thickened epithelium lining the dorsal and ventral surfaces of the posterior foregut endoderm. Some of thickened endodermal cells at embryonic day (E) 9.09.5 [5] express the gene Pdx1 (pancreas and duodenum homeobox factor 1, also known as IPF1, insulin promoter factor 1 in humans). The latter encodes a transcription factor of the parahox homeobox family and is required for the commitment of pancreatic progenitors [6]. A small fraction of these progenitors express a high level of the helix-loop-helix transcription factor neurogenin3 (Ngn3) and are known as islet progenitor cells [6 –8]. These progenitors give rise to all pancreatic endocrine cells [7] that assemble as islet-like structures around E17.5 [9].
The role of BMP signaling in the development of embryonic pancreas has not been clearly understood. Bmpr1a and other BMP ligand genes are detected in the secondary transitional phase pancreas [10] and BMPR1A protein is detectable in the developing islet cells [11]. In vitro, BMP-2, -4, -5, and -6 are each able to promote the formation of epithelial colonies containing insulin-positive cells from dissociated embryonic pancreas epithelial cells [10,12]. Corroborating with these, our recent study of pancreas-specific Bmpr1a-deleted islets suggests that MBPR1A signaling may regulate the development of prenatal islets rather than the function of adult islets [13]. Nevertheless, another previous study did not report a phenotype in the prenatal pancreas of insulin-driving Bmpr1a-deleted, or Pdx1-driving dominant negative (dn) Bmpr1a, mice [11].
Thus, we aim to confirm that BMP signaling plays a role in the development of prenatal endocrine pancreas. By using our pancreas-specific Bmpr1a-deleted mouse model [13], in this study we reveal that Bmpr1a deletion transiently disrupted islet cell aggregation by reducing in developing islet cells the expression of E-cadherin (E-cad) (also known as Slc2a2). The latter is a member of the classic cadherin family, mediating a homophilic Ca2+-dependent cell-to-cell binding for structural development and tissue integrity [14,15].
Materials and Methods
Bmpr1a-deleted mice
The generation of pBmpr1aKO mouse line was described previously [13]. Briefly, the compound heterozygous biogenic animal heterozygous for transgenic Pdx1-Cre and the floxed Bmpr1a allele (Bmpr1aflox/+), namely Pdx1-Cre; Bmpr1aflox/+ (known as pBmpr1aHet hereafter) was generated by breeding the Pdx1-Cre [9] with the Bmpr1aflox/flox mice [16]. The pBmpr1aHet mice were then crossed with the Bmpr1aflox/flox mice to generate homozygous conditional Bmpr1a-deleted (Pdx1-Cre; Bmpr1aflox/flox, namely pBmpr1aKO) mice. Bmpr1aflox/+, Bmpr1aflox/flox, and Pdx1-Cre mice were all phenotypically normal [9,17] and thus used as the controls (designated as Control hereafter). Cre-mediated DNA deletion of Bmpr1a exon 4 was previously confirmed by Southern blot analysis using pancreatic tissue DNA [13]. Tails were used for polymerase chain reaction (PCR)-based genotyping as described previously [13]. All mice were maintained on the C57BL/6 background for at least 10 generations in a 12-h light–12-h dark cycle and fed with normal chew. All experiments were performed in accordance with guidelines covering the care and use of animals in research, as approved by the Walter and Eliza Hall Institute of Medical Research Animal Ethics Committee and Animal Resources Center Animal Ethics Committee, Murdoch University.
Whole mount histology for β-galactosidase activity
The spatial and temporal expression of Cre was examined by crossing Pdx1-Cre [9] and Rosa26-Flox-Stop-Flox-LacZ (R26R) mice [18]. Noon of the day vaginal plugs were detected, and was designated as E0.5. E11.5 embryos (5–8/age group) carrying both Pdx1-Cre and R26R transgenes were harvested, washed twice in cold phosphate-buffered saline (PBS), and fixed in 4% paraformaldehyde (PFA). The stomach, duodenum, and pancreas complex from E13.5 or older fetuses were removed under a stereomicroscope (Olympus, Tokyo, Japan) and fixed in PFA. Tissues were permeabilized at 4°C in PBS containing 0.02% NP-40 and 0.01% 7-deoxycholic acid. Embryos and fetuses were then stained for β-galactosidase (β-gal) activity by incubation in a medium containing 5 mM K3[Fe(CN)6]/K4[Fe(CN)6] and 0.5 mg/mL 5-bromo-4-choloro-3-indolyl β-
Culture of embryonic stem cells
The undifferentiated mouse pluripotent embryonic stem cell (ESC) W9.5 line was maintained and passaged as previously described [19]. Briefly, ESCs were plated on gelatin-coated γ-irradiated primary mouse embryonic fibroblast feeder layers and cultured with 10% CO2 at 37°C in Dulbecco's modified Eagle's medium (4.5 g glucose/L) supplemented with 15% KnockOut Serum Replacement (Invitrogen, Melbourne, Australia), 103 U/mL leukemia inhibitory factor (Millipore, Melbourne, Australia), 1% nonessential amino acids, 1% nucleotides, 0.05 mM β-mercaptoethanol, and 1% penicillin/streptomycin.
Purification of islet progenitors
E15.5 pancreases (at which the expression of Ngn3 peaks [20]) from Ngn3-GFP/RIP-DsRed mouse line [21] were dissected under a stereoscope (Olympus) and enzymatically dissociated into single cells. The latter were washed twice with FACS buffer (0.1% bovine serum albumin in PBS) and resuspended in this buffer containing 1 μg/mL Propidium Iodide (PI) or 4′,6-diamidino-2-phenylindole (DAPI) prior for purification of islet progenitors with a fluorescence activated cell sorter (FACSAria II; Becton Dickinson LabWare) as described [21].
Isolation of fetal islet cells
Pancreases from E19.5 Control and pBmpr1aKO fetuses were microdissected and dissociated into single cells. The latter were then stained with mouse A2B5 monoclonal antibody (mAb) against islet ganglioside (Chemicon, Melbourne, Australia), marker of islet cells [22], and processed for isolation by FACS as described [21].
Isolation of adult islets
Islets of Langerhans were isolated from adult C57BL/6 male mice as described recently [13]. Briefly, the pancreas was injected through the bile duct with collagenase P solution (1.2 mg/mL dissolved in Hanks' balanced salt solution containing 2 mM Ca2+ and 20 mM HEPES). Islets were isolated by density gradient centrifugation, washing, and handpicking.
Gene mining and bioinformatics analyses
Gene mining and bioinformatics analyses were performed as described previously [21]. Briefly, the differential expression of genes (P ≤ 0.05; −1 ≤ log2 ≥ 1) based on the contrast between datasets at different time intervals was analyzed using the Limma package in the “R” environment (
Immunofluorescence analyses
E15.5, E17.5, and E19.5 fetuses and day 2 neonates with Control, pBmpr1aHet, and pBmpr1aKO genotypes were fixed in 4% PFA in PBS and processed for routine Hematoxylin and Eosin staining and for immunofluorescence histology.
Guinea pig antiserum to pig insulin and rabbit antisera to pig glucagon and human somatostatin were purchased from Dako (Glostrup, Denmark). Rat IgG 2a mAb to E-cad and mouse IgG1 mAb to β-catenin (β-cat) were from ZyMed (South San Francisco, CA). Rabbit immunoglobulin (Ig) to mouse Pdx1 was generated in-house [23] and elsewhere [24]. Rat anti-mouse glucose transporter 2 (Glut2) was gifted by Dr. B. Thorens or purchased from R&D Systems (Melbourne, Australia). Biotinated Dolichos Biflorus Agglutinin (DBA) was purchased from Vector Laboratories (Burlingame, CA). FITC-conjugated streptavidin, Texas Red- or FITC-conjugated goat anti-guinea pig Ig, Texas Red- or FITC-conjugated rabbit anti-rat or FITC-conjugated rabbit anti-mouse Ig, and Alexa 568-conjugated goat anti-mouse or Alexa 488-conjugated goat anti-mouse Ig were obtained from Caltag Laboratories (Burlingame, CA), Vector Laboratories, Molecular Probes (Eugene, OR), and Chemicon International (Temecula, CA), respectively. The immunofluorescence staining procedures were essentially as described previously [10]. Slides were observed and photomicrographed under the Inverse IX71 Olympus fluorescence microscope (Olympus).
Morphometric analyses
The fixed E19.5 fetuses with Control, pBmpr1aHet, and pBmpr1aKO genotypes were processed for serial sections. Five sections per pancreas were randomly sampled as we described previously [25] for glucagon (α), insulin (β), and somatostatin (δ) staining. The number of α, β, and δ cells, and of the ducts at E19.5 were counted manually in images with original magnifications of 20× or 40× and verified with the ImageJ software installed in the computer.
Real-time quantitative RT-PCR analyses
Specific PCR products (113–473 bp in length) for each gene analyzed were designed using Primer design tool (
Blood glucose concentration
Tail venous blood was collected and glucose concentrations were measured using a glucose meter (Roche Diagnostics) as described [13].
Statistics
Differences between groups were analyzed by nonparametric, Mann–Whitney U tests and analysis of variance. Data are presented as mean ± standard deviation of 3–25 independent experiments.
Results
BMP signaling genes were generally downregulated during development of prenatal islet lineage
We recently demonstrated at a transcriptomic level that numerous BMP signaling genes are differentially enriched during pancreas and islet progenitor specification and many become progressively negatively enriched from the development of prenatal islets to the function of postnatal endocrine cells [13]. To confirm their dynamic changes, we reanalyzed a cohort of 43 TGFβ signaling genes [13] in a more recent transcriptomic datasets generated from samples covering several time points of the differentiation of purified islet progenitors in the dual fluorescence-tagged Ngn3-GFP/RIP-DsRed mouse line [21]. Whereas majority unchanged, Bmp1, Bmp7, Bmpr1a, Smad1, Tgfrb1 (TGFβ receptor 1, also known as activin A receptor type II-like kinase), and Tgfrap1 (transforming growth factor β receptor-associated protein 1) were positively enriched, but Bmp4 and Tgfb1 were highly negatively enriched from the ESC to islet progenitor stage (Fig. 1A). Quantitative real-time reverse transcriptase (RT)-PCR analyses were used to verify these expression data with RNA samples extracted from purified islet progenitor cells as well as from undifferentiated ESCs and isolated adult islets for comparisons. These quantitative data generally corroborated with our bioinformatics analyses (Fig. 1A). Bmp4, Bmpr1a, Bmpr2, and Smad5 were all progressively decreased expression from the ESCs to adult islets; in contrast, Bmp6 expression was increased significantly in the islet progenitor cells and sustained thereafter (Fig. 1B). Moreover, the expression of Smad1 was peaked at the islet progenitor stage (Fig. 1B). These data collectively support previous evidence that the BMP pathway plays an active role on the development of prenatal endocrine pancreas [10,12,13].

Many representative genes in the BMP signaling pathway were developmentally downregulated.
Fetal pBmpr1aKO mice have transiently disrupted islet architecture
To establish the role of BMP signaling on the development of pancreatic islet lineages, we utilized our reported pBmpr1aKO mouse model [13]. We previously showed that floxed Bmpr1a exon 4 sequence is effectively deleted in the pBmpr1aKO pancreas [13], and here further visualized the temporal and spatial activity of Pdx1 promoter by the detection of Pdx1-Cre activity. This was made possible by crossing the heterozygous Pdx1-Cre mice [9] with the reporter R26R (Rosa26-Flox-Stop-Flox-LacZ) mice [18] that carry an activatable LacZ gene (encoding β-gal); and this transgene is activated when Pdx1-controlled Cre activity mediates the deletion of the floxed Stop codon sequence and promotes the expression of β-gal. The latter activity can be visualized with blue colors generated after a histochemical staining with X-gal (see Materials and Methods section for details). At E11.5, blue dorsal and ventral pancreases were clearly visible (Fig. 2A). From E13.5 onward, a typical pancreatic primordial branch tree was strongly stained with X-gal. Thus, we concluded that the functional Bmpr1a sequence will be effectively deleted permanently in the mutant pancreas once the Cre is active in the pBmpr1aHet and pBmpr1aKO mice, consistent with our previous study [13].

Fetal islets were not developed properly in pBmpr1aKO pancreases at E17.5.
To identify phenotypes in the prenatal islets after the deletion of Bmpr1a, we performed morphological and immunofluorescence analyses. The architecture of developing endocrine tissue was indistinguishable at E15.5 among Control, pBmpr1aHet, and pBmpr1aKO embryos, consistent with the report that the islet architecture is only formed and detectable around E17.5 [9]. Indeed, the islet-like structure was histologically visible in E17.5 Control fetuses (Fig. 2B). In contrast, the E17.5 endocrine pancreases in pBmpr1aHet and pBmpr1aKO fetuses displayed a disorderly morphological structure (Fig. 2B); immunofluorescence analyses revealed that β cells were not clustered in the core and α cells in the mantles (Fig. 2B).
The islet structures were well formed with endocrine cells in E19.5 Control pancreas (Fig. 3A, arrowed). However, such structures were not well assembled with endocrine cells that instead remained individually in pBmpr1aHet and even more dispersed in pBmpr1aKO fetuses (Fig. 3A, arrowed), although the development of α, β, and δ cells was comparable in all three genotypes quantitated after immunofluorescence staining (Fig. 3B, see Materials and Methods section in detail). The number of DBA-positive ducts was doubled in pBmpr1aKO pancreases (Fig. 3C, D), suggesting that pancreatic progenitors may have committed more to the ductal lineage. Nevertheless, the morphology of the acinar tissues was comparable in all stages of Control, pBmpr1aHet, and pBmpr1aKO pancreases.
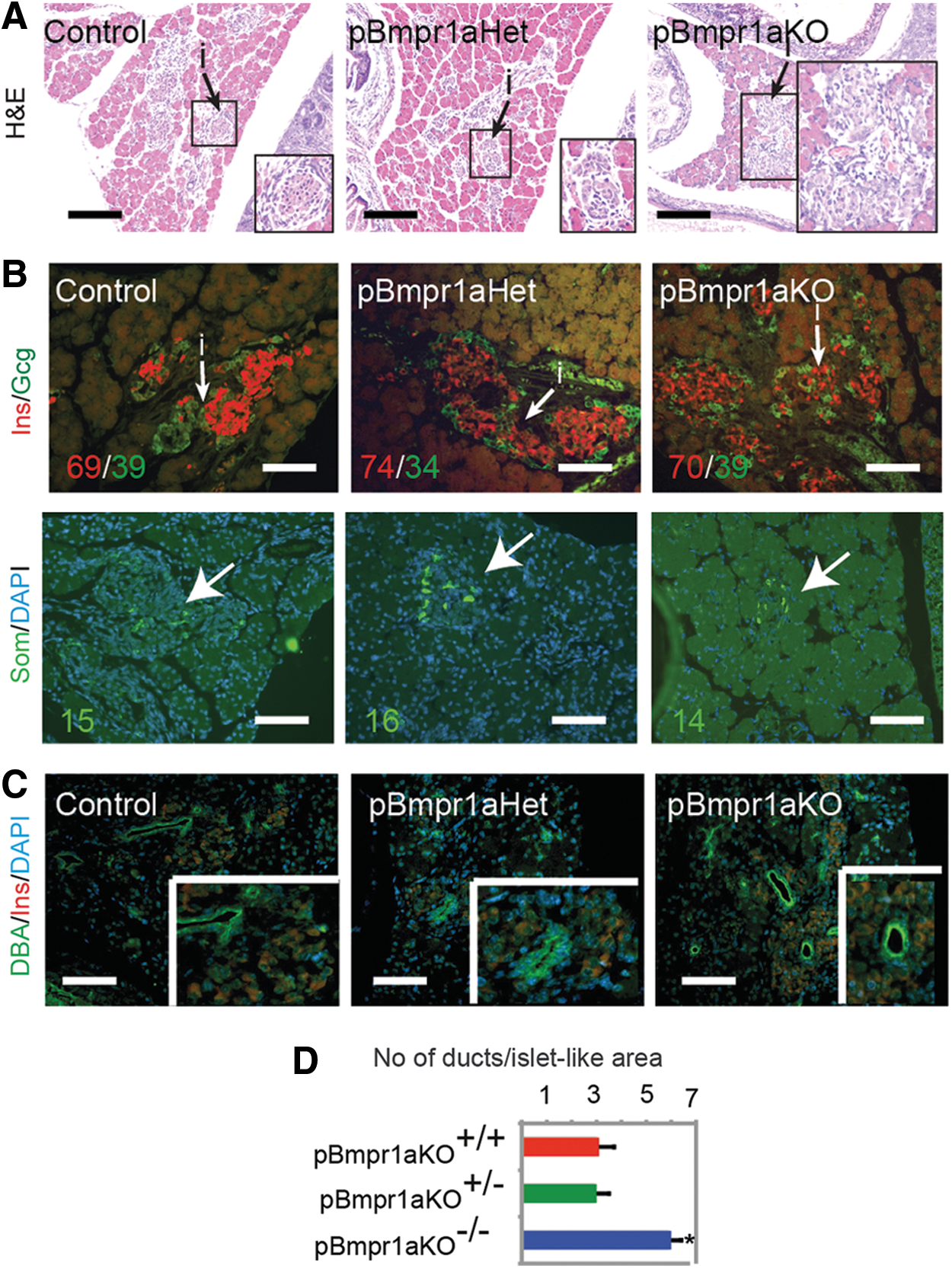
Islet architectural development was impaired in pBmpr1aKO pancreases at E19.5.
Islet architecture has recovered from day 2
To study whether the disorganized islet architecture would persist, we analyzed neonatal pancreases from the three genotypes. Both male and female neonatal Bmpr1a-deleted mice were given birth with a Mendelian ratio, suggesting that the Bmpr1a deletion was not embryonic lethal. Interestingly, the islet architecture was apparently recovered at 2 days of age. Consistent with the structural recovery of endocrine pancreas, the expression of PDX1 and GLUT2 was comparable in day 2 Control and pBmpr1aKO islet cells (Fig. 4A). In addition to the architectural recovery at day 2, the body weight and fasting blood glucose concentrations were also indistinguishable between day 2 Control and pBmpr1aKO mice (Fig. 4B).

Islet architectural impair was recovered in neonatal pBmpr1aKO pancreases.
Starting from day 2, the islet architecture was comparable among Control, pBmpr1aHet, and pBmpr1aKO mice at 3 and 12 months of age [13]. Nevertheless, glucose tolerance had impaired at 3-month pBmpr1aKO mice, and more severely impaired at 12 months of age [13].
Fetal pBmpr1aKO islet cells have transiently reduced expression of E-cad
To understand the molecular mechanism of why pBmpr1aKO fetuses had transiently disrupted development of islet architecture, we studied the expression of several relevant and selected molecules at the perinatal period. Interestingly, the islet architectural abnormality in Bmpr1a-deleted pancreases was strikingly similar to that observed in the insulin-driving dnE-cad mice [27], suggesting that the islet architectural disorder caused by Pdx1-controlled Bmpr1a deletion would be associated with the reduced expression of E-cad. Immunofluorescence analysis showed that the expression of E-cad's cytoplasmic binding partner, β-cat (also known as CTNNB1, located in both cell membrane and cytoplasm), and of insulin was similar to E19.5 Control (Fig. 5A). In contrast, the expression of E-cad was markedly reduced in E19.5 Bmpr1a-deleted islets (Fig. 5A). To confirm this protein expression data, we quantitated the expression of several genes, including E-cad encoding for selected functional proteins in fetal islet cells. The levels of these messenger RNAs (mRNAs) for β-cat, INS2, the glucose-sensing molecules, GLUT2 and GCK, and the ATP-sensitive potassium channel inward rectifier SUR1 (also known as Abcc8) were apparently indistinguishable between E19.5 Control and pBmpr1aKO islet cells enriched by FACS after dissociated pancreatic cells stained with the mAb A2B5 against an islet-specific ganglioside [22] (Fig. 5B). Whereas being abundant in islet progenitors (Fig. 1B), Bmpr1a expression was very low and comparable in E19.5 Control and pBmpr1aKO islet cells (Fig. 5B). In contrast, E-cad was significantly reduced (Fig. 5B) in E19.5 pBmpr1aKO islet cells, supporting our immunofluorescence data (Fig. 5A).
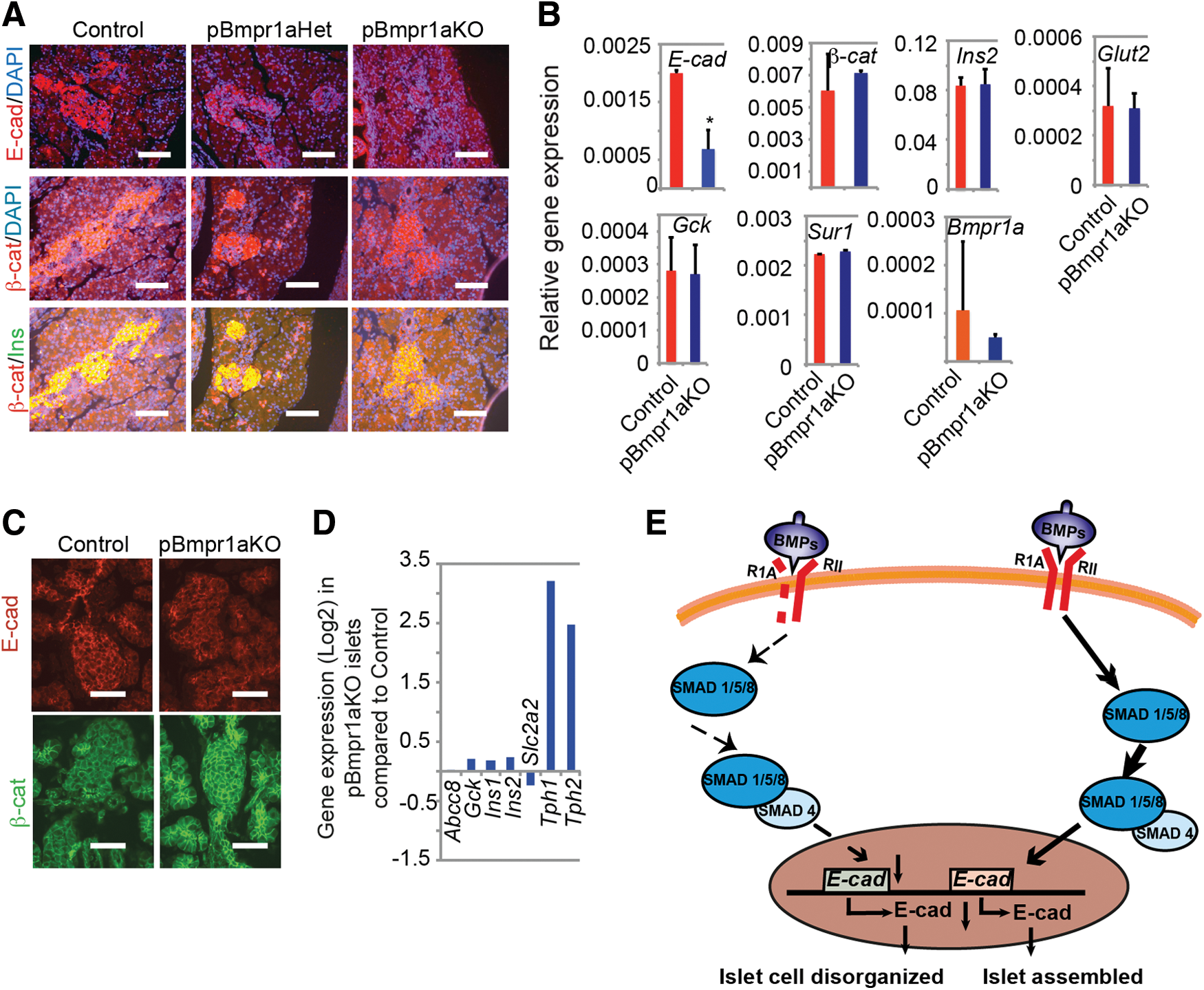
E-cad expression was reduced in fetal pBmpr1aKO islet cells.
As expected, the expression of β-cat was similar to day 2 Control islet cells and that of E-cad was largely recovered in day 2 pBmpr1aKO islet cells (Fig. 5C). The E-cad expression remained normal in pBmpr1aKO adult islets [13]. To confirm quantitatively the E-cad expression, we reanalyzed the transcriptome dataset generated from 3-month pBmpr1aKO and Control mouse islets [13]. Supportive to previous immunofluorescence data, E-cad expression along with above-selected genes in pBmpr1aKO islets was comparable to that in their Control counterparts (Fig. 5D), whereas the tryptophan hydroxylase 1 gene (Tph1) and Tph2 were highly upregulated and previously linked to glucose intolerance in pBmpr1aKO adults [13]. Collectively, there was no evidence of abnormal expression of E-cad, which may be correlated with the glucose intolerance in pBmpr1aKO adult mice.
Taken together, these data indicate that BMP signaling regulates islet architectural development by specifically promoting the expression of E-cad at a small temporal frame in islet progenitor cells, which was summarized in Fig. 5E.
Discussion
BMP signaling is implicated for pancreatic endocrine cell development [10,12], but its molecular pathway and the underlying mechanism were not previously identified. Our current study provides evidence that the BMP signaling modulates the development of islet architecture through regulating E-cad expression. This conclusion was drawn from three pieces of evidence presented in this study. First, pancreas-specific deletion of Bmpr1a transiently disrupts the development of islet architecture. Second, the dispersed Bmpr1a-deleted islet cells express less the critical cell–cell binding molecule E-cad. Third, the architecture of the islet structure recovered when the expression of E-cad recovered 2 days after birth.
We provide conclusive evidence that BMP signaling transiently regulates the expression of E-cad in the developing islets. First, Bmp6, Bmp7, Bmpr1a, and Smad1 are highly expressed before and in islet progenitors, and Bmpr1a expression was low and indistinguishable between enriched E19.5 Control and pBmpr1aKO fetal islet cells that are differentiated from the progenitors [21]. These data indicate that BMPR1A signaling is active before the formation of fetal islet cells. Second, the deletion of Bmpr1a in and before the islet progenitors did not alter the expression of many molecules but only reduce that of E-cad at the mRNA and protein levels in E19.5 fetal islet cells. Third, several BMP members are known to promote the in vitro formation of E-cad+ colonies containing insulin-positive cells from dissociated fetal pancreas cells [10,12]. Fourth, treatment with BMPs is shown to induce E-cad expression in definitive endoderm-derived liver cells and extraembryonic endoderm epithelial cells [28,29]. Finally, BMP signaling is required for the derivation and maintenance of hESC-derived PDX1+NKX6.1+ pancreatic progenitors [30]. Thus, although the exact molecular mechanism of how E-cad activity is activated remains not yet completely understood, we propose that BMP signaling activates Pdx1 and the latter promotes E-cad transcription by binding to two conserved sequences in the E-cad promoter region [31]. Future studies are therefore required to confirm or refute this molecular pathway, and to exclude other important molecules involved, in islet architectural development.
Our data indicate that BMP-regulated E-cad expression contributes to the development of prenatal islet architecture. The Pdx1 promoter-directed deletion of Bmpr1a in and before islet progenitor cells led to reduced expression of E-cad at mRNA and protein levels in Bmpr1a-deleted fetal islet cells and impaired the development of prenatal islet architecture. The very low expression of Bmpr1a in fetal islet cells allows the recovery of islet E-cad expression and of the islet architectural development at day 2 pBmpr1aKO mice. Mechanistically, the E-cad molecules expressed on adjacent cells, including islet progenitor cells undertake homophilic binding. The C-terminus of E-cad subsequently binds to the downstream molecule β-cat to regulate cellular development and tissue architecture [32,33], including islet architectural development. This also explains why directed reduction of E-cad by insulin-driving dnE-cad prevents the clustering of β cells and islet formation [27]. Nevertheless, the Pdx1-driving dnBmpr1a and insulin-driving Bmpr1a-deleted mice were not reported to have any abnormal development of fetal islet tissue [11]. The reasons for the discrepancy were not clear, but may be simply interpreted as the latter study [11] did not observe this prenatal phenotype. Nevertheless, the glucose tolerance being impaired mildly at 3 months and modestly at 12 months was previously linked to the high upregulation of (Tph1) and Tph2 in pBmpr1aKO adult islets [13]. Whether a molecular linkage between the transiently impaired fetal islet architecture, recovered postnatal E-cad expression and endocrine architecture, and adult glucose intolerance can be established needs future investigations.
In summary, we demonstrated that BMP signaling regulates E-cad expression for the development of prenatal islet structure as the disruption of which affects the clustering of developing islet cells. Further study of Pdx1-driving Bmpr1a-deleted mouse model might generate novel insights of how BMP signaling affects postnatal function of pancreatic endocrine through modulating prenatal development of the islet architecture.
Footnotes
Acknowledgments
The authors thank Dr. Illia Banakh, Dr. Kevin Li, and Mr. Gaetano Naselli for technical assistance; Prof. Pedro Herrera (University of Geneva, Geneva, Switzerland) for providing Pdx1-Cre mice; and Prof. B. Thorens (Universite De Lausanne, Lausanne, Switzerland) for providing antibody to Glut2. This study was supported by a Partnership Program Grant from the Juvenile Diabetes Research Foundation International and the National Health and Medical Research Council of Australia, by Victorian State Government Operational Infrastructure Support and Australian Government NHMRC IRIIS (to L.C.H.), by grants from Juvenile Diabetes Research Foundation International (4-2006-1025), Telephon Perth Child Health Research Fund (TPCHRF), Diabetes Australia Research Trust, Diabetes Research Western Australia, and Medical Research Foundation of Royal Perth Hospital Grant (to F.-X.J.).
Author Disclosure Statement
No competing financial interests exist.