
Abstract
Human embryonic stem cells (hESCs) represent a promising tool to study functions of genes during development, to model diseases, and to even develop therapies when combined with gene editing techniques such as CRISPR/CRISPR-associated protein-9 nuclease (Cas9) system. However, the process of disruption of gene expression by generation of null alleles is often inefficient and tedious. To circumvent these limitations, we developed a simple and efficient protocol to permanently downregulate expression of a gene of interest in hESCs using CRISPR/Cas9. We selected p53 for our proof of concept experiments. The methodology is based on series of hESC transfection, which leads to efficient downregulation of p53 expression even in polyclonal population (p53 Low cells), here proven by a loss of regulation of the expression of p53 target gene, microRNA miR-34a. We demonstrate that our approach achieves over 80% efficiency in generating hESC clonal sublines that do not express p53 protein. Importantly, we document by a set of functional experiments that such genetically modified hESCs do retain typical stem cells characteristics. In summary, we provide a simple and robust protocol to efficiently target expression of gene of interest in hESCs that can be useful for laboratories aiming to employ gene editing in their hESC applications/protocols.
Introduction
T
In this study, we describe an easy-to-use method of obtaining genetically modified hESCs using CRISPR/Cas9. Our data show that simple repeated transfection of undifferentiated hESCs leads to effective downregulation of expression of gene of interest (here p53) and very high efficiency (82%) of generation of knockout sublines. Our functional data further show that hESCs generated by this approach do retain their stem cell characteristics. We believe that our protocol can considerably simplify gene targeting in hESCs using CRISPR/Cas9 system.
Methods
Cell culture, cryopreservation, and differentiation
Undifferentiated hESC line CCTL 14 ([10]; and Human Pluripotent Stem Cell
Cryopreservation of cells was performed in the hESCs culture media supplemented with GIBCO® ES cell qualified fetal bovine serum [FBS; 30% (Thermo Fisher Scientific)] and dimethyl sulfoxide (DMSO; 10%). Cells were slowly frozen in ice-cold freezing media using Mr. Frosty™ Freezing Container (Thermo Fisher Scientific). During the thawing of hESCs, application of a selective Rho-associated kinase (ROCK) inhibitor Y-27632 (20 μM; Sigma-Aldrich) was used to increase survival of hESCs as described [12].
Differentiation of hESCs was induced by embryoid body (EB) formation in nonadherent cell culture conditions in MEF media. Briefly, hESCs from feeder-free cell culture were cut into small pieces using StemPro® EZPassage Disposable Stem Cell Passaging Tool (Thermo Fisher Scientific). These were then transferred to MEF culture medium (DMEM, 10% FBS, 1%
Transfection of cells with Neon® transfection system
Transfection experiments were carried out with plasmid px330-sgp53 (a generous gift from Oscar Fernandez-Capetillo, Karolinska Institute, Stockholm, Sweden), expressing both p53 gRNA (TGAAGCTCCCAGAATGCCAG) and Cas9 nuclease, using Neon transfection device (Thermo Fisher Scientific) according to manufacturer's instructions. Detailed experimental procedure is described in section “Results and Discussion”.
Western blotting
For western blotting, harvested cells were washed three times with phosphate-buffered saline (PBS), lysed in lysis buffer [50 mM Tris-HCl (pH 6.8), 1% sodium dodecyl sulfate (SDS), 10% glycerol], and stored at −80°C until use. The protein concentration was determined using the DC Protein Assay Kit (Bio-Rad) and lysates were supplemented with bromophenol blue (0.01%) and β-2 mercaptoethanol (1%). Equal amounts of total proteins were separated by SDS-polyacrylamide gel electrophoresis followed by transfer to a polyvinylidene fluoride membrane (Millipore). Proteins were immunodetected using the appropriate primary antibody followed by incubation with horseradish peroxidase-conjugated secondary antibody. Amersham ECL Prime western blotting Detection Reagent (GE Healthcare Life Sciences) was used to visualize antibody-antigen complexes. The following primary antibodies were used in western blot analysis: Oct-4 (sc-5279; Santa Cruz Biotechnology); GATA6 (No. 5851), Brachyury (No. 81694), Nanog (No. 3580), phospho γ-H2A.X (No. 9718), and β-actin (No. 3700; all from Cell Signaling); GDF15 (07-217; Millipore); α-tubulin (11-250-C100; Exbio); mouse monoclonal antibody against p53 (DO-1) and mouse monoclonal antibody against MDM2 (2A9) were generously provided by Bořivoj Vojtěšek (Masaryk Memorial Cancer Institute, Brno, Czech Republic) and Stjepan Uldrijan (Department of Biology, Faculty of Medicine, Masaryk University, Brno, Czech Republic), respectively. Alpha-tubulin or β-actin were used as loading controls.
RNA isolation and quantitative polymerase chain reaction
Total RNA was isolated using phenol-chloroform extraction. Harvested cells were washed with PBS and lysed in RNAblue reagent (Top-Bio) and immediately frozen and stored at −80°C. After thawing, 1-Bromo-3-chloropropan reagent (Sigma-Aldrich) was added to lysate in ratio 1:10 and solution was vortexed extensively and centrifuged for 15 min at 8,000 g at 4°C. Aqueous fraction containing RNA was transferred to a clean RNase free collecting tube and RNA was precipitated from the solution using 100% isopropanol for 20 min at −20°C. Precipitate was washed by 75% ethanol and eventually diluted in RNase free water. Concentration of RNA was measured using Nanodrop-1000 (Thermo Fisher Scientific) and stored at −80°C until use.
For the detection of microRNA expression, TaqMan microRNA Gene Expression Assays specific for selected mature microRNAs (hsa-miR-34a, hsa-mir-302a) were used according to the manufacturers' protocols. Synthesis of cDNA for quantification of microRNAs was performed using the TaqMan MicroRNA Reverse Transcription Kit and PCR was performed using TaqMan® Fast Universal PCR Master Mix 2× (all reagents from Thermo Fisher Scientific). All samples were analyzed in triplicates, DNA amplification was detected using Roche Light Cycler 480 (Roche), and data were analyzed using LC480 software (Roche). The relative gene expression was calculated by normalization to small nucleolar RNA RNU38B expression.
Fluorescence-activated cell sorting sorting
Cells growing on Matrigel were harvested by TrypLE Select (Thermo Fisher Scientific), washed with PBS, and resuspended in sterile fluorescence-activated cell sorting buffer containing PBS, 2% FBS, and 1% ethylenediaminetetraaceticacid. Cells were then pipetted through 40 μm Corning Falcon™ Cell Strainer (Thermo Fisher Scientific) and sorted using BD FACSAria™ cell sorter (BD Biosciences) as single cells per well into 96-well plate with fresh CM media supplemented with ROCK inhibitor (20 μM) to improve cell survival [12].
Indirect immunofluorescence
For immunodetection of Oct4 and Nanog proteins, cells were fixed in 4% paraformaldehyde (PFA) for 20 min on ice, washed 1× in PBS, permeabilized for 20 min with 0.1% Triton-X, blocked for 1 h with 1% bovine serum albumin (BSA) in PBS (pH 7.4) containing 0.05% Tween-20, and incubated overnight at 4°C with primary antibodies against Oct-4 (sc-5279; Santa Cruz Biotechnology), and Nanog (No. 3580; Cell Signaling). Final incubations with secondary antibodies were carried out for 1 h at room temperature. Cell nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich), and cells were mounted in Mowiol mounting medium (Sigma-Aldrich). Microscopy was performed using an Olympus FluoView 500 laser scanning microscope (Olympus C&S Ltd., Prague, Czech Republic).
DNA damage induction
For DNA damage induction, Etoposide diluted in DMSO was added to cell culture media for 8 h at the final concentration of 3.4 μM. Subsequently, all cells (including floating ones) were harvested and analyzed by western blotting as described above. DMSO in dilution of 1:10,000 was used as vehicle (negative) control.
Histology of EBs
EBs were fixed in 4% PFA for 20 min, washed in PBS, and gently inserted into 3% ultra-low melting agarose (Sigma-Aldrich). Agarose containing EBs was embedded into paraffin, sectioned paraffin blocks were placed on Superfrost plus slides (Thermo Fisher Scientific), rehydrated, and deparaffinized (Xylen-Ethanol). Antigen retrieval was performed in Target Retrieval Solution at pH 9 (Dako S2367; Agilent Technologies) at 92.5°C for 20 min. Sections were permeabilized in PBS with 0.025% Triton-X and incubated with primary antibodies diluted in Tris-buffered saline (TBS) with 1% BSA at 4°C overnight. The day after, the sections were washed in TBS, shortly washed in PBS, and incubated with secondary antibody diluted in TBS with 1% BSA for 1 h at 4°C. Following incubation, sections were extensively washed with PBS, cell nuclei were counterstained with DAPI, and slides were mounted in Mowiol mounting medium. The following primary antibodies were used: GATA6 (No. 5851), Brachyury (No. 81694), Sox2 (No. 4900; all from Cell Signaling), and TUJ (kindly provided by Peter Andrews, University of Sheffield, United Kingdom).
Teratoma formation assay
The ability to differentiate in vivo was assessed in two clones of p53 knockout (KO) hESC lines (clones No. A9 and No. A11). MOCK hESCs were used as a control. An inoculum of uniform clumps of undifferentiated hESCs (1 × 106 cells in 100 μL culture medium) was injected into the hind limb muscle of 8-week-old NOD/SCID/IL2Rg-null mice. When tumor diameter reached ∼1 cm, the mice were euthanatized by cervical dislocation. The tumors were dissected from the surrounding tissue, fixed in 4% PFA, embedded in paraffin, sectioned, and stained with hematoxylin and eosin to visualize the tissue composition. All animal studies were carried out according to approved ethical guidelines (Project No. 02/2010).
Statistical analysis
All statistical analyses were performed using two-tailed paired t-test and graphically visualized using GraphPad Prism Software v. 5.0 (GraphPad Software, Inc., La Jolla, CA;
Results and Discussion
Repeated transfection of hESCs using CRISPR/Cas9 system efficiently generates mixed population of cells with low expression of p53 protein while maintaining pluripotency
To rapidly and efficiently generate hESCs with disrupted expression of gene of interest (here p53), we used CRISPR/Cas9 construct expressing both Cas9 nuclease and gRNA (px330-sgp53). We initially performed transfections of hESCs using either Neon Transfection System (Neon TS) or Lipofectamine 3000 (L3000). However, transfection using L3000 proved to be inefficient (data not shown); therefore, we only describe the procedure that employs Neon TS.
As schematized in Fig. 1A, undifferentiated hESCs were grown in feeder-free conditions on Matrigel-coated culture dish. At the day of transfection, cells were enzymatically harvested, washed once with PBS, and resuspended in R-buffer at the concentration 10,000 cells/μL. A total number of 100,000 cells (volume of 10 μL) was mixed with 0.5 μg of px330-sgp53 construct and incubated for 10 min at room temperature. In case of MOCK transfection, equal volume of vehicle was used. Electroporation was performed in the “E” electroporation buffer using 10 μL Neon tip with one pulse set to the voltage of 1,100 V and 30 ms width (Neon Optimization Program No. 6). Subsequently, cells were transferred to a new Matrigel-coated dish with fresh cell culture media. The entire above described procedure was then repeated in 3–4 day intervals before cells reached the confluency. Each time, only 100,000 cells were used for electroporation. The remaining cells were harvested for protein analyses. As shown in Fig. 1B, our strategy resulted in notable downregulation of p53 protein already after two consecutive Neon transfections with the px330-sgp53 plasmid. The knockdown efficiency was further improved after additional rounds of transfection (up to five consecutive transfections performed).
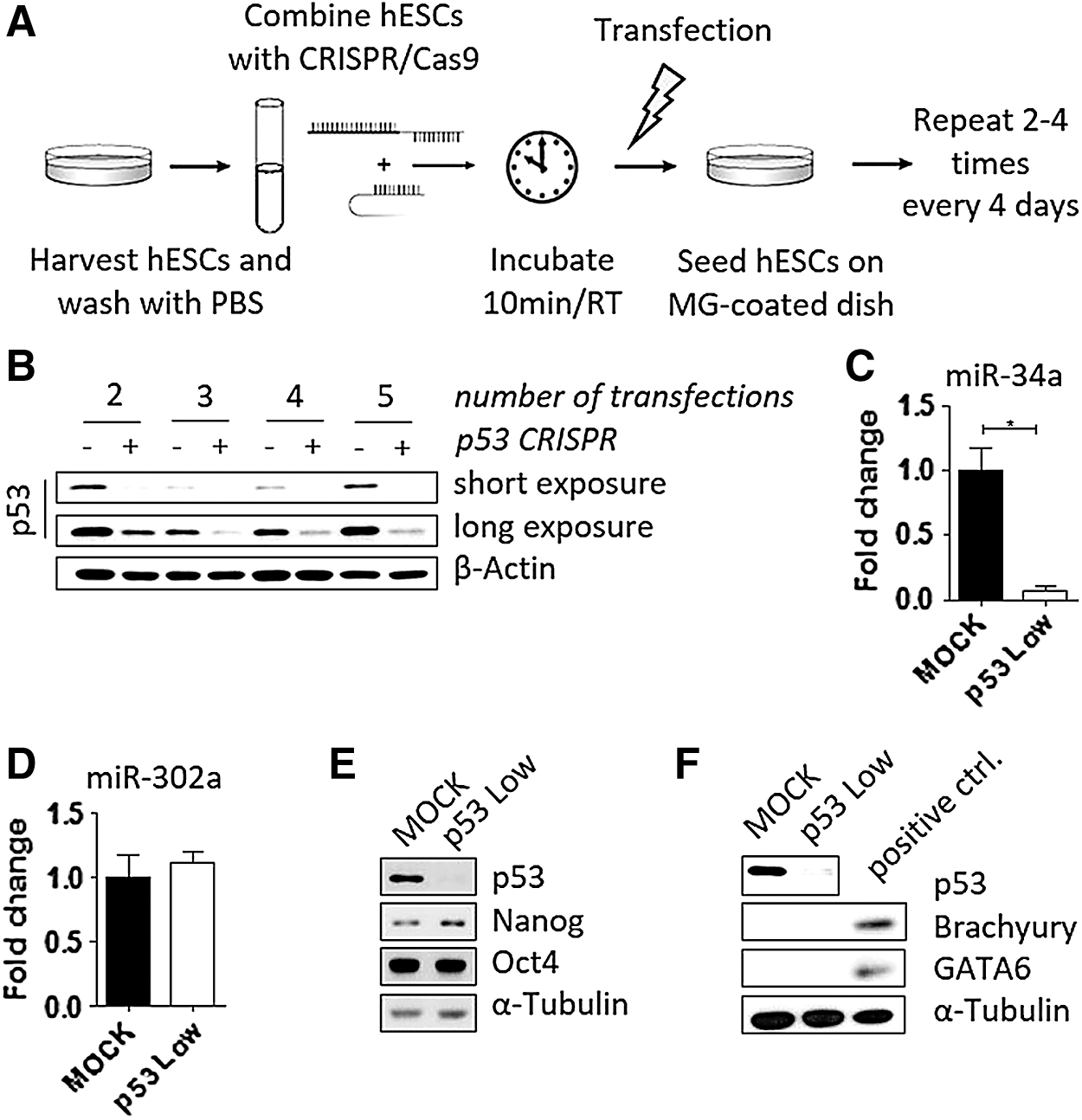
Repeated transfection of hESCs with p53-CRISPR/Cas9 shows efficient downregulation of p53 protein while maintaining pluripotency.
To assess both the efficiency and possible off-target activities of our protocol, following the fifth round of transfections the cells were passaged, approximately half of the total cell mass was harvested for analyses, while the rest was cryopreserved. Importantly, the expression of microRNA miR-34a, a direct transcriptional target of p53 protein [13], was significantly downregulated in those cells (Fig. 1C), thus providing functional evidence that a process, normally controlled by p53, becomes abrogated in hESCs transfected by px330-sgp53 plasmid. These cells are further referred to as “p53 Low” hESCs.
CRISPR/Cas9 system typically offers higher specificity (less off-targets) over downregulation of gene expression using RNAi strategies [14,15]. Nonetheless, prolonged expression of Cas9 nuclease might be associated with increased rate of off-target effects [4,8]. Therefore, we aimed to verify that hESC sublines, produced here by our approach of repetitive transfections, remain pluripotent and do not undergo spontaneous differentiation. To this end, we assayed these cells for the presence and absence of markers of pluripotency (Oct4, Nanog, and microRNA miR-302a; Fig. 1D, E) and differentiation (here represented by Brachyury and GATA6; Fig. 1F), respectively, by quantitative polymerase chain reaction and western blotting. We found that in p53 Low cells the levels of all markers tested are about the same as in controls, thus verifying the undifferentiated phenotype of hESCs with downregulated p53 protein.
Given our experimental design, the resulting population retained some expression of p53 protein (Fig. 1B). Most likely, this is due to the presence of small subpopulation of p53 heterozygotes [16]. Therefore, we decided to isolate individual clones of hESCs lacking expression of p53 (p53 knockout–p53 KO) to assess the efficacy of p53 targeting at single cell level.
Efficient clonal selection of p53 KO hESCs that retain their stemness
To select individual p53 KO clones from p53 Low hESC population, live cells were sorted to 96-well plates using standard tabletop cell sorter (Fig. 2A). Survival rate of hESCs at single cell level was around 20%, therefore, within the range of clonal survival typically seen in such experiments [9]. Surviving clones (total number of 33) were subsequently expanded, passaged, and approximately half of the total cell mass was harvested for protein analyses, while the rest was cryopreserved. Remarkably, out of the total number of clones analyzed, only six showed some minimal expression of p53 protein. The remaining 27 clones (82%) did not express any p53 protein as an evidence of efficient gene targeting and hence were considered p53 KO (Fig. 2B, C).

Efficient clonal selection of p53KO hESCs and evaluation of their stemness.
Three individual p53 KO clones (numbered A9, A10, and A11 in Fig. 2B) were characterized further. Under the standard culture conditions on MEFs, both MOCK and p53 KO hESCs retained morphology typical for undifferentiated hESCs-round smooth colonies with distinct edges (Fig. 2D, clone No. A11, data for clones No. A9 and No. A10 not shown). Also, both MOCK and p53 KO hESCs contained the same amounts of Oct4 and Nanog as demonstrated by indirect immunofluorescence. To functionally verify the absence of p53 protein in p53KO hESCs, we have induced DNA damage to both MOCK and p53 KO hESCs by treatment with Etoposide for 8 h. Results show that while the MOCK hESCs stabilized p53 protein and induced the expression of MDM2 and GDF15 proteins upon DNA damage, p53 KO hESCs failed to do so and the level of MDM2 and GDF15 proteins remained undistinguishable from the untreated cells (Fig. 2E; MOCK and clone No. A11, data for clones No. A9 and No. A10 not shown). Importantly, the level of phospho-γH2AX, a hallmark of DNA damage induction, was increased in similar manner in both the MOCK and p53 hESCs after etoposide treatment.
Finally, to assess their overall differentiation propensity both in vitro and in vivo, we induced the p53 KO hESCs to differentiate toward EBs and injected them to NOD-SCID mice and evaluated their ability to form teratomas.. As shown in Fig. 2F, both MOCK and p53 KO hESCs formed EBs of comparable size, frequently with typical cavity inside. After 40 days of differentiation we also examined expression of differentiation markers in EBs derived from p53 KO cells. As determined by indirect immunofluorescence, EBs contained ectoderm-specific TUJ and SOX2, mesoderm-specific Brachyury, and endoderm-specific GATA6. Teratomas comprising all three germ layers developed in vivo from both MOCK hESCs and two p53 KO clones analyzed (Fig. 2G; clone No. A11, data for MOCK and clone No. A9 not shown), further documenting uncompromised differentiation potential of hESCs derived by our protocol.
Conclusion
Altogether, our data show that repeated transfection of undifferentiated hESCs using Neon TS leads to dramatic downregulation of targeted protein (here p53) and provides an effective approach for establishment of p53 KO clones. We believe that our methodology represents an easy-to-use technique to obtain genetically modified hESCs, when the goal is to permanently disrupt expression of gene of choice. Given the increasing number of reports successfully and reproducibly utilizing CRISPR/Cas9 technology in high throughput screens [16 –19], without reporting significant differences in targeting efficiency for individual loci, we are confident that our protocol can be easily adapted for targeting of any gene of interest. Even though we cannot formally exclude that prolonged expression of Cas9 nuclease might have led to additional cuts in genomic DNA, in addition to the targeted p53 locus, our functional data clearly show that hESCs generated by this approach do retain the key stem cell features.
Footnotes
Acknowledgments
Authors would like to acknowledge Oscar Fernandez-Capetillo, Peter Andrews, Lumír Krejčí, Stjepan Uldrijan, Bořivoj Vojtěšek, and Karel Souček for sharing various reagents, Hana Kotasova for assistance with cell sorting, Milan Esner for assistance with microscopy, and Ondrej Bernatik, Michaela Klouckova, Klara Koudelkova, Katerina Mareckova, Veronika Sedlakova, and Katerina Vasickova for technical support. This work was supported by funds from the Faculty of Medicine MU to junior researcher D.B. and L.C. (ROZV/24/LF/2016, ROZV/25/LF/2017), funds from Masaryk University to A.H. (Tissue Engineering 7; MUNI/A/1369/2016) and M.B. (MUNI/C/1709/2016), grants from Czech Science Foundation to D.B. (15-18316Y) and L.C. (16-03269Y), Follow-up research fund from Federation of European Biochemical Societies (FEBS), and grant from Swiss National Science Foundation (IZ11Z0_166533) to L.C.
Author Disclosure Statement
No competing financial interests exist.