
Abstract
Today with the rapid advancements in stem cell studies and the promising potential of using stem cells in clinical therapy, there is an increasing demand for in-depth comprehensive analysis on individual cell transcriptome and epigenome, as they play critical roles in a number of cell functions such as cell differentiation, growth, and reprogramming. The development of single-cell sequencing technologies has helped in revealing some exciting new perspectives in stem cells and regenerative medicine research. Among the various potential applications, single-cell analysis for cardiac stem cells (CSCs) holds tremendous promises in understanding the mechanisms of heart development and regeneration, which might light up the path toward cell therapy for cardiovascular diseases. This review briefly highlights the recent progresses in single-cell sequencing analysis technologies and their applications in CSC research.
Single-Cell Sequencing Technologies
A
Single-cell transcriptome studies
Single cells can be isolated and obtained by using different available approaches such as micropipetting, laser capture, FACS, and microdroplet and microfluidic platforms, depending on the experimental requirements and the accessibility to the laboratories [5,6]. Once the single cells of interest have been captured, certain traditional methods such as single molecule mRNA fluorescence in situ hybridization and single-cell quantitative polymerase chain reaction (qPCR) can be used to measure gene expression. While these methods are reasonably sensitive and reliable, they are mostly used in analyzing the expression of a small set of target genes due to their low-throughput limitation [7]. On the other hand, the DNA microarray-based (see later sections for more discussion) [8] and the RNA sequencing-based single-cell analysis enable the interrogation of the whole transcriptome, which allows us to gain significantly greater insights into transcriptional regulations in individual cells that are often heterogeneous despite expressing the same set of identity markers.
In single-cell RNA-seq (scRNA-seq), mRNAs are converted into cDNAs after single cells being captured and lysed. Currently, both the 3′-transcript and the full-length transcript sequencing technologies are available (Fig. 1). The 3′-transcript sequencing technologies employ mainly poly-A tailing [9], CEL-Seq [10], and CEL-Seq2 [11] (Fig. 1). The recently developed technologies such as 10× Genomics and DropSeq system also use the 3′-transcript sequencing technologies (Fig. 1). Although they suffer from 3′-bias, with multiple barcoding techniques, these methods can analyze 1,000 and up to 10,000 of single cells per sample to allow a better characterization of the cell types within a given sample, with a great decrease in the cost per single cell studied. However, for the full length transcript analysis, template switching is used in several protocols, including SMART-Seq [12], SMART-Seq2 [13], and STRT-Seq [14] (Fig. 1). Another method is the Single Primer Isothermal Amplification (SPIA) used in Nugen Ovation RNA-Seq protocol [15] (Fig. 1). The major advantages of single-cell full-length transcript sequencing are to allow us to gain insights into the alternative splicing, the novel exon identification, and even the single-nucleotide mutations or variations (SNVs) within the open reading frame of transcripts, but the disadvantages and tradeoffs are their relatively high costs as a much deeper sequencing would be needed for the SNV identification and significantly smaller cell numbers being studied.
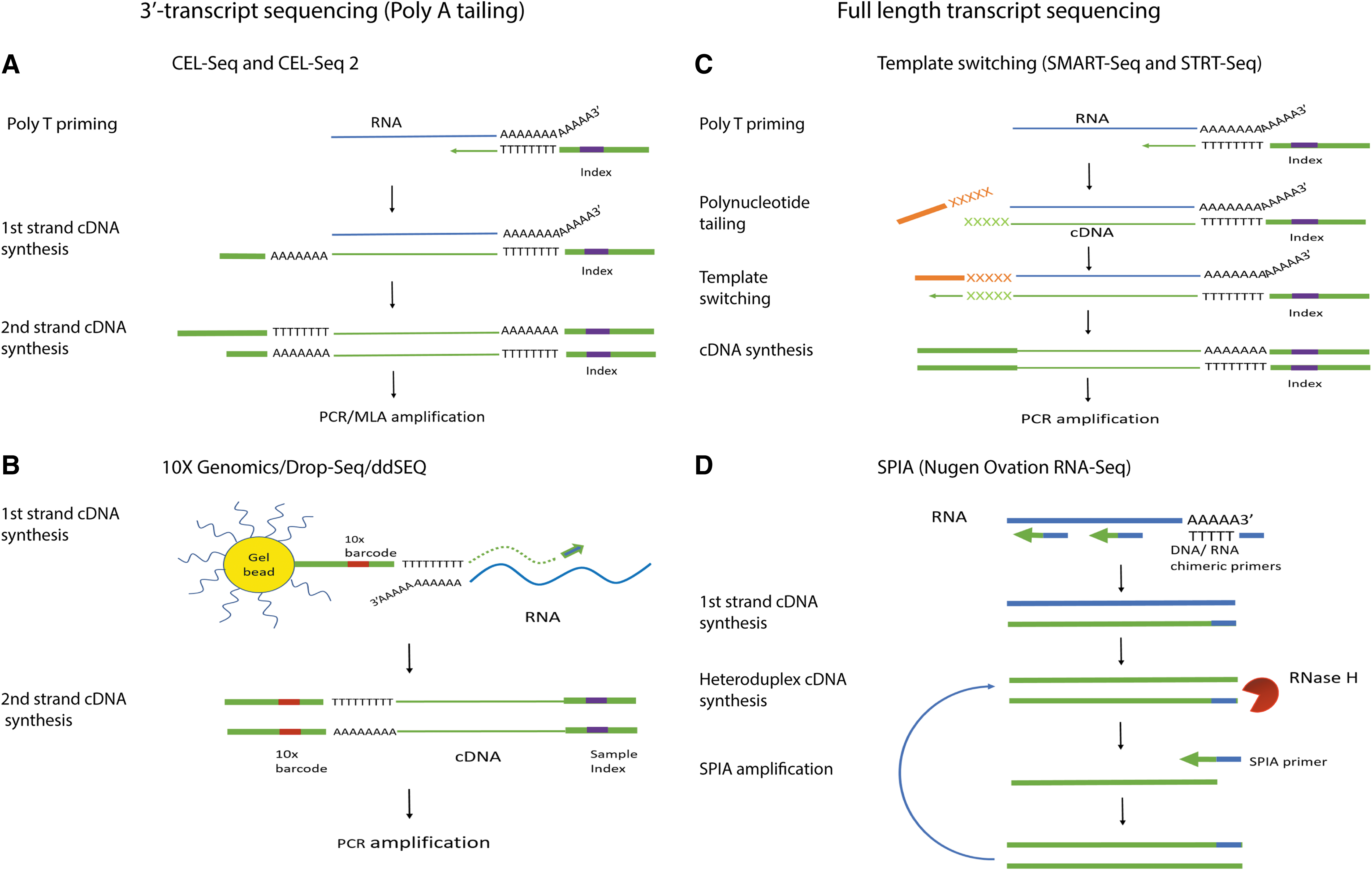
Schematic overview of single-cell RNA-seq library construction techniques.
It should be noted that because of the low amount of mRNA in a cell, a preamplification of the cDNAs after reverse transcription is required and achieved either by multiplexed linear amplification (CEL-Seq [10] and CEL-Seq2 [11]) or PCR amplification (Tang et al. [9], STRT-Seq [14]), SMART-Seq [12], and SMRT-Seq2 [13]). After the amplification, the next-generation sequencing-based scRNA-seq can be used to profile the transcriptome. In contrast to the population cell-based analysis, the high-throughput scRNA-seq can provide unprecedented insights and it allows us to answer many biological questions, whereas using the population cells would not have been able to do so. While physical compartmentation of single cells was used in single-cell studies, its retrospective identification of cell types based on whole transcriptome somehow mimics the principle of barcoding single cells followed by RNA-seq analysis. Nonetheless, when dealing with progressive events such as the cell cycle, one should be cautious in applying different algorithms, including near neighboring, and Monocle toolkit that can distinguish different cells at single-cell spatiotemporal resolution [16].
Nowadays, for the large number of samples in single-cell studies, there are several platforms such as Fluidigm C1 single-cell Auto Prep System [17 –19] and DropSeq system [20,21] that can automatically implement the processes of single-cell capture, reverse transcription, and preamplification of mRNAs/cDNAs in a chip, which has dramatically increased the efficiency and the accuracy of single-cell studies. However, the low process rate (<96 cells per chip) and the relatively strict requirements in the target cell size/shape place the Fluidigm C1 at disadvantage. On the other hand, microdroplet systems such as DropSeq system are capable of capturing and barcoding 1,000 and 10,000 of single cells simultaneously as well as allowing high-throughput reagent delivery into each individual droplet for subsequent reverse transcription [20,21]. The development of this technique significantly increased the speed and lowered the cost for single-cell sequencing. It is noted that the recent new advancements in single-cell technologies such as 10× Genomics Chromium, a gel bead system for 3′ scRNA-seq with fast workflow and high cell capture efficiency, and WaferGen's iCell8, with MultiSample NanoDispenser and a powerful imaging system, allow us to study the single cells with high throughput and flexibility, which may overcome the difficulty faced by some microfluidic platforms and further facilitate transcriptomic studies at single-cell level [22 –24]. More recently, the ddSEQ single cell isolator, developed by BioRad in collaboration with Illumina, provides the researchers with end-to-end single-cell sequencing solution from single cell capture to the data analysis.
Single-cell epigenomic studies
One aspect of the epigenetic regulations deals with the histone protein and genomic DNA modifications, in which the modifications often alter the transcription activity of proximal genes [25]. It is through at least, in part, the epigenomic reprogramming, that is, cells with the same genetic materials can have a dramatic different transcriptomic activity, thereby different cellular and molecular features in embryonic development and disease progression [26].
DNA methylation, one of the major aspects in epigenetic regulations, regulates the gene expression by methylating CpG islands in their promoter regions. Gene transcription could be either upregulated or downregulated depending on hypomethylation or hypermethylation level of the CpG islands. Conventional epigenomic analysis requires a large amount of starting materials due to low DNA recovery efficiency when being coupled with chromatin immunoprecipitation (ChIP), bisulfite conversion, or enzymatic digestion. This is also the most challenging part in single-cell epigenomic studies. Bisulfite sequencing (BS), by converting cytosine residue in DNA into uracil through bisulfite treatment, while leaving 5-methylcytosine unchanged, offers genome-wide DNA methylation analysis (Fig. 2). However, currently, it was reported that the single-cell BS (scBS) could cover only 50% of genomic CpGs due to the large amount of DNA degradation during the conversion [27]. Instead of targeting whole genome, single-cell reduced representation bisulfite sequencing (scRRBS) enriches CpG regions through restrictive enzyme digestion, followed by bisulfite conversion (Fig. 2). Nevertheless, there is still some limitation on scRRBS. It was reported that scRRBS could cover only around 10% of genomic CpGs, which was about 40% coverage of the CpGs compared to that acquired by reduced representation BS using population cells [28,29]. To sum up, there is still a large room for improvement in scBS and scRRBS and we expect great advances in next few years on these technologies.
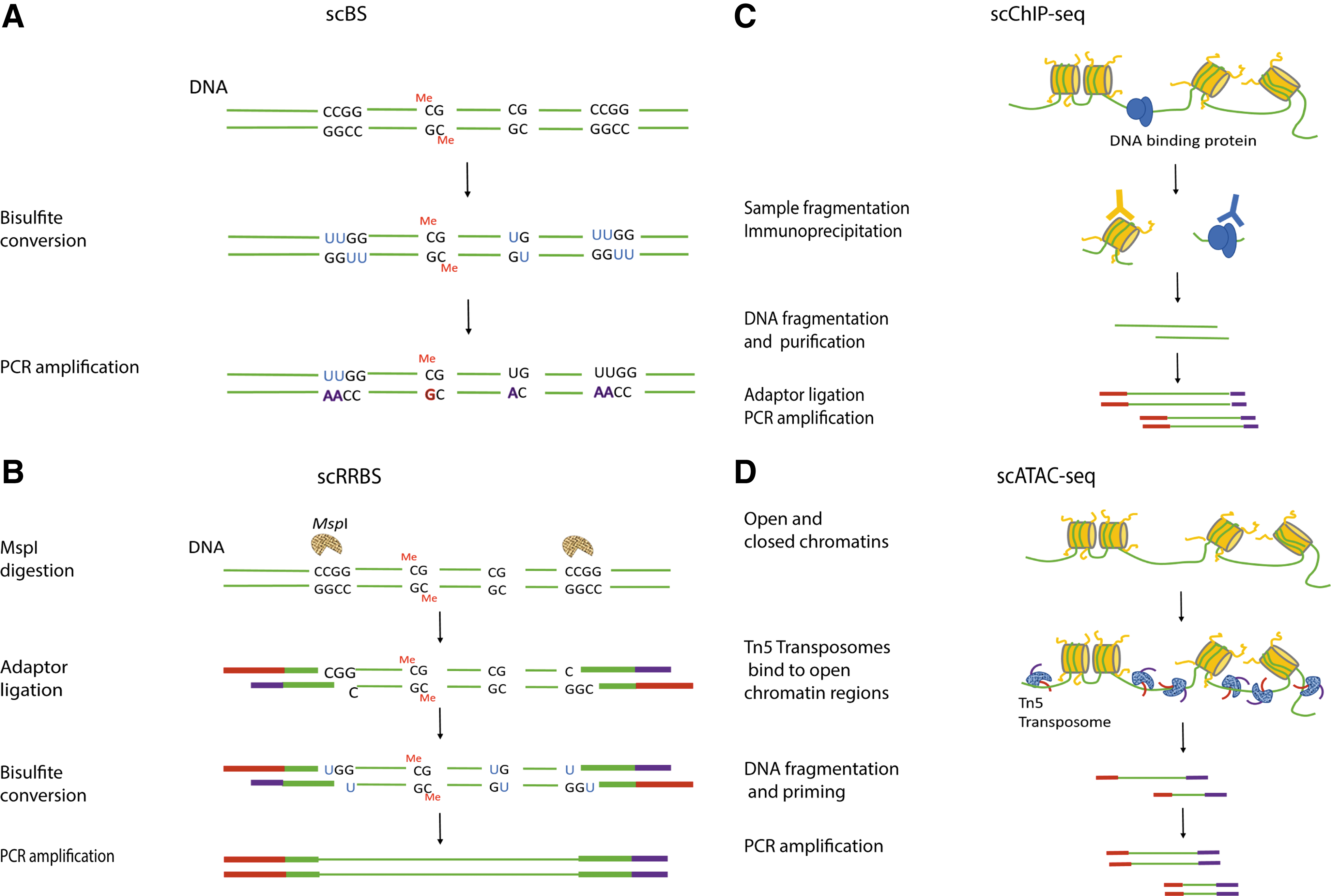
Schematic overview of single-cell epigenetic study library construction techniques.
In addition to DNA methylation, protein-DNA interaction such as histone modification, transcription factor binding, as well as chromatin structure are all the parts of the epigenetic regulations that influence cell transcriptomic activity and phenotypes [30]. Single-cell Drop-ChIP is a technique based on ChIP-seq coupled with drop-based microfluidics for analyzing chromatin-DNA interaction in single cells where DNA is barcoded in single-cell droplet first, then many single-cell drops are immunoprecipitated in the presence of “carrier” chromatin, and the enriched DNA is sequenced (Fig. 2). Although this single-cell Drop-ChIP technique covers sparsely about 1,000 peaks in each individual single cell, its specificity is high, with ∼50% reads aligning to known positive sites when histone modification marks H3K4me2 and H3K4me3 were tested in single mouse embryonic stem cells (ESCs), embryonic fibroblasts, and hematopoietic progenitors [31]. A new interesting technique recently reported is the single-cell assay for transposase-accessible chromatin-seq (scATAC-seq) [32] (Fig. 2). In principle, the ATAC-seq uses the prokaryotic Tn5 transposase to tag regulatory regions by inserting sequencing adapters into accessible regions of the genome, whereas in scATAC-seq, the individual cells are captured and assayed using a programmable microfluidics platform (C1 single-cell Auto Prep System; Fluidigm) with methods optimized for scATAC-seq epigenetic study [32] (Fig. 2). Recently, many additional new methods were reported for single-cell epigenomic studies, such as single-cell DNase I sequencing, offering genome-wide detection of DNase I hypersensitive sites [33], and single-cell DamID sequencing, studying chromosome organization by its interaction with nuclear lamina [34]; nevertheless, just like the scBS and scRRBS, all these methods offer low coverage and throughput. Thus, with current technologies, it is still limited to achieve a high-resolution whole-genome epigenomic profiling at single-cell level. However, as the demand for single-cell level analysis rises, the improvement in technology is expected.
Integrated single-cell genome, transcriptome, and epigenome sequencing
With the rapid development of single-cell technologies, several laboratories recently reported an integrated analysis of genome, transcriptome, and epigenome using the same single cells. Although the results are far from satisfaction, integrated single-cell analysis has brought some excitements in the scientific community. The scarcity of starting material in an individual cell still is the major roadblock.
In 2014, Han et al. first described a method for analyzing the single-cell genome and transcriptome-seq (scGT-seq) [35]. scGT-seq is a microfluidic-facilitated approach that separates cytoplasmic content from the intact nucleus in a single cell and then amplifies genomic DNA and mRNA by multiple displacement amplification (MDA). The controlled separation of mRNA-containing cytoplasm from genome-containing nucleus makes it possible to comprehensively analyze single-cell genome and transcriptome. Then, in 2015, Macaulay et al. reported another method called G&T-seq which utilizes biotinylated oligo-T primer to specifically amplify polyadenylated mRNA in which the biotinylated mRNA is amplified by SMART-Seq2, while the gDNA was amplified by MDA or multiple annealing and looping-based amplification cycles before sequencing [36]. Instead of physically separating the cellular content as applied in scGT-seq and G&T-seq, gDNA-mRNA sequencing (DR-Seq) uses a quasilinear amplification method in a one-tube reaction to quantify mRNA and gDNA, while minimizing their loss [37].
However, the gDNA amplified using the above-mentioned approaches cannot be used for DNA methylation profiling; thus it is limited in terms of parallel epigenomic analysis in single cells. Recently, a single-cell triple omics sequencing technique, scTrio-seq [38], was developed to simultaneously analyze genomic copy number variations, DNA methylome, and transcriptome of single human hepatocellular carcinoma cells. By using cellular content separation and single-cell RRBS sequencing, both methylome and genome sequence can be obtained. Together with mRNA sequencing, scTrio-seq enables triple omics sequencing in single cells and provides a comprehensive insight into molecular regulations in individual cells that present differently compared to the population cells as a whole. We foresee a rapid growth in applying these innovative technologies to delineating the complex molecular networks controlling cellular pathophysiology.
We would like to note that it is not our intention to give a very deep and detailed discussion on each above-mentioned single-cell sequencing technique because of the space limitation in this review. For a more detailed description of each single-cell sequencing profiling technique, we would recommend to refer to the cited reference articles related to each single-cell sequencing technique.
Single-Cell Technology Applications in Cardiac Stem Cell Studies
Heart disease remains to be one of the major causes of death around the world. Recent development in stem cell research and developmental cardiac biology unveils the mechanism of mammalian heart regeneration. Based on these discoveries, cardiac regenerative therapeutics, including stem cell therapy, holds great potential in cardiac tissue regeneration and myocardial function improvement caused by cardiac stresses and ischemic heart injury. However, the lack of comprehensive understanding in the cellular and molecular basis for human cardiogenesis and cardiac regeneration presents us with big challenges in developing efficient and safe therapies for heart injury treatment. There was great difficulty in studying heart cells at single-cell level because of their large size, unique shape of the cardiomyocytes (CMs), and the challenge in isolating and culturing the live CMs. The rapid advancement in single-cell technologies might address the issues in our current understanding toward heart development and heart regeneration, leading to the pathway toward clinical success of cardiac regenerative therapeutics (Fig. 3).

Applications of single-cell techniques in researches on cardiac development
Heart development and cardiac renewal
In cardiogenesis, part of the pluripotent ESCs becomes mesodermal precursors that in turn differentiate into the so-called cardiac progenitor cells (CPCs). These CPCs, referred to as first heart field (FHF) and second heart field (SHF) progenitors, are responsible for giving rise to different type of cells composing the heart, including CMs, conductive cells, vascular smooth muscle cells (SMCs), and endothelial cells (EDCs) [39,40]. In addition to FHF and SHF CPCs, it is suggested that epicardial cells also contribute to CM population by going through epithelial-to-mesenchymal transition, that is, epicardium-derived progenitor cells (EDPCs) [41 –43]. To have a better understanding of cardiac development regarding the lineage hierarchies and regulatory network, technologies with high resolution in characterizing new markers and identifying cellular heterogeneity even within an apparently same subpopulation are required. In 2014, Treutlein et al. reconstructed the molecular steps during the maturation of bipotential progenitors along with both alveolar lineages, of which they elucidated the full life cycle of the alveolar type-2 cell lineage using scRNA-seq [19]. This study serves as an inspiration to utilize scRNA-seq to study other tissues and organs, including cardiac development, to delineate the detailed molecular events at transcriptomic level. By unbiased transcriptome-wide sequencing of single cells at different locations across different developmental stages, cells can be classified into groups based on their distinct transcriptomes using newly developed bioinformatics algorithms. With scRNA-seq data and based on the known lineage-specific markers, cell populations can be assigned to specific lineages such as ESCs, CPCs, CMs, SMCs, EDCs, and fibroblasts by using new bioinformatics tools. By analyzing the gene sets for each population, novel lineage-specific markers or regulators can be discovered to distinguish different cell populations. To sum up, scRNA-seq, with capability for unbiased identification and molecular characterization of distinctive cell types, will allow us to define the progenitors and lineage hierarchies, phenotypes, and lineage-specific regulatory factors in cardiac development.
In terms of cardiac renewal, the prevailing dogma about mammalian CMs is their cease from replication soon after birth; so they do not seem to have regenerative ability or cellular plasticity. In recent studies, evidence by retrospective isotope dating suggests that human CMs have a slow turnover rate of about 1% per year, declining with aging [44]. Regarding the source of cardiac renewal or cardiac homeostasis, there are two major theories: (1) preexisting CMs reenter cell cycle and go through cell division to proliferate and (2) certain progenitor cells differentiate into CMs. These progenitor cells can be endogenous CPCs, EDPCs, or other population, yet their cardiac myogenic roles in injured adult hearts remained uncertain and controversial [45]. Growing evidence from genetic mapping experiments utilizing isotope labeling and GFP transgenic mice suggested that the dominant source for CM regeneration in normal or injured heart is through preexisting CM proliferation [46,47]. However, the results of those experiments do not exclude the involvement of progenitors in cardiac renewal. In addition, the molecular mechanism of how preexisting CMs generate new CMs is not known. The application of single-cell technology may hold promise to answer these questions. In 2016, an interesting study utilized scRNA-seq to reveal the core molecular signature of prehematopoietic stem cell, attempting to map out the stepwise mechanism of hematopoietic stem cell differentiations [48]. Similar strategy can be applied to the study of cardiac renewal in which cardiac cells will be subject to scRNA-seq at different time points during cardiac renewal. The molecular identity of cells can be characterized as mentioned earlier. scRNA-seq will enable us to reveal gene expression involving cell division or cell cycle regulation in each cell, as well as cell cycle heterogeneity within same cell type. The dynamic change of differentially expressed genes, especially transcription factors, between different cell types may help reconstruct the regulatory pathways for new CM generating. All this is less likely to be achieved by conventional bulk sample transcriptome analysis where the averaging effect might mask the pattern.
Heart disease and cardiac repair
Early studies suggested that urodele amphibians, such as salamanders and newts, have the ability to regenerate injured cardiac tissues and fully recover from heart injury induced by amputation, genetic ablation, and cardiac cryoinjury [49 –52]. It was revealed that the source for new CMs is from preexisting mature CMs that undergo partial dedifferentiation and then reenter the cell cycle to proliferate [53,54]. Unlike in zebrafish or other amphibians whose cardiac regenerative potential remains during normal cardiac maturation and possibly during aging, adult mammalian cardiac tissues have much limited regenerative capacity; therefore scar formatting is a major compensative repair after cardiac injury such as myocardial infarction (MI) [55]. Porrello et al. reported that a 1-day-old neonatal mouse heart subjected to a resection of ∼15% of apical ventricular tissue could regenerate completely at the day 21. However, the repair capacity decreases dramatically 7 days after birth and the injured heart develops fibrotic tissue instead [46]. Recent studies confirmed the regenerative capacity of neonatal mice and a rapid loss of regeneration capability within the first week of life, suggesting that the key pathways regulating the regeneration lie within those few days after birth [56]. To identify those key pathways, it is important to understand the molecular mechanisms driving cardiac repair after the injuries. Many hypotheses are brought up, including the roles of miRNAs, Hippo signaling, transcription factor Meis1, as well as paracrine signals from epicardial response [57]. High-throughput transcriptome analysis at single-cell level may allow us to identify the key genes and pathways dictating the cardiac regeneration.
Cardiac regeneration and therapeutic strategies
Due to the limitation of adult CM regeneration after cardiac injury, MI patients usually suffer from permanent CM loss and fibrotic scar formation en route to arrhythmias, unfavorable remodeling of ventricular walls, and eventually heart failure [58]. Various approaches were developed recently aiming to enhance cardiac tissue regeneration and regain normal function. Stem cell therapies based on the transplantation of in vitro-derived CMs or autologous somatic stem cells, and in vivo direct reprogramming, are being intensively studied for their safety and efficiency. In addition, “cell-free” therapies involving delivering specific paracrine factors to stimulate preexisting CM proliferation or activate endogenous CPC differentiation are also under investigation for cardiac regeneration therapies [59]. Programming and reprogramming a human heart cell is reviewed in much depth by Sahara et al., about the current attempts in cardiac regenerative therapies, which are far from a clinical practice [57]. Positive results are obtained from various therapies described above, nevertheless with much less satisfied outcomes in clinical trials [60]. One of the most outstanding problems is the lack of understanding the detail, complex cellular and molecular mechanisms underlying the CM regeneration in stem cell therapy.
Recently, Wu and colleagues demonstrated that animals transplanted with human induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) have significant functional improvement and attenuated cardiac remodeling. Their microfluidics single-cell qPCR results suggest that these beneficial effects are most likely explained by the paracrine activity because of significant levels of proangiogenic and antiapoptotic factors released by iPSC-CMs in the ischemic microenvironment [61].
In addition, recent researches demonstrated that adult mouse and rat CMs can spontaneously dedifferentiate, and reenter cell cycle in prolonged culture, and some eventually regain the property of CPCs. Importantly, these dedifferentiated cells can redifferentiate into functional CMs [62]. It is worthy to note that oncostatin M (OSM) is found to be a major mediator involved in CM dedifferentiation and remodeling during acute MI and in chronic dilated cardiomyopathy in human. The dedifferentiated myocytes also expressed a putative CPC marker c-kit as found in an earlier study using mouse CM genetic cell fate mapping system for myocyte dedifferentiation [62,63]. However, the questions are what kind of CMs can spontaneously dedifferentiate, and how these dedifferentiated cells redifferentiate during the cardiac injury. We believe that single-cell transcriptomic and epigenomic studies of these specific CMs and CPCs can allow us to gain deeper understanding on the molecular mechanisms dictating the dedifferentiation and redifferentiation, which eventually would allow us to design strategies for improving cardiac myogenesis in vivo using stem cells or cell-free therapies.
Similar to induced pluripotent stem cells (iPSCs), the molecular mechanism underlying the spontaneous dedifferentiation of CMs is associated with genome-wide epigenomic reprogramming [4,26,64]. However, dedifferentiation and reprogramming are often asynchronous, which means cells in bulk population may be at different stages of the processes. Thus, each individual cell will have its own unique cellular and molecular features despite some commonality such as surface markers of a certain lineage or disease state [65]. In addition, cell-to-cell variation in gene expression is essential [66,67] in tissue development, regardless of its controversy in measurement and data interpretation [68,69]. Therefore, transcriptomic and epigenomic studies at single-cell level may hold the key to addressing the molecular mechanism underlying CPC reprogramming. The ability to capture cell-to-cell heterogeneity in transcriptome and epigenome even within cell population of mutually homogenous genome gives the single-cell analysis technology much advantage over bulk population cell analysis [7,70]. The fact that this technology can be used in studies of cell state transition grants us in-depth view of cellular dynamics, as well as the mechanism within certain biological processes, including CPC dedifferentiation and reprogramming.
Current application of single-cell technology in CSC research
While qRCR-based single-cell analysis is reasonably sensitive and quantitative, it only allows analyzing a small set of target genes. Microarray-coupled single-cell analysis allows the full transcriptome analysis, but it still depends on preknown sequences for probe design [71]. Deep sequencing-based single-cell analysis promises a more unbiased genome-wide detection of differentially expressed transcripts or isoforms, and it allows the de novo discovery; nevertheless, the cost is still a major obstacle [72,73]. Today single-cell transcriptomic and epigenomic analysis has become popular in the areas of oncology and developmental biology, and the application of single-cell analysis in heart research has drawn bigger attentions from the scientific community. However, due to the challenges in the procedure involving CM isolation, its usage in the cardiovascular research is still not widely attempted. Up until now, there are only limited research articles in this field (Table 1).
FACS, flow-assisted cell sorting; iPSC, induced pluripotent stem cell; MSC, mesenchymal stem cell; qPCR, quantitative polymerase chain reaction.
In 2011, single-cell gene expression profiling for adult mouse CM was accomplished by Flynn et al., establishing the protocol of single-cell microarray for whole-genome transcriptomes or single-cell qPCR for specific genes [74]. Recently, our laboratory was able to use a microfluidics enabled multiplex qPCR array and gene expression microarray to analyze the transcriptome at single-cell level and DNA methylome of population cells of mouse CM-derived progenitor cells. We demonstrated that the transcriptomic changes in adult CM-derived CPC-like cells acquired stemness and displayed dedifferentiation and cell cycle reprogramming, which were orchestrated by genome-wide epigenomic reprogramming [4]. We used the Nugen's 3′ initiated and random primers in the reverse transcription protocol to achieve a whole transcriptome analysis, avoiding 3′ bias commonly associated with many single-cell transcriptomic studies.
In 2015, Li et al. performed multiplex single-cell qPCR on CPCs and CMs derived from mESCs and mouse embryos. The comparison of gene expression profiles revealed that the phenotypical similarity between mESC-derived CMs and embryo-derived CMs are in the neonatal stage. Combined with time-lapse microscopy, they were able to resolve the distinct cell phenotypes and lineage between single isolated CPCs [75]. In this study, a reference gene panel was established using five known lineage-specific markers in the developing heart, suggesting that the cell type identification is biased since only a set of selected preknown genes are analyzed.
Multiplex single-cell qPCR was also used in optimizing human iPSC cardiac differentiation strategy by examining and elucidating the molecular environmental requirements, as well as by characterizing the molecular mechanisms of human iPSC-derived CMs in contributing to cardiac function recovery under ischemic stimulation [61,76]. In addition, high-throughput qPCR was used after laser capture microdissection to profile the paracrine expression at the single-cell level of injected mesenchymal stem cell (MSC) in infarcted murine heart, revealing their roles in therapies by paracrine secretion [77]. To sum up, single-cell qPCR array proves to be very useful to determine the identity and the lineage relationships of each individual cardiovascular cells, and in identifying potential cell surface or intracellular markers [4,20].
Recently, scRNA-seq was performed in mouse embryo-derived CPCs from the neural plate stage to the head fold stage, which revealed a rapid and dynamic change of gene expression profiles in embryonic CPCs during the early phase after their segregation from cardiac mesoderm. By further exploring the distinct transcriptional signatures of Nkx2-5- and Tbx5-expressing CPCs, the study proposed a new early cardiac development model where CPC markers Nkx2-5 and Tbx5 play intricate roles in the cell fate decision and in driving the cardiac differentiation [78]. Using a microfluidics-based single-cell capture technique coupled with RNA-sequencing analysis, DeLaughter et al. were able to classify heart cell types (eg, CMs, EDCs, and fibroblasts) including different compartments of the heart during the early cardiac specification, differentiation, development, and growth. Their single-cell analysis revealed in greater detail on the molecular machinery leading to the human heart malformation due to Nkx2.5 mutations, such as haplo-insufficiency that impairs maturation of distinct heart cell lineages [79]. Using SMART-seq2 technique to analyze 1,205 single cells sorted, Scialdone et al. revealed a diversification of cells during early mesoderm development in mouse gastrulation [80]. They further tested the function of hematopoietic transcription factor Tal1 and demonstrated that the lack of Tal1 expression does not impact the differentiation of precursor cells into cardiac lineages, which was different from pervious retrospective analysis implicating that Tal1 could affect CM differentiation [80].
The above-mentioned studies served as examples of how the molecular mechanisms of cardiac development can be elucidated by scRNA-seq technologies. Nevertheless, other than the transcriptomic changes in the single cells during the development, epigenetic changes in the single cells should be analyzed to detect the possible correlations. Application of single-cell RRBS sequencing or single-cell tripleomics will be a promising future direction. In addition to human cardiac development, Fluidigm C1 coupled scRNA-seq was attempted in zebrafish epicardial cells to identify the epicardial lineage markers and mechanisms of heart regeneration [81].
In heart stem cell therapy, the donor MSCs possess a tendency to fuse with the recipient CMs, forming hybrids with uncertain influences toward the therapeutic outcome. To characterize the phenotypic diversification of MSC-CM hybrids, scRNA-seq coupled with Fluidigm C1 autoprep system allowed to gain insight into the hybrid transcriptome of the mouse MSCs [82]. It is noted that a certain analyzed cell is not a single cardiac cell per se, but a hybrid or fusion cell during stem cell regeneration therapy. This is an example of how single-cell technologies contribute to the understanding and development of stem cell and cell-free therapy for cardiac regeneration.
Last, although the mass of nonmyocytes in the heart accounts for much smaller fraction compared to CMs, their cell numbers are about two folds of the later. Therefore, it is critical to analyze the genomic and epigenetic landscapes of both cell types to get an unambiguous understanding of the heart development and disease remodeling. By using the concept of metagene entropy, Gong et al. developed a single-cell analysis algorithm named Dpath, which allows the ranking of cells based on their differentiation potential [83]. The key methods applied in their analysis were based on the self-organizing map and random walk with restart algorithms to separate the progenitors from the differentiated cells, thereby to reconstruct the lineage hierarchies in an unbiased manner. The scRNA-seq data set on mouse embryos analyzed by this method revealed specific molecular signaling pathways involved in the differentiation programming and specification of hematoendothelial lineages [83].
Conclusion
The rapid development of single-cell technologies has provided unprecedented tools for studying the molecular mechanisms underlying cardiac development and renewal, along with the intrinsic cellular heterogeneity. In addition, they provide powerful solutions in understanding gene regulations in rare cells, which were difficult to achieve previously. In research of CSC-based regeneration and therapeutic strategies, the capacity of cardiac-derived stem cells reprogramming into functional CMs can be dramatically improved by deciphering the molecular mechanisms of cell reprogramming and differentiation using single-cell technologies. We anticipate that a deeper insight can be gained in the molecular regulation of CSCs by coupling single-cell genomic and epigenetic analyses with functional assays covering single-cell protein, metabolism, and cell functions [4]. It is expected that single-cell analysis technologies will continue to improve and will contribute greatly to our better understanding of CSC research.
Footnotes
Acknowledgment
Authors would like to thank Dr. Yiqiang Zhang for his review on the article.
Author Disclosure Statement
No competing financial interests exist.