
Abstract
Although human embryonic stem cells (hESCs) were first derived almost 20 years ago, it was only recently acknowledged that they share closer molecular and functional identity to postimplantation lineage-primed murine epiblast stem cells than to naïve preimplantation inner cell mass-derived mouse ESCs (mESCs). A myriad of transcriptional, epigenetic, biochemical, and metabolic attributes have now been described that distinguish naïve and primed pluripotent states in both rodents and humans. Conventional hESCs and human induced pluripotent stem cells (hiPSCs) appear to lack many of the defining hallmarks of naïve mESCs. These include important features of the naïve ground state murine epiblast, such as an open epigenetic architecture, reduced lineage-primed gene expression, and chimera and germline competence following injection into a recipient blastocyst-stage embryo. Several transgenic and chemical methods were recently reported that appear to revert conventional human PSCs to mESC-like ground states. However, it remains unclear if subtle deviations in global transcription, cell signaling dependencies, and extent of epigenetic/metabolic shifts in these various human naïve-reverted pluripotent states represent true functional differences or alternatively the existence of distinct human pluripotent states along a spectrum. In this study, we review the current understanding and developmental features of various human pluripotency-associated phenotypes and discuss potential biological mechanisms that may support stable maintenance of an authentic epiblast-like ground state of human pluripotency.
Developmental Capacities of the Murine and Human Preimplantation Embryo
T
In most mammals, totipotency sensu stricto is limited to the zygote and to 2-cell blastomeres (although there have been successful reports of functional totipotency from 4- or 8-cell blastomeres) [2]. The cleavage and blastula stages of development mark the loss of totipotency and the subsequent specification of the epiblast, which is a transient embryo-forming structure that undergoes species-specific morphogenetic reorganization before gastrulation [4] (Fig. 1).
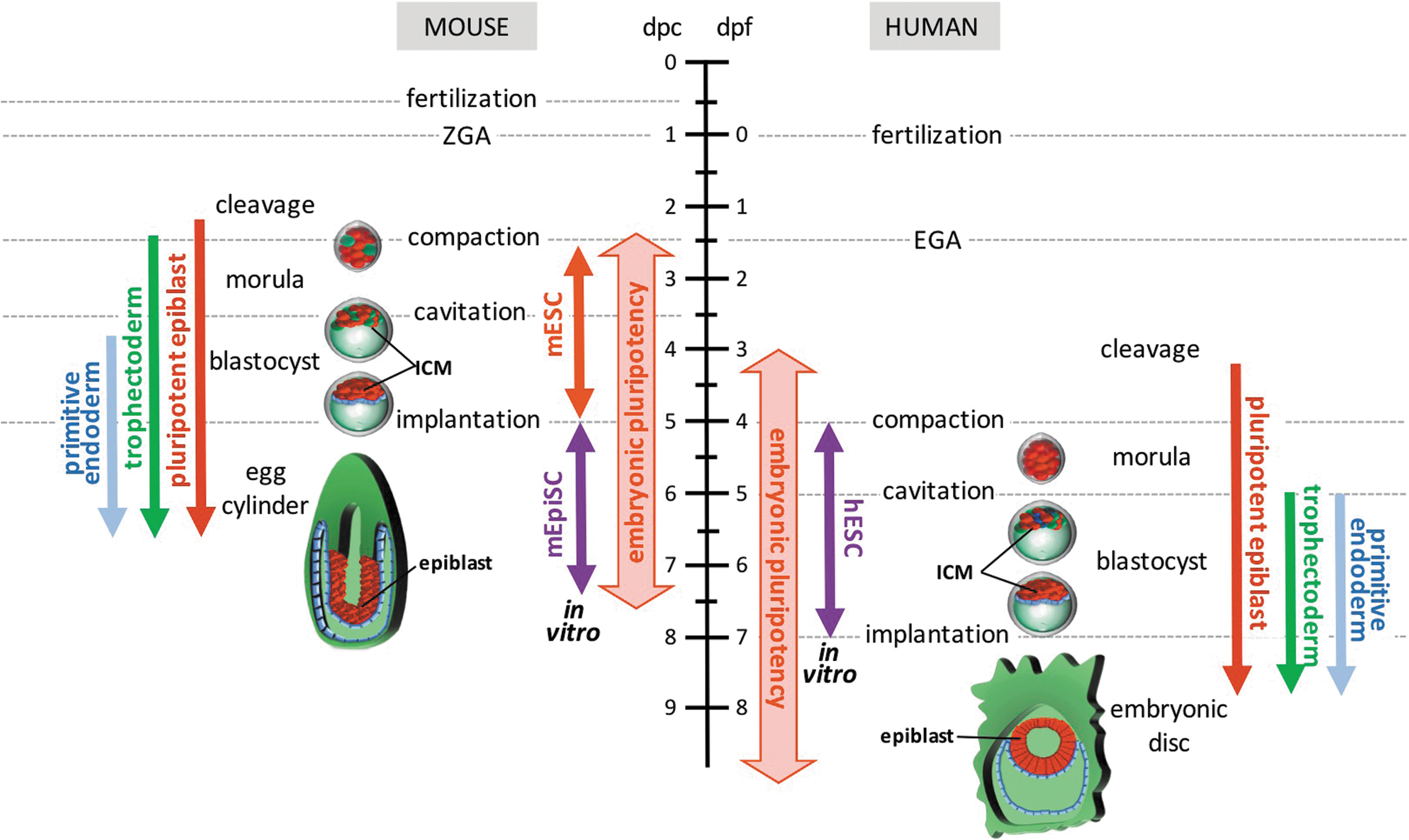
Embryonic pluripotency in early mouse and human embryonic development. Left: Pluripotent cells arise in the murine embryo during the cleavage stage, following loss of totipotency. Functional capacity to form all three germ layer lineages is retained up to the postimplantation egg cylinder epiblast. Two categories of PSCs have been isolated from murine embryos: mESCs and mEpiSCs. mESC lines can be isolated from postcleavage preimplantation embryos and model the ground state of pluripotency in the ICM of the blastocyst. In contrast, mEpiSC lines can be isolated from postimplantation epiblasts and mimic the continuum of lineage-primed developmental states that proceed to gastrulation. Right: Human embryonic pluripotency follows slower developmental kinetics than the mouse, but can be classified by analogous morphogenetic changes. Similar to mESCs, hESCs have been isolated from postcleavage preimplantation embryonic ICMs, but hESC lines share closer phenotypic and functional similarity with mEpiSCs than mESCs. hESCs may represent an equivalent of the developmentally more advanced human embryonic disc rather than the preimplantation epiblast cells they originate from. Red: pluripotent cells, green: trophectoderm, blue: primitive endoderm. hESCs, human embryonic stem cells; ICM, inner cell mass; mESCs, mouse embryonic stem cells; mEpiSCs, mouse epiblast-derived stem cells; PSCs, pluripotent stem cells.
Following zygotic activation, embryonic development follows defined rapid successions of ontogenetic phases that can be classified through standardized systems (eg, Carnegie or Hamburger–Hamilton stages) (Fig. 1). The morulae and early blastocyst stages of preimplantation development (up to the fifth cleavage division in the mouse [5]) conserve pluripotent capacity for differentiation into most, if not all, lineages. However, their capacity to self-organize into an integrated body plan is limited and has been accomplished only through artificial methods such as multicellular aggregation or tetraploid complementation [2]. The term pluripotency was originally employed by Haecker in 1914 [6] as the potential for several different developmental options [7].
The rodent preimplantation inner cell mass (ICM) (Fig. 1) transiently embraces a naïve ground state of pluripotency phenotype that is captured in vitro by ICM-derived self-renewing embryonic stem cells (ESCs) [8]. In contrast, the mouse postimplantation epiblast and its derivatives [eg, epiblast-derived stem cells (EpiSCs)] adopt primed pluripotent states with variable degrees of lineage commitment [9] and defective chimeric contribution following injection into recipient blastocysts, although limited contribution can be achieved using postimplantation embryos [10].
Current consensus dictates that putative pluripotent (pluripotential) cells should demonstrate, at a minimum, a differentiation capacity in all three germ layers (although this may extend to differentiation capacity in some or all extraembryonic tissues); although without requirement for competence of self-organization into a coherent embryo. The most widely utilized assay to validate the functional pluripotency of pluripotent stem cells (PSCs) remains teratoma formation, which is a method that was originally developed using single embryonal carcinoma cells [11].
This assay detects differentiation in all germ layers following the subcutaneous, intramuscular, intrarenal, or intratesticular injection of putative pluripotent cells into mice. However, pluripotency is more rigorously validated through potency for chimera formation and germline incorporation following morula aggregation or injection of PSC test cells into a blastocyst-stage embryo. This assay was first described following the injection of murine teratocarcinoma [12] or murine ICM [13] into mouse blastocysts or interspecifically between rat ICMs into mouse blastocysts [14]. Unlike teratoma formation, the capacity for functional chimeric incorporation into a murine blastocyst is lost by murine blastocyst ICM cells following embryo implantation [15]. Thus, this divergence in functional chimera-forming capacity broadly represents a critical delineation of at least two functional classes of pluripotent cells in early rodent embryos [16].
A critical distinction between mouse and human postimplantation embryos is revealed by the progression of the human ICM into an embryonic disc, which contrasts with the developmental structure of the well-described mouse egg cylinder (Fig. 1) [4]. However, the general nonaccessibility of implanted human embryos restricts detailed in vivo studies of this process. Recent descriptions of in vitro systems for ex utero culture and development of human embryos may provide information about human-specific cues governing human epiblast development, epithelialization, and proamniotic cavity formation throughout these poorly accessible early postimplantation phases [17,18]. However, although determination of human functional pluripotency in pre- and postimplantation embryos is limited by ethical and availability constraints, it can be extrapolated from nonhuman primate studies.
For example, using nonhuman primate embryonic cells as surrogates for human PSCs, whole rhesus ICMs and rhesus ESCs both failed to robustly chimerize with rhesus monkey host embryos with the ease routinely observed with rodent PSCs [19]. Interestingly, these studies revealed that rhesus ICMs generated reproducible chimerism only in the extraembryonic compartment and a limited engraftment in fetal liver and spleen that possibly reflected blood cell exchange through placental perfusions [19]. In contrast, monkey chimeras were efficiently generated from totipotent cleavage-stage 4-cell embryos [19], suggesting that preimplantation epiblast pluripotency may follow different functional kinetics in primates and rodents.
Distinct Molecular and Functional Pluripotencies of the Rodent Epiblast
Pre- and postimplantation epiblast cells both possess the capacity to form all three germ layers in most species, and rodent PSC lines can be successfully derived from both developmental stages. Mouse ESCs (mESCs) were originally derived as ICM-derived explants that were expanded over mitotically inactivated mouse embryonic fibroblast (MEF) feeder cells in undefined culture systems (eg, employing specific lots of fetal bovine serum (FBS) [20] or conditioned media from teratocarcinoma cultures [21]). mESC lines were subsequently revealed to exploit gp130/LIF/STAT3 [22 –25], WNT [26], and bone morphogenetic protein (BMP) [27] signaling for their self-renewal. In contrast, EpiSCs derived from the postimplantation epiblast of murine egg cylinders were stably propagated through FGF2/MEK/ERK [28 –30] and WNT-β-catenin pathway [31] signals.
Serum-based cultures of mESCs produced heterogeneous populations of lineage-primed subsets [32], and a more stringent culture system was ultimately developed using small-molecule inhibition to sustain a more primitive self-renewal [33]. This system utilized two small molecules (2i) that augmented WNT/β-catenin activation while simultaneously diminishing extracellular signal-regulated kinase (ERK) signaling [via GSK3β and mitogen-activated protein ERK (MEK) inhibition, respectively] [33]. This 2i culture system proved sufficient for stably maintaining a naïve pluripotent state in mESCs [34,35] that was biologically akin to the ground state of pluripotency of the murine preimplantation ICM [36,37].
mESCs and EpiSCs are both derived from embryonic cells separated by only several cell divisions. However, they reproduce distinct pluripotent states (ie, naïve and primed) representing major peri-implantation transcriptomic, epigenomic, and metabolic transitions of the pluripotent epiblast (Figs. 2 and 3) [8,16]. Indeed, while both mESCs and EpiSCs share a similar core pluripotency molecular network [38,39] and can differentiate into derivatives of the three germ layers in teratoma or directed differentiation assays [29,30], they retain distinct molecular and functional characteristics.

Functional phenotypes of primed and naïve pluripotent states.
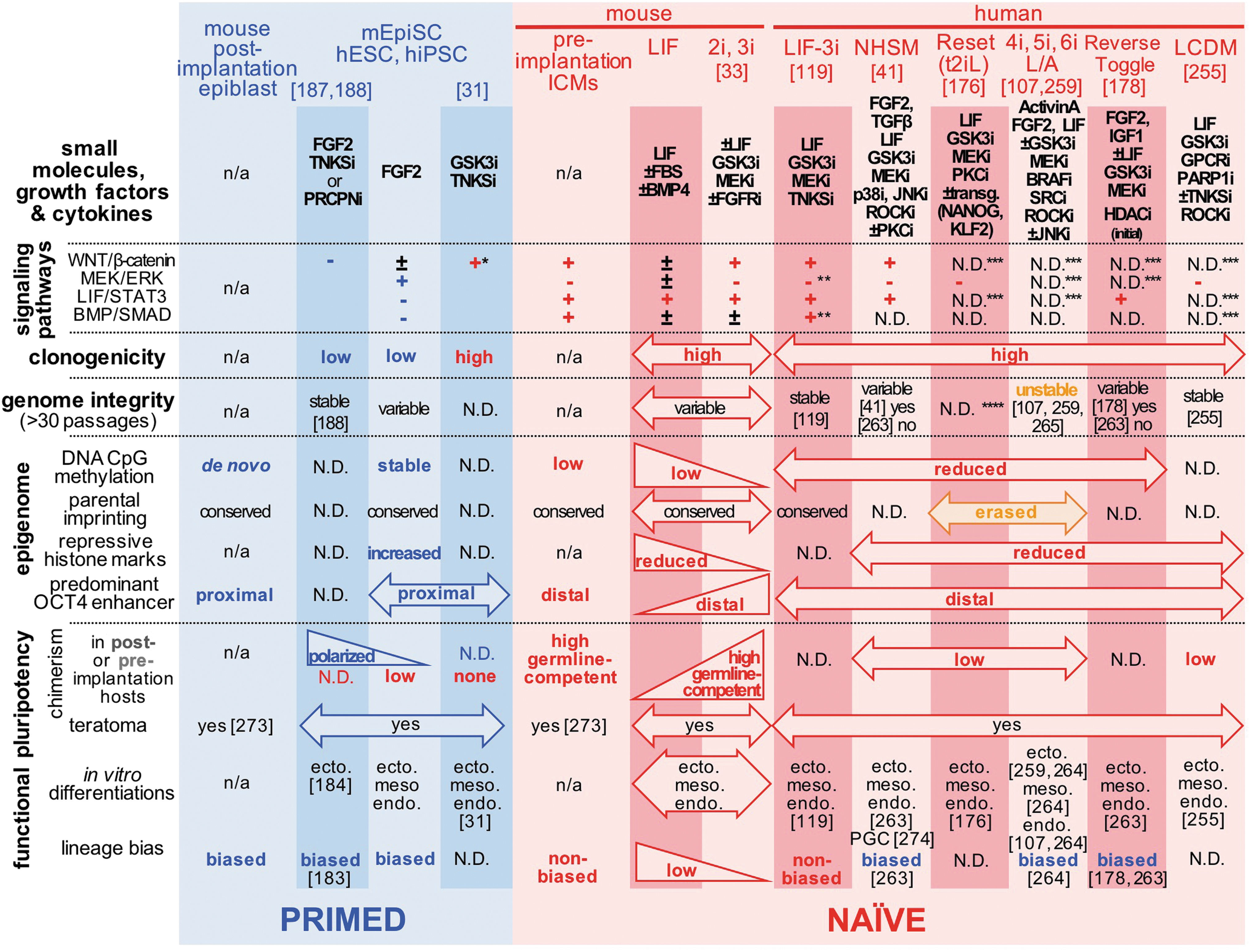
Summary of epigenetic and functional phenotypes that are detected in distinct human and mouse pluripotent states. Select human and mouse PSC culture systems are presented with their downstream outcomes on WNT/β-catenin, FGF2/MEK/ERK, LIF/STAT3, and BMP/SMAD circuitries. + and − indicate signaling activities that have been verified to be, respectively, up- and downregulated using the aforementioned cocktails of small molecules, growth factors, and cytokines. The figure lists a series of epigenomic and functional hallmarks that have been associated with and distinguish between primed and naïve pluripotent cell populations. *Non-nuclear β-catenin only, **unpublished data (MEK/ERK) or subject to interline variability (BMP/SMAD), ***directly targeted by culture conditions, but nonverified, ****normal chromosome preparations were only verified between 5 and 17 passages. n/a, not applicable; N.D., not determined. ecto., meso., endo., PGC, and TE indicate reported detections of neuroectoderm, mesoderm (ie, cardiac, hemato-vascular), definitive endoderm, primordial germ cell, and trophectoderm lineages in directed differentiation assays. BMP, bone morphogenetic protein; ERK, extracellular signal-regulated kinase; LIF, leukemia inhibitory factor; MEK, mitogen-activated protein ERK.
For example, EpiSCs exhibited higher levels of epigenetic repressive marks (eg, increased CpG promoter DNA methylation [40,41] and bivalent/repressive histone marks [41,42]) that were partially erased following reversion to naïve culture [34,35,41]. The reduced levels of classical repressive chromatin marks in naïve PSCs may potentiate a distinct epigenetic regulation of transcriptional activities [43,44] that includes seed enhancers [45], miRNA networks, RNA-induced silencing complex-mediated control of chromatin [46], and post-transcriptional regulators (eg, PARsylation by PARP1 and PARP7 [47], YAP/TAZ signaling [48 –50], and regulation of retrotransposon elements [51,52]).
The postimplantation epiblast (and thus EpiSCs [53 –55]) is characterized by increased lineage-primed gene expression relative to the naïve ground state [9,41,54] that also correlates with a functional lineage differentiation bias [9,54]. This functional discrepancy between primed and naïve pluripotencies in the mouse is revealed by their chimeric contribution capacity in pre- or postimplantation embryos (Fig. 2a). ICM-derived mESCs show robust capacity for contribution to chimeric animals when aggregated with morulae or injected into blastocysts, including efficient contribution to germline lineages. This chimera-forming capacity is further reinforced with LIF-2i-cultured mESCs [56]. In contrast, mEpiSCs expanded under standard culture conditions are not capable of significant chimeric contribution when injected into preimplantation embryos [29,30].
However, this deficiency can be improved by injection into stage-matched postimplantation epiblasts [10]. Even though EpiSCs may be artificially conditioned for engraftment into preimplantation ICM [57], their baseline inefficiency of contribution to the germline supports the notion that naïve reversion of EpiSCs is necessary for such contribution. Thus, functional and molecular pluripotencies may overlap between the two states and may not necessarily be stringently compartmentalized. For example, specific EpiSC subsets have been reported to retain naïve-like phenotypes, including chimera contribution [58]. Additionally, hybrid EpiSC culture systems using FGF2, Activin, and leukemia inhibitory factor (LIF) [59] or alternatively FGF2, Activin, and a GSK3β inhibitor [60] were shown to produce pluripotent stem cells that retained capacity for chimeric germline contribution.
Importantly, primed mEpiSCs can be successfully reverted into a naïve-like pluripotent state by exposing them to LIF/STAT3 signaling [61] or transgenic expression of key naïve inducers (eg, E-Cadherin [62], Esrrb [63], the Krüppel-like factors Klf2 (in synergy with Prdm14) [64], Klf4 [65,66] or Klf5 [67], Mbd3 [68], cMyc [66], Nanog [69], or the nuclear receptors Nr5a1/Nr5a2 [70]). Discrepancies in reversion efficiencies have been attributed to either advanced developmental progression of the starting primed state [9] or genetic background [66].
Interestingly, although LIF/STAT3 activation may be sufficient to revert specific EpiSC lines [71,72], poor reversion efficiencies or strain-specific requirements of other lines may be circumvented by employing chemical WNT modulation by the ATP-competitive cyclin and GSK3β inhibitor kenpaullone [66], the tankyrase inhibitor XAV939 [62], inhibition of the histone H3K4 methyltransferase MLL1 using MM-401 [73], nonspecific histone deacetylase (HDAC) inhibition with sodium butyrate [74], or a combination of WNT, MEK, FGFR, and TGFβ pathway inhibitions and epigenetic erasure involving inhibition of histone demethylase LSD1 [75].
The multiplicity of mouse pluripotency states captured in vitro appears to correspond to a spectrum of dynamic shifts in molecular and cellular identities in vivo that naturally progress within epiblast cells during the peri-implantation period (Fig. 2a). Pluripotency briefly persists through the developmental progression of the ICM by continuous expression of core pluripotency regulators. Thus, maintenance of a stable naïve pluripotent state in vitro may similarly require sustained reinforcement of WNT, BMP4, and LIF/STAT3 signaling [27,76 –84]. Such reinforcement likely requires a stable orchestration of events that incorporate repressive and bivalent epigenetic marks and subsequent downstream expression of epiblast lineage specifiers.
For example, this epigenetic transition is known to involve dynamic reorganization of chromatin enhancer signatures for regulating developmental factors [45,85 –87], most notably a shift of distal to proximal OCT4 enhancer usage [29,88]. Furthermore, the ICM undergoes dramatic metabolic transitions, including an interruption of its use of oxidative phosphorylation, and exclusive alternate use of glycolysis for sustaining its energy expenditures [89 –91]. Additional developmental shifts include changes in activities responsible for DNA repair [92,93] and telomere maintenance [94,95]. For example, before implantation, the embryo relied predominantly on homologous recombination for DNA repair, whereas in the subsequent postimplantation period, there is a transition toward increase of more error-prone, but more efficacious, nonhomologous end joining for double-stranded DNA repair [96].
Determinants of Human Molecular and Functional Pluripotency
Human ESCs (hESCs) were originally derived from human blastocysts in FBS-containing medium on mitotically inactive MEF feeders and human LIF [97] in conditions similar to those sustaining mESC self-renewal. However, unlike mESCs [24,25], LIF was found to not be essential for sustenance of self-renewal of human ICM cells, including in the absence of feeders [97]. Similarly, while BMP4 and LIF/STAT3 pathways synergized to support clonal growth of mESCs, supplementation of hESC cultures with BMP4 led to trophoblast lineage differentiation [98]. Subsequent hESC culture conditions adopted supplementation with FGF2, serum replacer (ie, knockout serum replacer), and mitotically inactive MEF feeders or MEF-conditioned medium for more uniform propagation and expansion of undifferentiated hESCs [99]. These culture conditions were further optimized using feeder-free and more defined medium formulations (eg, mTESR [100] and E8 [101]).
Differences in morphology, gene expression, cell cycle regulation, telomerase activity, and functional performance between mouse and human PSCs were originally attributed to species-specific attributes [102 –104]. However, subsequent isolation of EpiSCs from postimplantation mouse epiblasts [29,30] revealed that despite being a derivative of the human preimplantation ICM, human PSCs shared greater molecular, epigenetic, and functional pluripotency similarities with mEpiSCs than with mESCs (Fig. 3) [29,30,105].
In the absence of an ethically conceivable human chimera assay, the functional pluripotency of conventional (primed) human PSCs was extrapolated from surrogate nonhuman primate PSC experiments (Fig. 2b). Conventional (primed) cultures of rhesus monkey ESCs failed to participate in chimera formation when injected into rhesus blastocysts [19]. Moreover, cross-species chimera studies using host mouse blastocysts revealed that even though injected conventional monkey ESCs could transiently associate with the mouse ICM, they did not significantly contribute to developing murine fetal tissue [106]. Similar interspecies chimera approaches with human cells confirmed that even though conventional, primed hPSC cultures could not survive injection into mouse preimplantation blastocysts [41,107], limited, but measurable, integration occurred within injected gastrula-stage mouse embryos [108].
Although functional pluripotency can be validated by teratoma or directed differentiation across germ layers, interline genetic variability between conventional hESC lines [109] can result in differentiation lineage bias and skewing in response to microenvironmental cues [110,111]. Indeed, marked differentiation disparities have been documented extensively between hESC lines [112 –115]. Furthermore, hESC lines comprise heterogeneous populations [116] with epigenetically distinct coexisting subsets showing variable differentiation capacities [117].
One study involving a cohort of 20 independent hESC lines revealed that discrepancies in functional pluripotency reflected variations in both epigenetic and transcriptional profiles, including a high disparity in genes regulating development and differentiation [118]. Our own studies revealed disparities between conventional human PSCs based on highly variable lineage-primed gene expression that directly impacted functional pluripotency [119]. These studies supported the notion that conventional, primed human PSCs embrace diverse states of primed pluripotency in a manner similar to mEpiSCs [9].
Various factors among conventional human PSCs have been hypothesized to contribute to this functional variability; these include genetic background [120], acquisition of mutations in key developmental genes, and differences in derivation and culture methodologies [109]. For example, derivation of hESCs under physiological oxygen concentration may result in acquisition of naïve-like X chromosome activation, which may reinforce ground state pluripotency by suppressing spontaneous differentiation [121].
Yamanaka's discovery of transcription factor-mediated cellular reprogramming [122] for generating human induced pluripotent stem cells (hiPSCs) [123,124] revolutionized the study of pluripotency and regenerative medicine. However, hiPSC reprogramming further accentuated the variability observed in functional pluripotency between conventional PSC lines. Most hiPSC lines were noted to display more augmented lineage skewing [119,125 –128] than standard hESC lines despite their strong overlap of transcriptional and epigenetic signatures with conventional hESCs [118,129,130]. While optimization of differentiation methods partially erased these functional discrepancies in directed differentiation [131 –137], epigenomic aberrations were identified in a number of hiPSC lines that included retention of donor cell-specific somatic memory and reprogramming errors [138 –141] and were shown to be transmitted to differentiated progenies [138].
Some studies argued that such reprogramming errors resulted in differentiation bias toward their respective cell of origin lineage in mouse [142 –145], human [146 –150], and dog [151] iPSCs. It may be worth noting that most of these studies involved genome-integrating methods that introduced reprogramming factor transgenes through retroviral [142,144 –147,150,151] or lentiviral [148,149] vectors (including a lentiviral doxycycline-inducible secondary system [143]). Such viral reprogramming methods are now known to promote transcriptional and epigenetic errors [141] that were not detected using somatic cell nuclear transfer [141] or nonintegrative episomal derivation methods [119,120].
Transgene-integrating reprogramming methods may also have potentiated an increased frequency of genomic aberrations in established hiPSC clones [152 –154], which likely compromised functional pluripotency [155] through transgene reactivation in differentiated cells [156 –158].
Factors Determining the Quality of the Pluripotent State in Reprogrammed hiPSCs
A series of nonintegrative reprogramming strategies have been developed (eg, Sendai virus, episomal, and mRNA) to avoid the risks associated with viral transgene integration, but with notable disparities of their aneuploidy rates, reprogramming efficiency, reliability, and workload that have been discussed by Schlaeger et al. elsewhere [159]. We previously demonstrated that optimized episomal reprogramming was uniquely superior in activated myeloid progenitors (MPs) and could consistently achieve bulk reprogramming at high efficiencies across variable donor genetic backgrounds. Moreover, human MP-iPSC lines possessed hESC-like transcriptomes closely with significantly fewer reprogramming errors than hiPSC lines obtained from standard episomally reprogrammed adult skin fibroblasts [119,160].
This myeloid reprogramming method exploited a stromal priming activation step that delivered various signals (eg, Toll receptor/NFκB, JAK/STAT3 signaling) responsible for decreasing reprogramming efficiency barriers [119,160]. Analogous interactions with the mesenchyme may contribute to aberrant reprogramming of tumor cells toward invasive cancer phenotypes [161,162] and have also been shown to induce epigenetic changes that favor cellular reprogramming using retroviral vectors [163]. Such deterministic reprogramming generated hiPSC clones with stable genomes and reduced lineage bias [119,160] that translated into the generation of highly functional progenitors across germ layers in independent studies (ie, hemato-vascular [164], cardiac [132], and photoreceptors [165]).
Importantly, the specialized cell populations obtained from such directed differentiation assays displayed enhanced functional capacities with lower senescence, superior DNA repair capacity, or improved long-term engraftment [164], underlying a correlation between terminal differentiation and the initial pluripotency state. As such, functional pluripotency requires evaluation of terminally differentiated progenies, even though most studies limit their characterization to intermediate progenitors. To this end, we and others have developed a battery of in vitro and in vivo directed differentiation assays that were included for our group hematopoietic [131,166] (eg, macrophages [167]), vascular [164], cardiac [132,168,169], or retinal [165] lineages.
A number of comparative strategies were also employed between isogenic hiPSC lines reprogrammed from distinct cell types [170 –173] or from isogenic donor hESCs/iPSCs [120,174]. These studies have suggested that donor-specific genetic background rather than cell of origin or reprogramming system plays a more dominant role on the differentiation capacities of hiPSC lines [170,171,174]. These studies employed both integrative and nonintegrative derivation methods for cellular reprogramming (eg, Sendai virus [120,170 –172,174], episomal plasmids [171,173], lentiviruses [174], or retroviruses [171,172,174]).
A publication from the Progenitor Cell Biology Consortium analyzed a large repertoire of 58 hiPSC lines from 10 independent laboratories and reported a segregation of DNA methylation profile signatures based on their cell type of origin, but these differences could not be directly attributed to somatic donor memory [130]. Taken together, these studies have revealed that multiple complex determinants collectively impact the differentiation potency of conventional, primed human PSCs [119,120].
Conventional mEpiSC-Like Human PSCs Can Be Chemically Reverted to Highly Variable Naïve Preimplantation Epiblast-Like Pluripotent States
Several groups have developed various culture systems to revert EpiSC-like conventional human PSCs [41,48,107,119, 175 –183] or derive de novo hESCs [41,107,178,184] to pluripotent states resembling the human preimplantation epiblast (Fig. 3). These studies revealed that the classical mESC 2i cocktail of inhibitors targeting MEK and GSK3β [33] was insufficient for stable sustenance of a human ICM-like state.
LIF/STAT3 signaling, a critical self-renewal signaling pathway in mESCs [22 –25], can promote naïve reversion of both mEpiSCs [61] and hESCs [181]. However, such reversion of mEpiSCs in 2i was relatively inefficient even with transgenic STAT3 reinforcement (ie, ∼1%–2% efficiency) [61], and a number of EpiSC lines required sustained transgenic STAT3 expression in 2i culture [65]. Despite LIF supplementation, forced transgenic expression of STAT3 was essential for achieving naïve reversion of both human and mouse primed pluripotent states [61,181].
In mEpiSCs, several factors were shown to potentiate LIF-dependent STAT3 responsiveness and naïve reversion. These variables included colony size [72], increased BMP/SMAD signaling [185], and FGF/ERK inhibition [28,66], as well as stimulated [66,186] or, alternatively, reduced [62,187 –189] WNT signaling. Interestingly, these determinants were previously reported to impact lineage priming and differentiation [27,28,31,72,187,190 –194]. These studies also exposed an interline molecular and functional variability among mEpiSC lines [28,66] that impacted naïve reversion efficiencies in LIF-2i [9,186]. Our studies similarly observed a similar pattern when reverting a cohort of variably lineage-primed human PSC lines to an LIF/STAT3-dependent naïve pluripotent state in LIF-3i [119].
Four Main Molecular Axes Intersect to Balance the Maintenance and Exit of Molecular Pluripotency
The BMP/SMAD, LIF/STAT3, FGF2/MEK/ERK, and WNT/β-catenin pathways have all been recognized to regulate the self-renewal of pluripotent stem cells (Fig. 4). These molecular axes are strongly intermingled and not only control the pluripotency states but also initiate differentiation if their balanced circuitry is altered. Even though these signaling pathways regulate multiple independent downstream transcriptional targets, they also converge to a few shared effectors (Fig. 4).
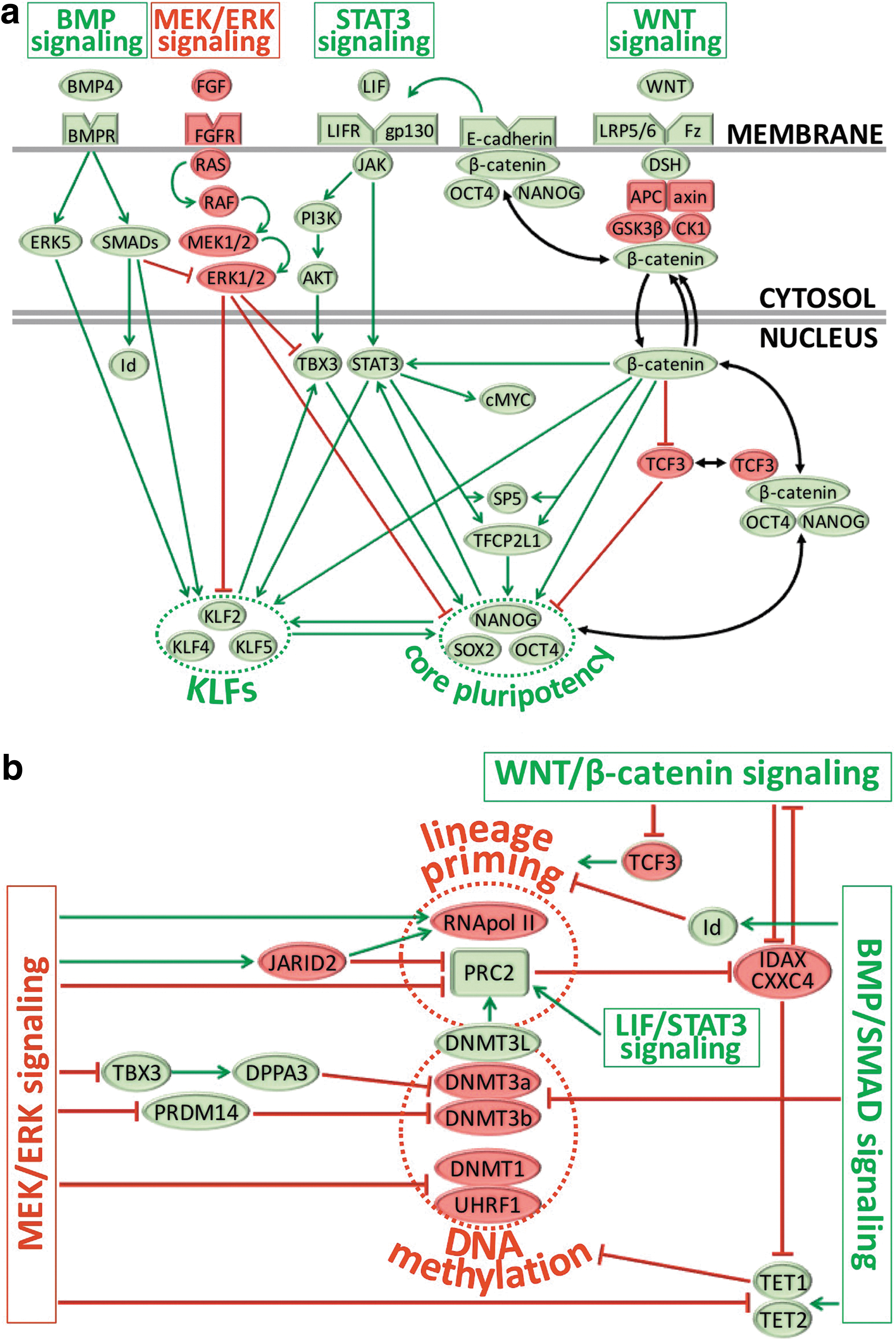
Schematic summary of four main signaling pathways that regulate the naïve pluripotent state.
For instance, the LIF/STAT3 and MEK/ERK pathways antagonistically regulate TBX3 [76], a known transcriptional activator of the pluripotency-associated genes Nanog and DPPA3/Stella [195,196]. The LIF/STAT3 and WNT/β-catenin pathways also synergistically converge onto the trans-acting protein 5 (Sp5), a transcription factor that closely relates to the Klf gene family and can reprogram mEpiSCs to the naïve state [197]. Another transcription factor named Tfcp2l1 is also similarly and independently reinforced by STAT3 and WNT signaling [77,198] and facilitates EpiSC reprogramming to naïve pluripotency by interacting with KLFs and Nanog [198] (Fig. 4).
The BMP/SMAD pathway stimulates ESC self-renewal by endogenously inhibiting MEK/ERK signaling [82], recruiting multiple Kruppel-like factors [83], or suppressing differentiation by stimulating expression of the inhibitor of differentiation (Id) genes [27] (Fig. 4). While BMP4 signals cooperate with the LIF/STAT3 axis to stabilize the naïve state [27], they can also promote differentiation of primed pluripotent stem cells under control of WNT signaling [191].
LIF/STAT3 activity is essential for maintenance of the mouse naïve state [22 –25] and promotes transition from primed to naïve pluripotency [61,181]. STAT3 signaling regulates multiple pluripotency-associated targets (Fig. 4), including reinforcement of the core pluripotency factors Nanog and SOX2 through activation of Tbx3 and Klf4 in two parallel circuitries [76], or augmented transcriptional expression of Rex1, Stat3 itself, and the epigenetic modifiers, Lin28, Ezh2, and Mbd3 [84]. Nanog expression has been shown in return to amplify STAT3 and KLF4 activities to reinforce naïve pluripotency [199]; conversely, STAT3 activity may suppress mesoendodermal differentiation in cooperation with Nanog [200].
Forced chemical rewiring of MEK/ERK and WNT/β-catenin has been shown to bypass at least partially LIF/STAT3 and BMP/SMAD signaling and sustain the naïve ground state in mESCs [33]. MEK inhibition promotes naïve ESC self-renewal by blocking lineage priming toward primitive endoderm and differentiation in naïve cells [201] through dynamic remodeling of polycomb repressors and H3K27me3 repressive marks [202]. While suppression of the ERK1/2 pathway stabilizes mESCs [203], it will promote differentiation of mEpiSCs and hESCs [8,16]. Moreover, MEK/ERK signaling may be indispensable for self-renewal of primed pluripotent cells [28,204].
Unlike the pivotal role of ERK signaling in naïve-to-primed transition, the role of WNT/β-catenin is more ambiguous since either reinforcement [66,186] or inhibition [62,187 –189] of WNT signals can augment naïve reversion of mEpiSCs. In addition, β-catenin targets have been involved in promoting both self-renewal [26,78,194,205 –207] and differentiation [187,188,191,208,209] of primed and naïve PSCs. This ambiguity may reflect the importance for a synergetic balance between the different pathways and is highly context specific. For example, in the absence of LIF and without ERK inhibition, WNT stimulation will prime mESCs toward the primitive endoderm [210].
In addition, the availability of β-catenin for interaction with distinct factors directly affects the balance between propluripotency or differentiation cues. As such, the accessibility of β-catenin to the transcription factor TCF1 in the nucleus antagonizes long-term self-renewal and functional pluripotency of mESCs [211].
MEK/ERK Inhibition Suppresses Differentiation and Reinforces Naïve Pluripotency
In mouse cells, ERK inhibition potentiates naïve reversion in cooperation with WNT and LIF/STAT3 signaling (Fig. 4) [28]. MEK/ERK inhibition also antagonizes primitive endoderm differentiation of naïve cells [201], while FGF stimulation promotes differentiation [191]. In contrast, FGF promotes pluripotency in primed cells, partially by inhibiting neuroectodermal commitment [28]. The involvement of ERK inhibition in suppressing neural commitment is not clear since ERK-mediated effects have also been shown to direct differentiation toward primitive endoderm, but not neural, lineages [201]. In this study, ERK inhibition was actually reported to reinstate neural capacity of a differentiation-compromised EpiSC line [201].
Direct isolation and expansion of mouse naïve ICM using MEK/ERK inhibition support the idea that MEK signaling blockade replicates the signaling circuitry in the mouse preimplantation epiblast [37]. Furthermore, ERK-mediated phosphorylation of Nanog was shown to promote differentiation by inhibiting Nanog transactivation and compromising Nanog stability [212]. ERK also negatively regulates the Kruppel-like factors, including KLF2 [213] and KLF4 [214], which regulate maintenance of ground state pluripotency by reinforcing core pluripotency signaling [215,216] (Fig. 4). MEK inhibition protects KLF2 phospho-degradation in mouse naïve cells and cooperates with the GSK3/TCF3 cascade to establish ground state pluripotency [213].
MEK/ERK inhibition also cooperates with GSK3β inhibition in establishing global DNA hypomethylation in mESCs to reach levels that are similar to early embryos [217]. Maintenance of a hypomethylated epigenome in mESCs due to forced MEK blockade has been shown to rely on molecular mechanisms that are redundant to those exploited by primordial germ cells and early embryos: transcriptional repression of Dnmt3A and Dnmt3B [34,217], recruitment of the polycomb repressive complex 2 (PRC2) complex [218] and the ten-eleven translocation (TET)-mediated base excision repair pathway [219], and impaired recruitment of DNMT1 due to low levels of the E3 ubiquitin-protein ligase UHRF1 [220] (Fig. 4).
ERK1/2 also contributes to establishing lineage priming in mESCs by binding to DNA sequence motifs at developmental genes that are typically accessed by polycomb repressors [221]. ERK inhibition directly interferes with PRC2 promoter occupancy and contributes to decreased phosphorylation of RNA polymerase II (RNApol II) at lineage commitment genes [221]. These ERK-mediated activities mirror epigenomic features that were previously detected in 2i cultures (ie, reduced H3K27Me3 repressive marks, fewer bivalent domains and RNApol II pausing at developmental genes, and reduced lineage priming in mouse naïve ESCs) [35]. ERK1/2 activity also regulates access of the PRC2 repressor JARID2 to developmental promoters [221]. Overall, these studies support that ERK signaling inhibition may directly or indirectly play an essential role in repression of developmental genes within a naïve epigenome to maintain pluripotency.
Subcellular WNT/β-Catenin Fluctuations May Orchestrate the Naïve Pluripotency Molecular Network
The WNT/β-catenin pathway has been linked to multiple mechanisms that ensure the maintenance of naïve pluripotency in mESCs (Fig. 4). These mechanisms include upregulation of Stat3 mRNA levels [205], augmented expression of Klf2 and Tfcp2l1 [222], and downregulation of Tcf3 to suppress neuroectodermal differentiation [223]. The canonical WNT pathway also directly affects DNA methylation by regulating TET proteins through the TET-negative regulator IDAX/CXXC4 [224]. This activity is regulated in a feedback loop inhibition since IDAX can also repress WNT by binding to DVL [225].
The involvement of WNT signaling in mediating pluripotency states is complex and may depend on its synergy with other pathways, including TGFβ/Activin [186], BMP/SMAD [191], and FGF/ERK [28]. Alternatively, intrinsic regulation of the WNT pathway itself by altering β-catenin subcellular distribution may regulate pluripotent states [31,119]. Reinforcement of WNT signaling by inhibition of GSK3β not only reinforces naïve pluripotency in mESCs [192] but also promotes acquisition of naïve features in some EpiSC lines [60,66]. Conversely, the use of inhibitors of the WNT pathway facilitates derivation of EpiSCs [188,226] and reinforces primed pluripotency in EpiSCs with [31] or without [187,226] the presence of a GSK3β inhibitor (Fig. 3). GSK3β is the kinase that initiates the cascade of phosphorylation targeting β-catenin that will ultimately lead to β-catenin proteolysis.
Stabilization of members of the destruction complex of β-catenin, while preventing β-catenin phosphorylation, can alter subcellular localization of β-catenin [227 –230], but not impede its expression [31], and will particularly reinforce β-catenin levels in the cytoplasm [230,231]. As a result, diverse WNT signal responses will be determined by the cellular distribution of β-catenin and the Axin/APC/GSK3β complex [230]. For instance, at a high nuclear β-catenin concentration in the on-state of the pathway, Axin/APC/GSK3β shuttling can maximize the response to WNT signaling by reducing fluctuations [230].
Interestingly, a membrane-associated β-catenin/OCT4 complex marks the mouse ground state of pluripotency [232] and supports the notion that a subcellular reorganization of β-catenin may participate in stabilizing the naïve state in EpiSCs. Further investigation will be needed to clarify the role of subcellular β-catenin levels in modulating naïve versus primed pluripotencies.
β-Catenin fluctuations were also observed to tightly correlate with Nanog expression levels in LIF/serum mESC cultures [233]. These fluctuations were retained in LIF-2i naïve conditions [233], presumably in a Nanog-independent manner since Nanog expression is homogenized in the latter system [35]. WNT stimulation was also shown to mitigate Nanog expression variability in mESCs [232], and β-catenin upregulated Nanog expression through its interaction with OCT4 [78]. Since shuttling of β-catenin participates in reinforcing the WNT response, continuous β-catenin fluctuations may be inherent to the naïve pluripotency state and may dynamically orchestrate the stabilization of the core pluripotency factors, OCT4 and Nanog. OCT4 participates in a shuttling complex with β-catenin and Axin that typically potentiates β-catenin degradation in the absence of GSK3β inhibition [234].
The detection of small amounts of both Nanog and OCT4 in mESC membrane containing fractions supports a possible regulation of these pluripotency factors through complexes with β-catenin [232], although further studies are still required to elucidate the role of these intricate interactions across subcellular compartments. These complexes at the membrane involve E-cadherin, β-catenin, Nanog, and OCT4 and are believed to be specific to the ground state [232]. Several other protein complexes comprising OCT4, Nanog, β-catenin, and TCF3 were also detected in the nucleus and were proposed to stabilize the mouse naïve ground state mainly by regulating the amount of free OCT4 [235]. Dynamic rearrangements of these complexes between β-catenin and core pluripotency factors may be induced by fluctuations of β-catenin shuttling and were reported to be augmented when mESCs were cultured in LIF-2i [235].
The subcellular distribution of the mESC proteome is complex, and even though the interactome of the three core pluripotency factors concentrates onto chromatin-bound factors, it also extends to a variety of non-nuclear targets, suggesting regulatory mechanisms involving rearrangements between compartments [236]. For example, subcellular relocalization of proteins between naïve and primed pluripotent states has been described for Tfe3, an important bHLH transcription factor that regulates Esrrb expression in mESCs and that relocates to the cytoplasmic compartment upon exit from ground state pluripotency [237].
Interestingly, by using a cocktail of small inhibitors targeting MEK, GSK3β, and tankyrase, our group (Zimmerlin et al.) achieved a rearrangement of activated β-catenin in human primed PSC lines [119]. The tankyrase inhibitor XAV939 stabilizes Axin, the presumptive limiting factor of the β-catenin destruction complex [238]. In the LIF-3i culture system, simultaneous exposure to GSK3β and tankyrase inhibitors permitted simultaneous stabilization of β-catenin and Axin and paradoxically reinforced active β-catenin levels in both nuclear and cytosolic compartments [119].
A similar approach involving GSK3β and tankyrase inhibition (but without concomitant MEK inhibition) was employed to stabilize hESC and mEpiSC lines in a primed pluripotent state [31] and also augmented cytoplasmic (at the expense of nuclear) levels of β-catenin. Simultaneous exposure to CHIR99021 and XAV939 permitted clonal propagation of mouse and human primed cells, although without any detectable acquisition of a naïve phenotype (ie, no upregulation of naïve pluripotency genes, switch of OCT4 enhancer usage, or blastocyst chimera potential) [31]. Interestingly, the effects of the CHIR99021/XAV939 inhibition combination in this study appeared to be independent of E-Cadherin since an E-cadherin-depleted EpiSC line could still be propagated in culture [31].
The CHIR99021/XAV939-induced primed state may primarily benefit from nuclear exclusion of β-catenin. Indeed, while promoting self-renewal [26,239] or derivation [240] of mESCs, transcriptional activity of β-catenin promotes emergence of lineage-specified progenitors in EpiSCs and hESCs [187,191,241,242].
Thus, although molecular rewiring in the 2i condition has been shown to be sufficient for maintaining the naïve ground state in mESCs [33], a customized tuning of the WNT/β-catenin signaling output may be required to sustain naïve molecular pluripotency in human PSCs. Such WNT regulation has been achieved in distinct mouse genetic backgrounds by further reinforcing [66,186] or attenuating [62,187 –189] WNT signals. This may potentially be accomplished through marked reduction (eg, 3–10-fold) of the standard 3 μM concentration of the GSK3β inhibitor CHIR99021 in 2i-based cultures, which may prevent the spontaneous differentiation of rat ESCs [243,244] or human PSCs [107,176,178,184] possibly resulting from excessive amounts of nuclear β-catenin.
E-Cadherin May Regulate Naïve Pluripotency by Regulating the Intracellular Levels of β-Catenin
As outlined above, the balance between self-renewal and differentiation may be regulated through subcellular levels of β-catenin, and this shuttling between cytoplasmic and nuclear compartments may be controlled at the level of the actin cytoskeleton (Fig. 4). At the membrane, β-catenin promotes intercellular adhesion by complexing with E-cadherin and facilitating the binding of cadherins to the actin cytoskeleton. In contrast, in the nucleus, β-catenin serves as a transcriptional cofactor to activate target genes of the canonical WNT signaling pathway either through repression of members of the TCF protein family [192,245,246] or through TCF-independent mechanisms that involve direct targeting of pluripotency factors such as Nanog [78], OCT4 [81], and KLF4 [247].
At the membrane, β-catenin promotes cadherin-mediated intercellular adhesions by binding the cytoplasmic domain of E-cadherin and linking the adherens junction to actin filaments through interaction with α-catenin. β-Catenin may first bind E-cadherin within the endoplasmic reticulum (ER) membrane, which initially protects it from proteolytic degradation [248] and subsequently facilitates its exit from the ER before its transport toward the cell membrane [245]. Maternal E-cadherin and β-catenin are present until early morula stages in mouse embryos and promote blastomere adhesion and morula compaction [249].
The observation that the absence of maternal E-cadherin can restore the developmental deficit induced by a truncating β-catenin mutant suggests that the interactions between E-cadherin and β-catenin at the membrane may directly regulate the availability of nuclear β-catenin during embryonic genome activation [249]. A similar interdependence between adhesive and cotranscriptional roles of β-catenin has also been implied in cancer cells [250]. Moreover, E-cadherin is required for proper activation of LIF receptor/gp130 signaling and STAT3 phosphorylation in mESCs [251]. This E-Cadherin-mediated STAT3 signaling has been shown to contribute to elevated Nanog expression [80] and stabilizes naïve pluripotency.
The importance of β-catenin-induced E-Cadherin reinforcement at the membrane in safeguarding functional pluripotency was highlighted in studies exploiting a TCF/LEF signaling defective β-catenin variant that independently restored β-catenin-mediated adhesion [252]. These β-catenin-deficient mESCs exhibited impaired mesendoderm formation and neuronal differentiation, and introduction of a β-catenin variant without TCF-mediated nuclear activities partially rescued adhesion and endoderm (although not mesoderm) formation as well as neuronal differentiation [252].
Other functional data substantiating the importance of E-cadherin levels to support naïve pluripotency have been obtained from genetic manipulation of primed EpiSCs. For example, ectopic E-cadherin expression in mEpiSC lines enhanced chimerism efficiency in blastocyst injection experiments [253], although without germline contribution. In contrast, disruption of E-cadherin at the membrane following either genetic loss of β-catenin or through tankyrase inhibition (XAV939) augmented a biased integration of primed EpiSCs into postimplantation embryos [187].
Functional Validation of Human and Nonhuman Primate Naïve Pluripotent States: A Work in Progress
Conventional human PSCs were recently reverted to a highly variable spectrum of naïve-like pluripotent states that partially replicated the molecular circuitry of mESCs and human preimplantation embryos (Fig. 3). These culture systems generally not only relied on utilization of classical mouse naïve 2i conditions (GSK3β and MEK inhibition), but also required additional chemical modulation for stabilizing inherently unstable or metastable human naïve states.
These methods included (1) hybrid culture systems that costimulate primed pluripotency circuitry with exogenous FGF2 [41,107,177,178,182,254], Activin/TGFβ [41,107,177, 254], or the BMP inhibitor dorsomorphin [177]; (2) forced transgene expression of OCT4, SOX2, and KLF4 [175], OCT4 and KLF4 [175], KLF2 and KLF4 [175], NANOG and KLF2 [107,176], YAP [48], or STAT3 [181]; (3) global epigenetic erasure using HDAC inhibitors [178]; (4) chemical rewiring of antiapoptotic signaling pathways (eg, activation of adenylyl-cyclase [48,175] and/or YAP [48] or inhibition of B-RAF [107], JNK [41,107], p38 [41] PKC [41,176], ROCK [41,107,176,177,180,181], and SRC [107]); and (5) multipathway biochemical and epigenomic rewiring of uncharacterized naïve stabilizing pathways through tankyrase [119] or PARP1 [255] signal inhibition.
Many of these strategies stemmed from previous efforts of mEpiSC naïve reversion or potentiation of mESC derivation conditions. For example, (1) hybrid EpiSC culture systems (eg, FGF2, Activin and LIF [59], FGF2, Activin, and inhibition of GSK3β [60]); (2) forced transgenic expression of E-Cadherin [62], Esrrb [63], Klf2 [64], Klf4 [65,66], Klf5 [67], Mbd3 [68], cMyc [66], Nanog [69], Nr5a1 [70], Nr5a2 [70], or Stat3 [61]; (3) global epigenetic remodeling using the H3K4 methyltransferase MLL1 inhibitor MM-401 [73], the HDAC inhibitor sodium butyrate [74], or the histone demethylase LSD1 [75]; and (4) reinforcement of naïve pluripotency molecular circuitry by augmentation of LIF/STAT3 [61], BMP/SMAD [185], WNT/β-catenin [66,186], or inhibiting FGF/ERK [28,66] signaling.
Similar to mEpiSCs, conventional human PSCs have demonstrated measurable, but limited, capacity for engraftment into postimplantation mouse developing embryos [108], yet only limited chimerization in mouse blastocysts [256]. The chimeric contribution of human and nonhuman primate cells into preimplantation embryos was assessed for naïve-reverted cells obtained using the naïve human stem cell medium (NHSM) method developed by Hanna's team [41,254,257
–259], variants of the 5i/L/A cocktail from the Jaenisch's group [107,259] and the t2iL technique [254,259] developed in Austin Smith's laboratory [176], and more recently the FGF2, Activin, CHIR990211 (FAC) [254] and
Reliable detection of transgenic green fluorescent protein (GFP) within murine embryos was technically challenging with such reduced levels of chimerism [107,254,259], and several groups employed sensitive polymerase chain reaction (PCR)-based methods (ie, human mitochondrial DNA, human Alu sequence) [254,255,259] to determine chimerism for NHSM [41], t2iL [176], 5i/L/A [107], FAC [60,254], and LCDM [255] human naïve or intermediate culture methods.
Using a mitochondrial DNA PCR detection assay, Theunissen et al. reported rare and sporadic chimerism in less than 1% of E10.5 embryos following injection of NHSM, t2iL, or variants of 5i/L/A human naïve cells into murine morulae and blastocysts [259]. A similar approach was employed by Yang et al. for the LCDM culture system and the authors similarly measured limited (≤1%) human chimeric integration in E10.5 embryos, but with significantly higher frequencies of human cell contribution within the murine trophectoderm [255].
Interestingly, the authors of the LCDM method recommend the addition of the tankyrase inhibitor IWR-1-endo to their chemical cocktail when culturing human cells [255], and as such, their results further corroborate our own studies establishing that tankyrase inhibition stabilizes a human naïve-like state with improved functional pluripotency [119].
Belmonte's group similarly reported extremely low levels of human–animal chimerism using a PCR-based genomic assay of human-specific Alu sequences that detected human cells in the chimeric offspring of human–pig blastocyst injection experiments. Collectively, these experiments demonstrated that robust levels of interspecies chimerism with human naïve PSCs have not yet been achieved [254].
However, it remains unclear whether limited chimerism of human naïve PSCs in mouse blastocysts [41,107,257,259] represents a poorly obtained human naïve functional pluripotency or alternatively reflects obstacles of genetic distance, phenotypic differences, and/or developmental divergences of human and mouse embryos (eg, early postimplantation epiblast morphogenesis and ontogenesis variations in size, shape, and speed). Efforts to improve host compatibility in chimera assays have included allogeneic (monkey-to-monkey) transfer [258] and injection of human naïve PSCs into ungulate (ie, pig and cattle) blastocysts [254]. Unfortunately, both strategies have still yielded particularly low levels of chimerism that did not match the successes of rat–mouse interspecies [254,260,261] or large animal (ie, pig) allogeneic [262] blastocyst complementation.
Strategies that provide a selective advantage for donor PSCs may augment the incorporation of human cells into host embryos. Future studies will test candidate naïve PSCs with such improved methodologies for assaying the functional pluripotency of putative bona fide naïve human PSCs.
Several reports have now demonstrated impaired functional pluripotency from selected human naïve-reverted pluripotent states [263,264], including the NHSM [41,263], 5i/L/A [259,264,265], reverse toggle [178,263], and naïve conversion medium (NCM) [182,263] systems. Moreover, the 5i/L/A method produced hPSCs in a naïve-like state that either required prolonged repriming for proper differentiation [259,265] or displayed pronounced neuroectodermal bias and impaired terminal differentiation [264]. Other human naïve reversion methods similarly displayed limited capacity to undergo terminal differentiation toward functional phenotypes when compared with primed isogenic cultures [263].
Notwithstanding the hypothesis that these states may embrace a more primitive and paradoxically less competent pluripotent state [266], the impaired functional pluripotency that has been detected using these human naïve states is in clear contradiction with results obtained from mouse [9,35,267,268] and rabbit [269,270] PSCs, especially in regard to neural differentiation. Primed mEpiSC lines display disparate lineage priming and differentiation capacity [9,54], while naïve cultures not only exhibit restored or augmented neuroectodermal capacity [9,35,267] but also generate terminally differentiated neural populations that resemble more closely the mouse adult brain [267].
Unlike the aforementioned variable human naïve-like states described above, chemical naïve reversion that supplements classical MEK/ERK and GSK3β inhibition with a tankyrase inhibiter (LIF-3i) is the only method described thus far that improves functional pluripotency across germ layers of a large repertoire of hESC and hiPSC lines [119]. Importantly, karyotypic and epigenomic imprinting aberrations were not detected in LIF-3i-reverted naïve-like hPSCs [119].
In contrast, the impaired functional pluripotency in other naïve reversion methods described thus far may derive from either the reported chromosomal instability or aberrant erasure of genomic imprints following chemical manipulation from these systems. For example, abnormal karyotypes were reported in 5i/L/A [259,264,265], NHSM [263], and reverse toggle [263] methods, while aberrant parental imprinting was inherent to the 5i/L/A [259,271] and t2iL [271] methods. The further optimization of tankyrase inhibitor-utilizing LIF-3i methods in defined feeder-free, xeno-free culture conditions may allow efficient and clinically useful generation of functional and engraftable adult-like cell types for therapeutic use, including for hemato-vascular regeneration [272].
Footnotes
Acknowledgments
This work was supported by grants from NIH/NHLBI [U01HL099775 (E.T.Z.), NIH/NEI R01EY023962 (E.T.Z.)], NIH/NICHD R01HD082098 (E.T.Z.), RPB Stein Innovation Award (E.T.Z.), The Maryland Stem Cell Research Fund 2013-MSCRF-III-114936 (L.Z.), 2013-MSCRFII-0032-00 (E.T.Z.), 2014-MSCRFE-118153 (T.S.P.), and Novo Nordisk Science Forum Award (E.T.Z.).
Author Disclosure Statement
No competing financial interests exist.