
Abstract
Deubiquitinating enzymes may play a major regulatory role in pluripotent stem cells (PSCs), but few studies have investigated this topic. Within this family of enzymes, we found that the ubiquitin-specific peptidase USP44, is highly expressed in embryonic stem cells, induced PSCs (iPSCs), and testes as compared with differentiated progenies and somatic organs. Analysis by quantitative polymerase chain reaction and 5′ RACE showed that alternate promoters are responsible for expression in PSCs and organs. We noticed seven regions of transcription initiation, some of them with cell- or tissue-specific activity. Close analysis showed that one of the promoters involved in stem cell- and testis-specific activity is differentially regulated in those tissues. At the epigenetic level, USP44 transcription was correlated with DNA methylation of a CpG island close to the main promoter region. These data imply a complex picture where regulating factors such as OCT4 may interact with other epigenetic mechanisms to regulate USP44 expression in PSCs and testes.
Introduction
I
These three closely interacting factors bind to specific DNA patterns and control gene expression through multi-protein complexes integrating extra- and intra-cellular signals (for reviews, see Young [6]; Chambers and Tomlinson [7]). This core triumvirate maintains the pluripotent state through three complementary mechanisms: (1) by functioning together on their respective promoters through positive regulatory loops; (2) by positively regulating the expression of genes involved in stem cell-specific physiology; and (3) by maintaining a low or even null level of expression of genes involved in differentiation. Understanding this core concept was a key step in the invention of cell reprogramming and derivation of induced PSCs (iPSCs) [8,9]. In humans, POU5F1, NANOG, and SOX2 together or independently regulate several hundred genes [10 –12], but very few of them are exclusively expressed in PSC and the way their expression is driven remains mostly unknown.
Ubiquitin is a 76-amino-acid residue protein that can be covalently conjugated to targeted proteins via a multistep enzymatic cascade [13]. Ubiquitylation adopts various forms such as the addition of either one peptide (monoubiquitylation) or multiple independent ubiquitin monomers or polymers, with or without branched chains [14]. When linked to polymers, ubiquitin peptides can be anchored to each other by several residues (lysine 11, 48 or 63), making the ubiquitin code very complex. Depending on the modification, the consequence of ubiquitylation can be the destruction of targeted proteins, the regulation of their function/interaction with partners, or their relocalization. The level of ubiquitination is tightly regulated by the action of deubiquitinating enzymes (DUBs) [15], which belong to five protein families comprising the Ubiquitin Specific Peptidases (USPs). DUBs specifically interact with their target to remove ubiquitin in a controlled manner. Moreover, some linkage-specificities exist among DUBs, but they are still poorly defined [16].
Because ubiquitination-deubiquitination cycles are increasingly known to take part in many key cellular mechanisms, we decided to address the expression of DUBs in PSCs. Within this family, we identified USP44 as highly expressed in PSCs and germinal organs/cells but mainly repressed on differentiation and in somatic tissues. Using 5′ RACE, we characterized new promoter regions that are responsible for this specific expression whereas others are involved in weak transcription in a few somatic organs. Interestingly, expression levels correlated with methylation ratios of a CpG island (CGI) identified within the promoters area. Our data suggest that USP44 is under control of a complex machinery resulting from interactions between pluripotent core genes POU5F1, NANOG, and Sox2 and epigenetic factors.
Materials and Methods
Biological material
H9 human ESCs were provided by the ESteam platform (Villejuif, France) under agreement from the WiCell Institute. HUES24 and HUES24-derived EB RNAs were kindly provided by M. Pucéat, INSERM, Marseille, France. Isolation of CPRE2 human fibroblasts and derivation and characterization of iPSC lines 1 to 6 are described elsewhere [17]. In the latter publication, iPSC lines 1 to 6 correspond to iPSC lines LV-MONO-1 and LV-MONO-2, RV-Mono-1 and RV-Mono-2, and LV-ONSL-1 and LV-ONSL-2, respectively, derived by using monocistronic and polycistronic lentivirus and retrovirus vectors. Human PSCs were amplified in mTeSR1 medium (Stemcell Technologies, Grenoble, France) on Matrigel-coated dishes (BD Biosciences, Pont-de-Claix, France).
A human tissue RNA set was provided by Life Technologies (Villebon sur Yvette, France). Every organ mRNA sample was a mix from three donors. Genomic DNAs were from Biochain (Newark, USA).
Reverse transcription quantitative polymerase chain reaction
Cell RNA was extracted by using Qiagen RNeasy kit according to the manufacturer's conditions (Qiagen, Courtaboeuf, France). One microgram of RNA was analysed by reverse transcription quantitative polymerase chain reaction (RT-qPCR) for gene expression. Briefly, each sample was first split into two tubes (0.5 μg each). The first tube was treated as previously described [17] whereas the second tube was similarly treated but without addition of reverse transcriptase, as a control for gDNA contamination of RNAs. Then, this cDNA control was submitted to qPCR amplification of an internal sequence of the POU5F1 gene by using Hs00742896-s1 Taqman assay (Thermo Scientific, Illkirch, France). Samples with Ct >40 were considered negative for gDNA contamination. Primers are listed in Table 1. All qPCR data were standardized according to the double Δ method, first relative to the internal reference GAPDH and then to the undifferentiated HUES24 sample that served as a calibrator. Results were considered as not significant when the Ct value was more than 35 cycles and when variations among triplicate were ≥1.
RT-qPCR, reverse transcription quantitative polymerase chain reaction.
5′ RACE analysis
5′ RACE was performed according to the provider's instructions (Roche, Meylan, France) by using primers described in Table 1. Briefly, USP44 mRNAs were retro-transcribed from total RNAs in the presence of a first USP44-specific primer (called SP1) hybridizing in exon 2 coding region. cDNA were purified (Qiagen MinElute purification kit), and a polyA tail was added by the terminal transferase enzyme. Two subsequent PCRs were performed to amplify USP44-specific sequences: a first one using a USP44-specific primer (SP2) and an oligo dT-anchor primer and then a second one using a nested USP44-specific primer (SP3) and the anchor primer. Terminal PCR products were visualized on an agarose gel, extracted (Qiagen MinElute gel purification kit), cloned by using TOPO kit (Thermo Scientific), and, finally, sequenced for identification of 5′ USP44 sequences.
DNA methylation analysis
Online MethPrimer software [18] was used to identify CGIs and draw primers for amplification of bisulfite-treated sequences (primers in Tabe 1). Genomic DNA was bisulfite treated by using the EpiTect bisulfite kit (Qiagen). Then, 0.5 to 1 μg of gDNA was treated and 50 ng of treated DNA was used for each PCR. PCR products were purified by using a PCR purification kit (Qiagen), cloned into a Topo TA vector (Thermo Scientific), and sequenced (GATC, Mulhouse, France).
Results
In silico analysis reveals putative PSC-specific expression of USP44
To identify PSC-specific USP, we first checked the expression profile of each USP on the Amazonia! web site [19]. This web site provides a meta-analysis of available gene expression databases from stem cells and a panel of tissues [20]. We specifically looked for USP encoding genes highly expressed in human embryonic stem cells (hESC) and human induced pluripotent stem cells (hiPSC), but repressed in hESC-derived embryoid bodies and in somatic organs, that is, similar to POU5F1 [21], NANOG [22], or DPPA4 [23] profiles. Each USP gene profile was visually analyzed and only USP44 was identified as specifically expressed in PSCs, and also in testes and oocytes [24]. In a second step, we confronted the Amazonia! USP44 expression profile to NCBI EST data (UniGene Hs.646421; [25]). The latter showed strong USP44 expression in testes and in blastocysts (from which hESC are derived), suggesting that the gene could be highly espressed in PSCs, and in germinal organs and/or cells. Therefore, USP44 was retained for further analysis.
USP44 is expressed in undifferentiated hESC and hiPSC
In a first experimental approach, we aimed at confirming data available online about USP44 expression. We first focused our analysis on human PSCs, that is, ESCs and iPSCs, and on their directly differentiated derivatives, embryoid bodies. We performed coding region-specific qPCR by using two primer pairs: a first pair in exon 2, and a second spanning the exon 5 and 6 junction (respectively, light and dark gray bars in Fig. 1A). In both instances, USP44 was readily detected in both populations of PSCs, with closely related values (Fig. 1B). In contrast, expression in embryoid bodies was much lower than in their parental hES cells when using E2-specific primers, and under the detection threshold with E5–E6-specific primers, confirming Amazonia! web site analysis.

USP44 gene is expressed in undifferentiated PSCs, hESCs, and hiPSCs.
When this study began, the Ensembl database described three USP44 mRNAs, called USP44-001, USP44-004, and USP44-002, differing by alternative first exons (from 5′ to 3′ on the genomic DNA sequence; Fig. 1A) and transcribed from alternative promoters. We, thus, checked whether USP44 expression could be due to dedicated promoter(s) by performing independent qPCR with USP44-001 and USP44-004-specific forward primers in tandem with a common reverse primer positioned in exon 2 (our attempt to elaborate a USP44-002-specific forward primer remained unsuccessful, probably because of the composition of the primary sequence). In either instance, USP44 mRNAs remained undetectable in embryoid bodies and in dermal fibroblasts (Fig. 1C, D). In contrast, hESC and hiPSC expressed similar levels of USP44-001 (Fig. 1C) and USP44-004 (Fig. 1D) mRNAs.
USP44 coding region is highly expressed in PSCs and in testes
USP44 gene transcription was then investigated in various organs by testing the expression of the USP44 coding region using the same exon 2 and exons 5–6-specific primers (respectively, light and dark gray bars in Fig. 2A). In both cases, the results clearly showed that USP44 mRNA was highly expressed in hESC and in testes; whereas the ovary, thymus, lung, and thyroid gland displayed significant but much lower expression. All other organs tested in our panel showed almost no expression of USP44.
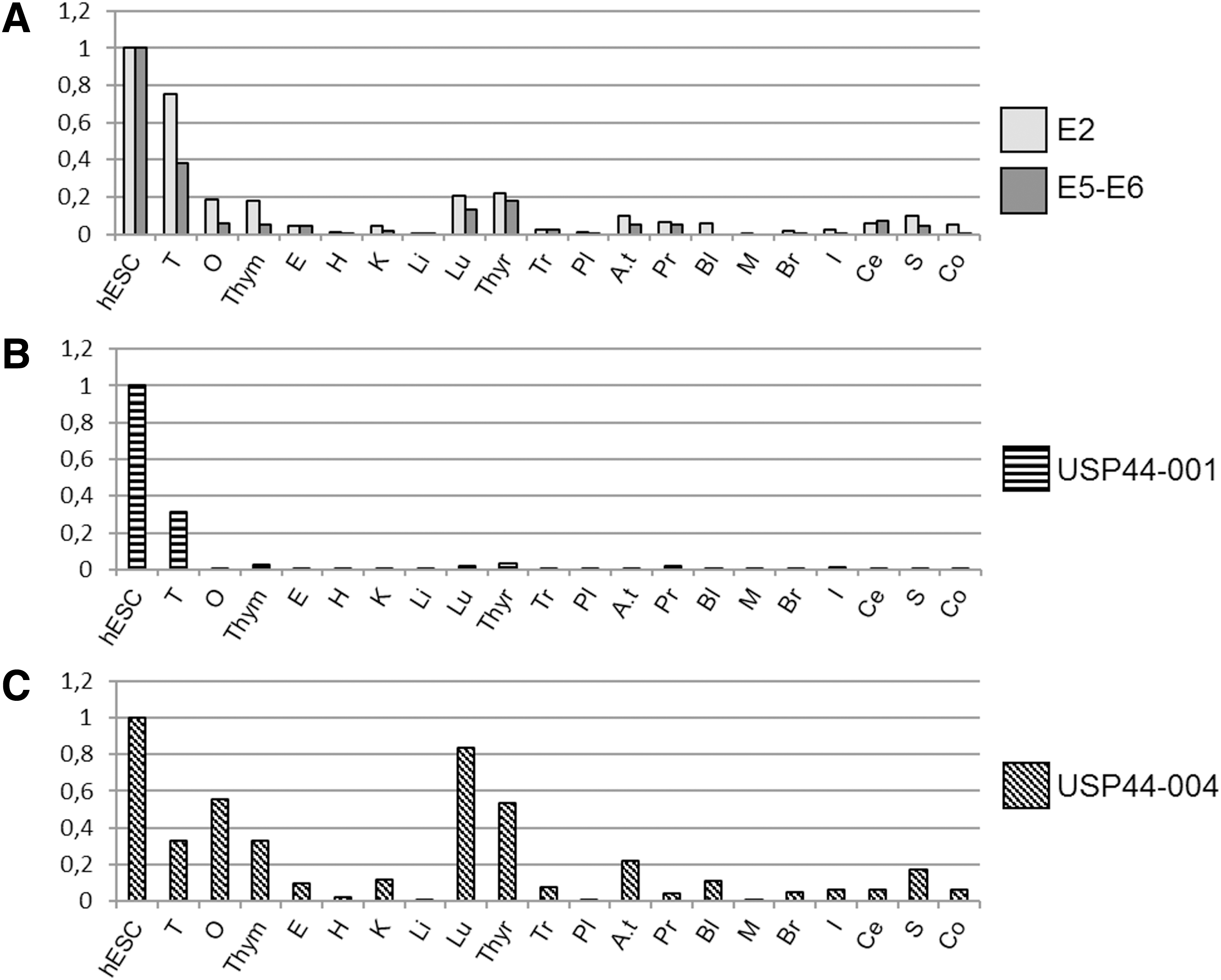
USP44 gene is mainly expressed in PSCs and testes.
Expression in PSCs is under control of a specific promoter region
To identify PSC-specific promoters, we performed RT-qPCR analysis on hESC and organ RNAs by using USP44-001 and USP44-004-specific primer pairs described earlier. When using USP44-001-specific primers, quantitative analysis showed that the corresponding promoter is almost exclusively active in pluripotent hESC and in testes (Fig. 2B). No other organ expressed USP44-001 mRNA. In contrast, USP44-004 mRNA could be detected in several organs (Fig. 2C), that is, testes, ovary, thymus, thyroid gland, and lung where we previously detected USP44 coding regions (Fig. 2A), and in adipose tissues.
These results suggest that USP44 transcription may be differentially regulated in various organs/cells. The absence of quantitative data on the USP44-002 expression profile prevents us from drawing a definitive conclusion about the origin(s) of USP44 expression in respective organs, but our data strongly suggest that independent promoters actually regulate USP44 expression. One promoter ensures that USP44-001 mRNA transcription is definitely specifically active in hESC and testes, whereas another at the origin of USP44-004 may more widely initiate transcription in somatic organs. These results also suggest that in hESC and testes, USP44 is mainly produced through USP44-001 mRNA.
5′ RACE analysis highlights several promoter regions with tissue-specific activities
During the course of this work, more mRNAs transcribed from alternative promoters were published on database web sites. To get a deeper insight into USP44 gene transcription, we performed a 5′ RACE assay to identify transcription start sites (TSS) and regions of promoter activity, using a two-step PCR system with nested primers situated in exon 2, more than 80 bp downstream of the ATG codon. Our 5′ RACE approach produced qualitative data on exon presence or absence, with no insight on quantitative aspects.
5′ RACE profiles of the two hESC lines investigated herein, HUES24 and H9, looked very similar, with transcription initiation occurring at four common sites (Fig. 3A). The vast majority of sequenced clones obtained from hESC were a part of USP44-001 and USP44-004 mRNAs. USP44-002 mRNA was observed in H9 and testis, but not in HUES24, maybe because of differences in cell culture procedures. Analysis of TSS in human somatic tissues demonstrated that USP44-001 and USP44-002 were only detected in hESC and testes, suggesting that their respective promoters could be specifically active in pluripotent cells such as ESCs and germinal cells (the data confirmed qPCR results on USP44-001 expression; Fig. 2C).
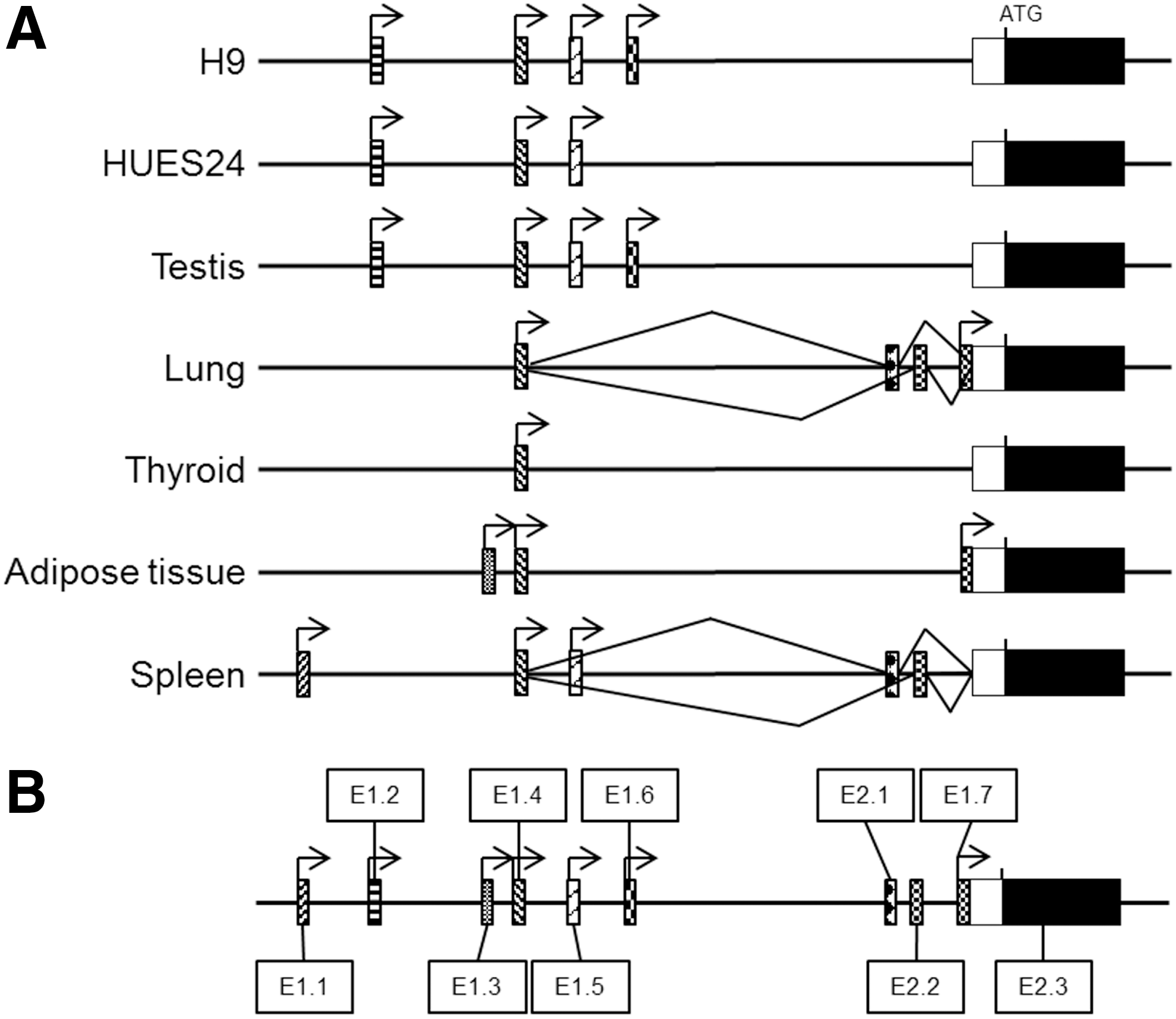
5′ RACE reveals a complex organization of the USP44 gene, including new alternative exons 1 and 2.
While this article was being written, a total of eight mRNAs were identified and displayed on the Ensembl web site. Two of them were truncated mRNAs (USP44-006 and USP44-008), and six were full protein-coding RNAs. Their transcription was initiated from four promoters (ie, four different “exon 1”); they all contained exon 2 (ie, the first coding exon), and they were all potentially detectable in our assay. Our 5′ RACE data described seven different alternative first exons, including the four described in Ensembl. This more complex situation prompted us to adopt a new nomenclature to identify each first exon. We simply numbered them from 5′ to 3′ along the locus, from exon 1.1 (E1.1) to exon 1.7 (E1.7) (Fig. 3B; Supplementary Fig. S1; Supplementary Data are available online at
For E1s: the farthest 5′ initiation nucleotide and the last nucleotide of each exon. For E2: first nucleotide to last nucleotide of each exon.
ND, not determined.
The promoter used for transcription of E1.4-containing mRNA was clearly active in all tissues examined, whereas promoters for E1.5 and E1.7 transcription were less broadly active, even though they were not exclusive to one tissue. On the other hand, E1.1 and E1.3 were definitely exclusively transcribed in spleen and adipose tissue, respectively. In a few clones, two exclusive non-coding second exons (that we called E2.1 and E2.2) were detected between E1.4 and exon 2 (ie, the first coding exon we finally renamed E2.3 and that became in this case an “exon 3”!). Surprisingly, we also characterized transcription initiation in the 5′ region of this E2.3. Transcripts from this region (their first exon is E1.7+E2.3) are described in the
Exon 1.2 transcription initiation could be differently regulated in hESC and testes
To specifically investigate E1.2 initiation (USP44-001), we tried to precisely define the position of TSS in the three samples containing this mRNA, that is, HUES24, H9, and testes. Even if our strategy could not produce robust quantitative data, we compared the E1.2 TSS profile in those samples to identify major initiation tendencies. To this aim, we first partitioned the first exon sequence upstream of the splicing donor site in 20 bp-long segments from the 3′ end (splicing donor site numbered as 0) to 5′, as depicted in Fig. 4A. We then counted the number of clones in each segment for every sample (Fig. 4B). The two hESC showed a similar profile occurring between −40 and −180 bp. Only one clone came from an initiation occurring between −220 and −240. In contrast, TSS distribution in testes showed two peaks: a first one at the same position as in hESC, and a second one further upstream, between −180 and −260 bp. Surprisingly, 75% of the clones in the −220 to −260 region did not display a splice from the previously described donor site (position 0), but from an alternative splicing site in position −195. This donor site was never detected in hESC samples. This new donor site actually splices with exon 2.3 (coding exon) acceptor site. Altogether, the data show that the promoter driving E1.2 transcription (USP44-001) is regulated differently in testes and in hESC.
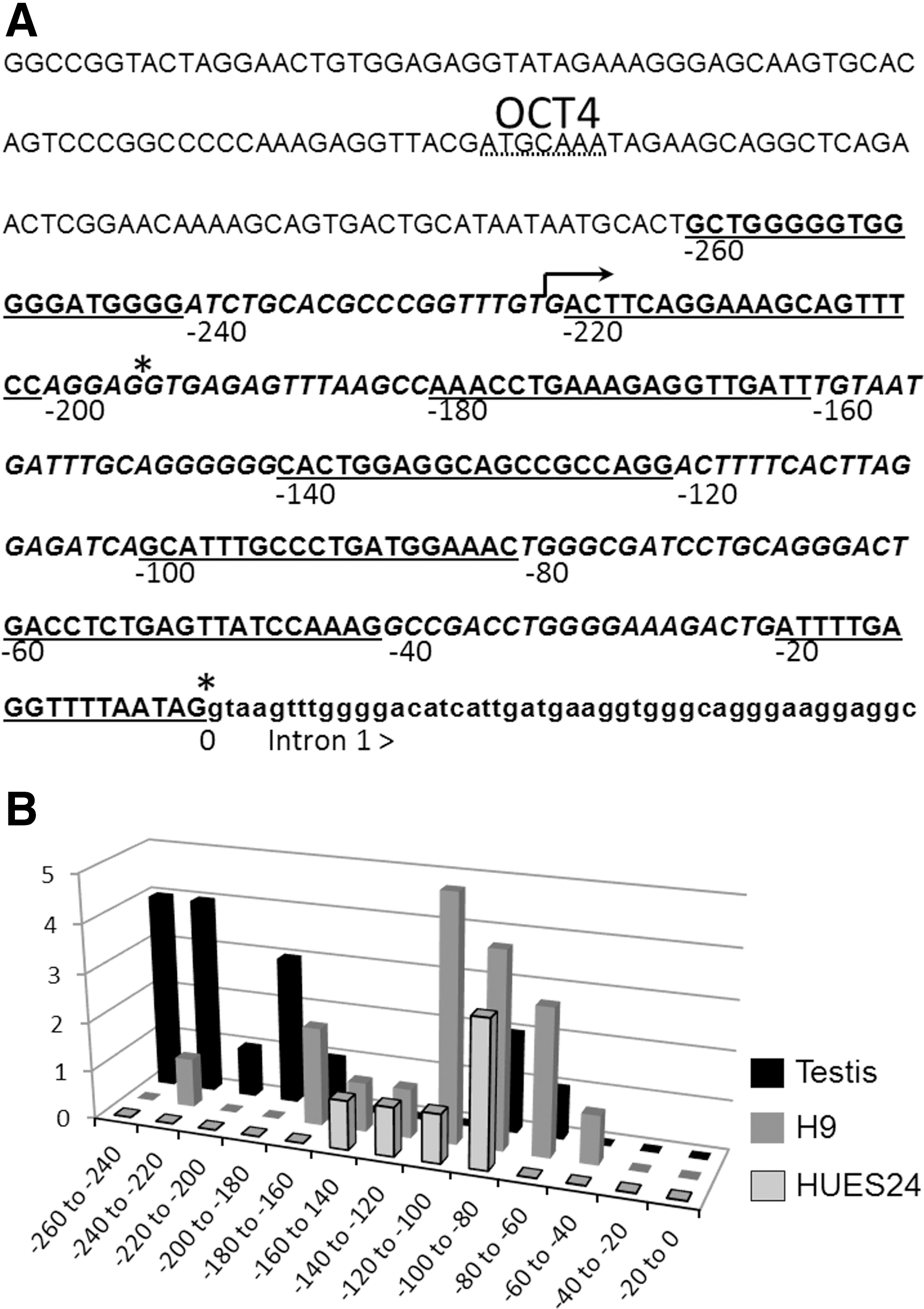
Close-up on E1.2 transcription start and splicing sites in hESC and testes.
A CGI methylation ratio correlates with expression level
Differences among tissues in transcription initiation and processing of a specific gene are often associated with variations in epigenetic marks such as DNA methylation. In eukaryotes, genomic DNA can be methylated on the C base of a CpG doublet grouped in CGI. We identified three CGI (Fig. 5A): CGI1 (100 bp long) is situated between E1.2 and E1.3; CGI2 (1,000 bp) covers E1.3 to E1.6 (partially); and CGI3 (300 bp) is situated between exons 5 and 6. We hypothesized that such CGIs may be involved in transcription regulation and/or promoter selection. As a first step to test this hypothesis, we addressed the methylation status of CGI1, CGI2, and CGI3 in genomic DNA extracted from USP44-expressing and -non-expressing tissues/cells, and also in sperm, by using the bisulfite sequencing method (Fig. 5B and Table 3). We designed primers allowing an analysis of the entire CGI1 and CGI3 sequences, and of a CGI2 core zone (unfortunately, amplification of the whole CGI2 zone remained unsuccessful despite our attempt). We found the latter to be almost 100% unmethylated in all samples. In contrast, CGI1 methylation varied from 1% to 50%. CGI3 analysis revealed a completely different situation with methylation ratios near 100% in all tissues, except in testes and sperm (71% and 46% methylation, respectively). The testis is a complex tissue that is composed of multiple cell types (germinal cells at different steps of maturation; Sertoli and Leydig cells; endothelial cells; … ) that may influence diversely the CGI3 methylation ratio. In contrast, the sperm is almost 100% composed of germinal cell-derived spermatozoids (with a very low fraction of macrophages, epithelial cells, and others). Our results intriguingly suggest that about half of the spermatozoid population may carry a fully methylated CGI3, whereas the other half may carry a fully unmethylated island.
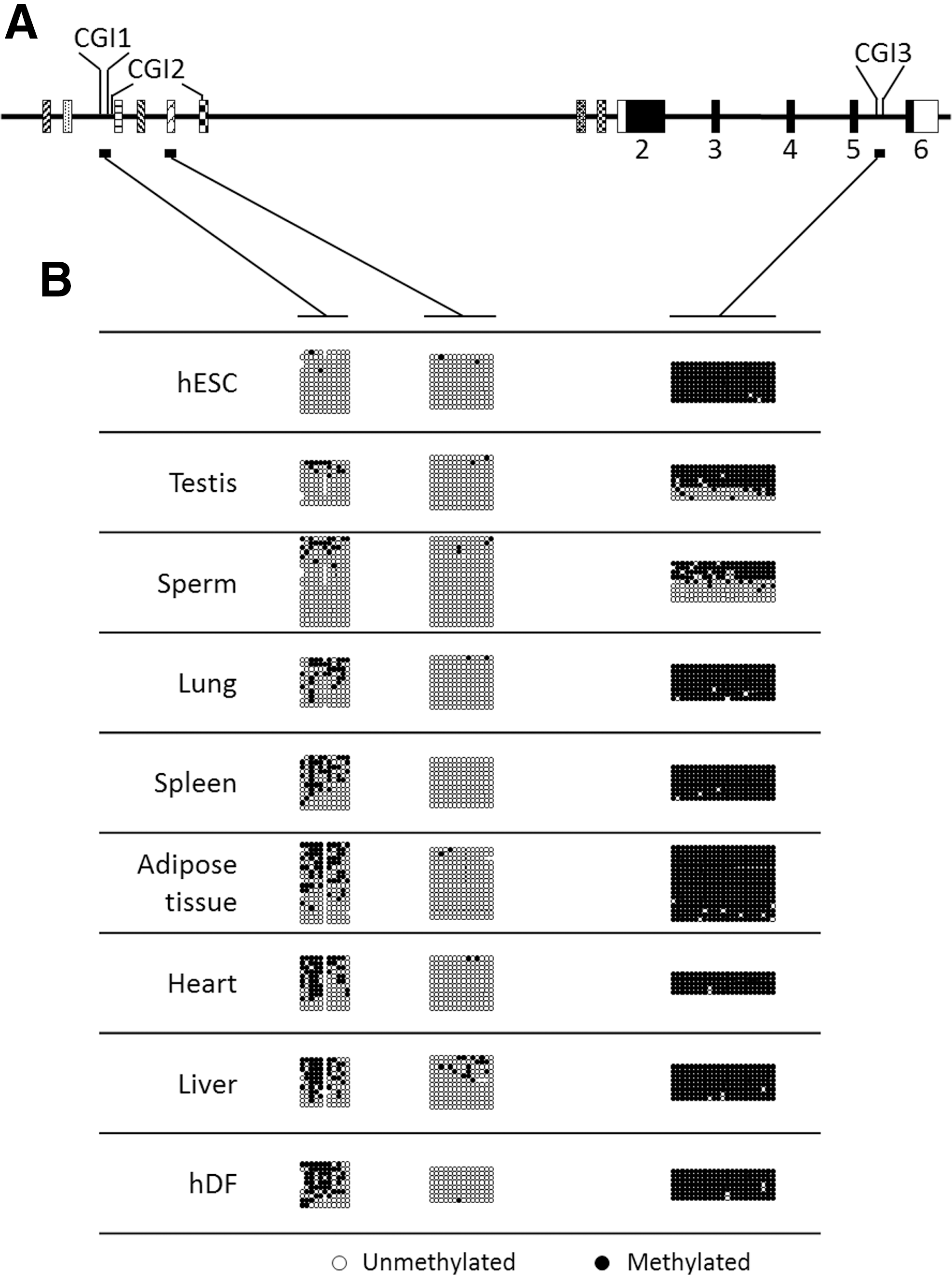
CGI methylation status in cells and tissues.
hDF, human dermal fibroblasts; hESC, human embryonic stem cells.
Another experiment aimed at correlating gDNA methylation and USP44 gene expression. The CGI2 core region and CGI3 were clearly not good candidates as they did not show any variation in sequence methylation. In contrast, CGI1 methylation displayed clear variations between tissues. To test USP44 expression versus DNA methylation, we plotted qPCR data previously obtained against the percentage of methylation in the same organs (Fig. 6). This approach clearly showed a strong correlation between the level of methylation of CGI1 and USP44 transcription. E1.2 transcriptional initiation seemed to be more tightly correlated with CGI1 methylation than initiation of the E1.4 exon.
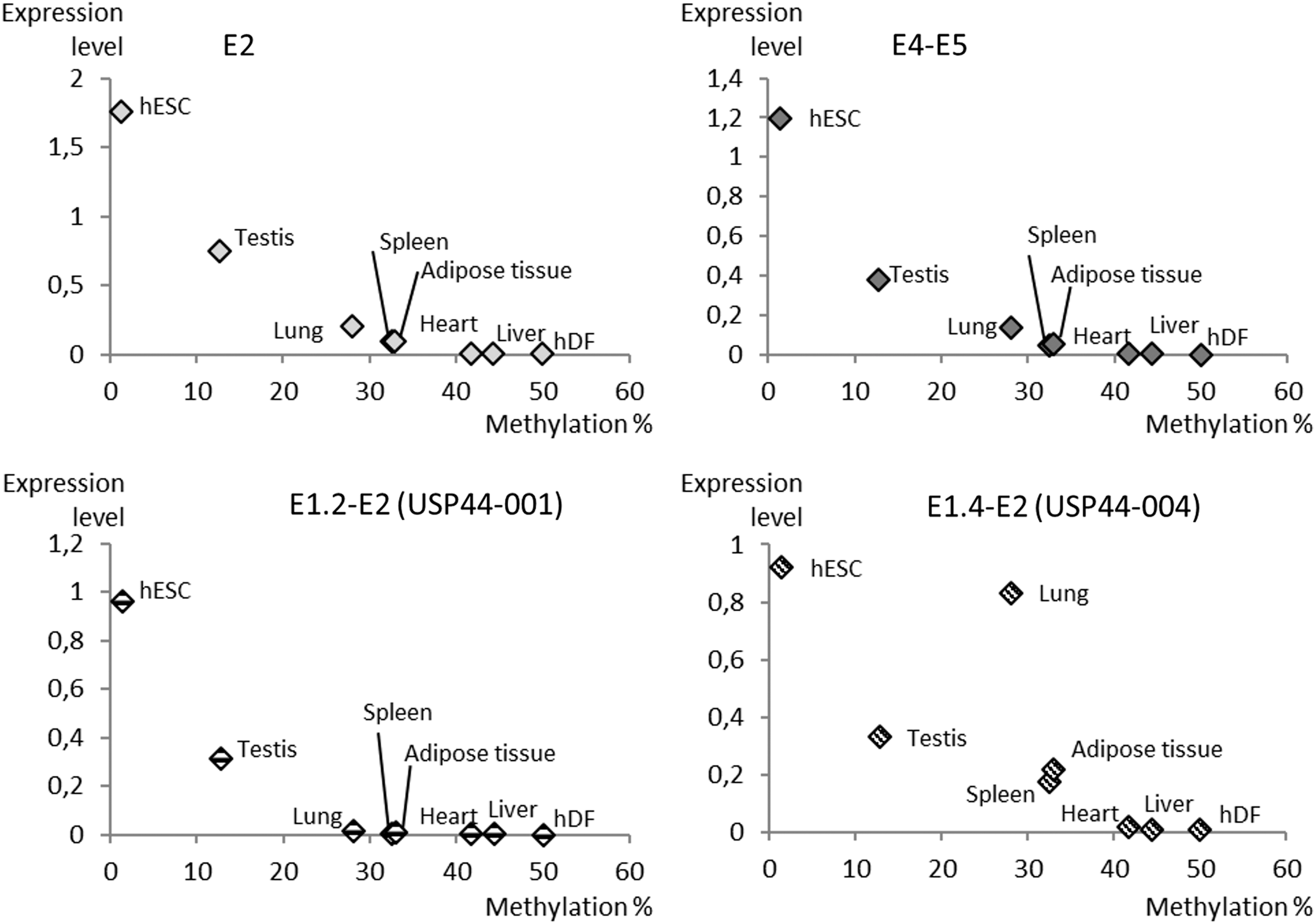
CGI1 methylation correlates with USP44 expression driven by E1.2 promoter. Expression qPCR data obtained for the four regions tested, that is, E2, E4–E5, E1.2–E2 (USP44-001), and E1.4–E2 (USP44-004) (please refer to Figs. 1 and 2), are plotted against methylation percentage of CGI1 in various cells and tissues (please refer to Fig. 5 and Table 3).
Discussion
To identify new genes involved in stem cell physiology, we adopted a two-step screening strategy. In a first step, we took advantage of available online databases to compare the transcription levels of DUB family members in human tissues and cells. Using two independent technologies, microarray and EST, the browsing of INSERM Amazonia! and NCBI EST databases allowed us to identify USP44 as fulfilling the expected criteria, that is, expression in PSCs but not in somatic tissues/cells. In-house RT-qPCR analyses confirmed USP44 expression at similar levels in both human ESCs and iPSCs regardless of the region of mRNA tested. USP44 expression dramatically fell under the detection threshold in differentiated hESC, comforting previously published data [26]. In human tissues, USP44 is mainly detected in testes, and it can also be detected in the ovary, thymus, lung, and thyroid. Adipose tissues and prostate also expressed low but detectable levels of USP44 mRNA.
A 5′ RACE approach in several USP44-expressing tissues made the picture far more complex than what it was earlier. At the time our work started, databases indexed three alternative first exons. We identified four more alternative first exons, showing that the gene is potentially under control of at least seven different promoters. Expression in PSCs and testes seems mainly due to the promoter controlling the transcription of the E1.2 exon (yielding the USP44-001 mRNA described in databases; Table 2). The stem cell-specific activity of this promoter may involve OCT4 (product of the gene POU5F1), which interacts with a DNA motif (ATGCAAA) situated 313 bp upstream of the E1.2 splicing site [27], 92 bp upstream of the “official” TSS (according to Ensembl, the TSS is 221 bp upstream of the donor splicing site). Statistically, almost 38% of OCT4 binding sites are located within 1 kb upstream of TSS [27], indicating that the motif detected in the USP44 promoter is a very good candidate. OCT4 interaction with the USP44 gene was confirmed by others [10,12,28] who also demonstrated that SOX2 and NANOG bind to the same region. The simultaneous interaction of these three key transcription factors may initiate the expression of USP44 in PSCs since knock-down of each factor in ESCs leads to downregulation of the gene [11,29]. The major part of USP44 gene activity in PSCs could actually be directly controlled by the stemness core regulatory network. Nevertheless, our attempt to activate the endogenous USP44 gene in human dermal fibroblasts with ectopically expressed POU5F1, SOX2, NANOG, and LIN28 genes remained unsuccessful (unpublished data), suggesting that other epigenetic mechanisms may control chromatin opening.
Previous investigations in colorectal cancer suggested that USP44 gene expression could be controlled by DNA methylation [30]. We, thus, investigated DNA methylation of the three CGIs identified in the UPS44 gene. CGI1 and CGI2 are spatially close to each other, separated by 115 bp. The sequences we could test by bisulfite sequencing were the entire CGI1 and CGI3 (containing 9.7% and 7.7% CpG site density, respectively), and a 106 bp segment in the central part of CGI2 (13% CpG density). Among the three regions tested, only CGI1 methylation was found to be tightly correlated to USP44 expression, more particularly with the activity initiating from the E1.2 promoter. It should be noted that CGI1 and CGI2 displayed distinct profiles while they were spatially close (the tested sequences were about 700 bp apart). A similar configuration was previously described as CpG “shores” (see Ref. [31] for review). The most dense CGIs located near gene promoters are generally unmethylated, whereas some low-density CGIs display transcription-correlated methylation profiles. Maunakea et al. [32] clearly showed that intragenic CGIs regulate gene expression in the brain through modulation of alternate promoters. In the USP44 context, CGI1 can be considered as either an internal CGI (regarding E1.1 and E1.2 positions) or an external CGI (regarding other promoters), introducing confusion in classifications and comparisons. The same authors as well as others [33] correlated lack of CGI methylation to H3K4me3 histone methylation, an epigenetic mark of promoters. With respect to the USP44 gene, the ENCODE database confirmed the presence of H3K4me3 marks in the same area. CGI1 displays a unique picture throughout the literature because of its evolving methylation status correlated to gene expression in adult tissues. Whether promoter activity impacts DNA methylation or DNA methylation determines transcription initiation remains an open issue. In the case of USP44, our inability to induce USP44 transcription in dermal fibroblasts after transduction of reprogramming factors (unpublished results) suggests that CGI1 methylation and/or another epigenetic switch may preclude inappropriate gene expression.
CGI2 shares the characteristics of unmethylated regions (UMRs) described elsewhere [33,34]. The latter authors described several consensus sequences that may direct UMRs demethylation in the early embryo. We identified 31 of those sequences (sometimes overlapping each other) concentrated in the CGI2 area (about 1,000 bp wide) and only 12 dispersed in the 45 kb wide region covering the whole USP44 gene, suggesting their involvement in the lack of methylation of the CGI2 core zone. In contrast to both CGI1 and CGI2, CGI3 displayed a 100% methylation profile in all somatic organs. The region analyzed in CGI3 spans two head-to-head Alu sequences (Short Interspesed Elements, short interspersed nuclear elements (SINEs)). Alu family members in intragenic regions are mainly methylated in somatic organs (see Ref. [35] for review). This methylation precludes PolIII-based transcription, and subsequent retrotranscription and insertional mutagenesis. In the embryo, the genome of primordial germ cells goes through a demethylation/methylation cycle. At birth, 50% of mouse SineB1 DNA sequences (derived from 7SL RNA, such as human Alu sequences) are methylated in male germ cells [36]. Our results shows that a similar situation may occur in human germ cells. After developmental methylation erasure in human primordial germ cells, Dnmt3 may methylate only one out of two alleles of the sequence we analyzed in the USP44 gene. Alternatively, CGI3 methylation may not be conserved through meiosis during spermatogenesis. Whatever the mechanisms, spermatozoa split into two populations: one carrying a fully methylated CGI3 allele, and a second carrying a fully unmethylated sequence. This dichotomic situation questions the function of intragenic Alu methylation in male germ cells and also how the original methylation profile we highlighted in USP44 gene impacts this gene and Alu expression in the very early embryo.
A previous publication suggested that G1-S transition is regulated in PSCs by a direct interaction of NANOG with key cell cycle factors [37]. NANOG and coworkers, that is, OCT4 and SOX2, may also control another cell cycle checkpoint, metaphase to anaphase transition, through the regulation of the USP44 gene. USP44 was, indeed, first characterized as preventing premature anaphase onset by deubiquitinating inhibitory factors of the Anaphase Promoting Complex [38]. This activity may explain why USP44-depleted cells displayed both a higher incidence of lagging chromosome during anaphase and a high level of aneuploidy. Others demonstrated that such an outcome may result from disorganization of USP44-centrin interaction, leading to centrosome positioning abnormalities [39,40]. In agreement with these findings, USP44 was found to be misregulated in various cancers [30,41,42]. In ESCs, USP44 was described as an N-CoR complex component counteracting H2B monoubiquitination by RNF20, to maintain chromatin in an open state favorable to the proper activation of genes along differentiation [43,44]. H2A is also a substrate for USP44, when monoubiquitinated by RNF8 and RNF168 in response to DNA double-strand breaks [45]. H2A monoubiquitination is important for gene repression in ESC [46], but the involvement of USP44 in the regulation of this precise function has not been addressed. USP44-mutant mice are prone to cancer but do not display any obvious abnormalities [40], a finding that may require further embryological investigations to be confirmed or rejected [47].
In summary, we identified USP44, encoding a DUB, as a highly expressed gene in ESCs, iPSCs, and testes, whereas a few other somatic organs expressed lower levels. This expression profile results from the activity of a complex network of seven promoters, two of them being specifically active in stem cells and testes. The promoters may be under the influence of epigenetic factors, as we clearly showed that the methylation status of a nearby CGI was strictly correlated to the level of expression of USP44. An Oct4-binding site and published results also strongly suggest that stem cell-specific initiation may be under the control of stemness factors. Even if few results are currently available on USP44 function in PSCs, our data indicate that this gene may be a central player in the genome field of PSCs.
Footnotes
Acknowledgments
The authors thank Dr. Michel Puceat for HUES24 and HUES24 EB RNA samples. This work was supported by a collaborative project called “Ship-In” and funded by the Fond Unique Interministériel (FUI), a subsidiary of the French Government.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.