
Abstract
Cardiac progenitor cells (CPCs) are being developed as a promising treatment for heart failure. Although clinical trials have predominantly used donor cardiac biopsies to derive CPCs, a better solution could be to use previously cryopreserved human heart tissue. This would enable timely and convenient access to healthy and young heart samples for CPC production. However, few studies have attempted to isolate CPCs from previously cryopreserved heart tissue. In this study, we isolated CPCs from eight nondiseased human heart samples previously cryopreserved as part of the Sydney Heart Bank. Resulting cells were strongly positive for known fibroblast (DDR2, Vimentin), mesenchymal/CPC (PDGFRα, CD90) markers, and for pluripotency genes (SOX2, NANOG, MYC, KLF4), whereas being negative for the pan-hematopoietic marker (CD45). Outgrowth cells from aged hearts had decreased proliferative and self-renewing capacity that correlated with shorter telomere lengths compared with cells from young hearts. No telomerase activity was detected in any cells isolated. Colony-forming assays and fluorescence-activated cell sorting were used to enrich PDGFRα+/CD90+/CD31− CPCs. Multipotent potential was confirmed using in vitro differentiation assays with smooth muscle (MYH11+), endothelial cell (vWF+), and cardiomyocyte-like (cTnT+, α-actinin+) cell formation. Single cell assays demonstrated clonogenicity of PDGFRα+ CPCs with maintenance of prolonged self-renewing capacity (>2 months), and pluripotency gene expression at both early and late culture passages. Our results demonstrate that multipotent PDGFRα+ CPCs can be harvested and expanded from previously banked cryopreserved human heart samples. These data support cardiac tissue banking as a strategy for improved access to CPCs for future clinical therapies.
Introduction
T
We have recently reported a platelet-derived growth factor receptor alpha (PDGFRα)-expressing cardiac progenitor cell population (PDGFRα+ CPCs) in both rodent [11] and human [7] hearts. These PDGFRα+ CPCs are similar to mesenchymal stem cells (MSCs) derived from adult bone marrow (BM), but are isolated from the adult heart. They originate from the proepicardium during development and exhibit distinctive properties determined by their cardiac origin [11,12]. In contrast to BM-MSCs, PDGFRα+ CPCs highly express cardiovascular markers, thus differentiating into cardiovascular lineages better than BM-MSCs [11 –13]. They share similar properties of BM-MSCs, including adherence to tissue culture plastic and the expression of mesenchymal markers, such as CD90, but not hematopoietic markers, CD45 or CD34 [14].
CPCs can be derived from freshly isolated cardiac biopsies [7,8,10], obtained during interventional procedures on patients with cardiac disorders. However, CPCs obtained from these tissues may not be optimal for cardiac cell-based therapy because pre-existing cardiac disease is often found in these donors. Furthermore, generating the high numbers of CPCs required for cell therapy requires time-consuming isolation, expansion, and characterization. This may not be possible immediately before cell transplantation due to clinical urgency. As an alternative, recent clinical trials (ALLSTAR NCT02293603, DYNAMIC NCT01458405) have used allogeneic cardiac-derived cells. However, allogeneic donor tissue still requires invasive and potentially harmful procedures for tissue collection. To ethically justify these procedures, the donor would usually be already undergoing such a procedure for clinical treatment or diagnosis. Therefore, these hearts will also often have underlying cardiac pathology.
A preferable source of tissue for cardiac regenerative cell therapy is nondiseased human cardiac tissue that is collected rapidly, snap frozen, and maintained at −196°C until required. Such samples can be obtained from consenting donors whose hearts may not be suitable for cadaveric whole heart transplantation for other reasons and one donor heart will be sufficient to generate enough cells for many different allogeneic cardiac cell treatments. In this light, we sought to isolate and characterize PDGFRα+ MSC-like CPCs derived from heart tissue cryopreserved for up to 27 years. This sets the stage for the formation of cardiac tissue banks to serve as a ready source for “off-the-shelf” CPCs.
Materials and Methods
Donor tissue samples
Human left ventricular (LV) tissue samples from donor hearts were provided by the Sydney Heart Bank (SHB). The SHB, established in 1989, is a substantial Bank of tissues from all chambers and parts of the heart. The Bank comprises about 150 healthy donor hearts and three times as many failing human hearts [15]. This tissue was collected soon after a transplant coordinator declared the donor to be brain dead. Donor hearts were perfused with ice-cold cardioplegic solution and LV transmural tissue samples were rapidly sampled, frozen, and stored in liquid nitrogen (−196°C) [15,16]. Cause of death and clinical characteristics of the donor are summarized in Table 1. Hearts were selected only when the donor was declared brain dead by the transplant coordinator, but did not involve cardiac arrest. The collection process of human cardiac tissue was approved by the University of Sydney Human Research and Ethics Committee, and the conditions required that the donors be deidentified and that their associated clinical data be stored securely. Freshly isolated samples were collected from patients at the time of cardiothoracic surgical procedures. This study was approved by the Western Sydney Local Health District Human Research Ethics Committee. All patients provided written informed consent for the collection of samples.
Isolation and culture of CPCs were achieved for eight donor left ventricle samples. Echocardiography result was reported to be normal for all donors. None of these donors experienced cardiac arrest before removal for transplantation.
LV, left ventricular; CPC, cardiac progenitor cell; MVA, motor vehicle accident; SIDS, sudden infant death syndrome.
Isolation and culture of cryopreserved human heart-derived cardiac cells
Cell isolation
Explant-derived CPCs were isolated from fresh and frozen human hearts (Tables 1 and 2) as previously described [10] with minor modifications. Some of the frozen cardiac samples were stored for periods of 19–27 years and others for 3–5 years (Table 1). CPCs were isolated from frozen cardiac samples (∼100–200 mg) by explant culture. Samples were removed from liquid nitrogen and quickly thawed by washing twice with ice cold DPBS solution (Lonza). The tissue was then minced into small segments (diameter <1 mm) (Fig. 1A). The tissue pieces were placed in six-well plates coated with 0.1% (wt/vol) gelatin. All explants were cultured in MEMα (Sigma-Aldrich) supplemented with 20% FBS (Sigma-Aldrich), 2 mM

Isolation of explant-derived cells from cryopreserved LV samples.
Samples were collected from patients at the time of cardiothoracic surgical procedures and divided into two sections for fresh cell isolation and tissue cryopreservation. Isolation and culture of CPCs was achieved for four fresh and four cryopreserved samples.
Cell culture
Outgrowth of CPCs was enriched by subculturing for an additional two passages in MEMα supplemented with 20% FBS, 2 mM
Karyotype analysis
Karyotype of outgrowth and purified PDGFRα+/CD90+/CD31− cells was analyzed by G-banding by Trypsin using Giemsa (GTG-banding). The cells were harvested, fixed, and karyotyped at metaphases using a chromosome imaging analysis system.
Immunofluorescence staining of outgrowth cells
The cells were seeded onto gelatin-coated glass coverslips in 24-well plates. After 2 days in culture, the cells were washed twice with PBS and then fixed with 4% (wt/vol) paraformaldehyde (PFA) for 15 min. After permeabilizing with 0.3% Triton X-100 and blocking with 10% goat serum (Sigma-Aldrich), the cells were incubated with primary antibodies: vimentin (Abcam; 1:200), DDR2 (Abcam; 1:200), PDGFRα (Abcam; 1:1000), SMA (Dako; 1:500), CD90 (BioLegend; 1:200), Nkx2.5 (Abcam; 1:1000), CD45 (Abcam; 1:200), and cTnT (DSHB; 1:800) for 1 h at room temperature. The cells were then stained with appropriate fluorochrome-conjugated secondary antibodies for 1 h. Nuclei were stained with DAPI (1 μg/mL, Sigma-Aldrich). Slides were analyzed using an Olympus FV 1000 Confocal Laser Scanning microscope with FV10-ASW 1.7c software (Olympus, Japan). The positively stained cells were counted under 200-fold magnification. Ten fields per sample were randomly selected for quantitative analysis.
Bromodeoxyuridine (BrdU; Sigma-Aldrich) was used to measure cell proliferation. After the cells were incubated with 20 μM BrdU for 2 h, immunofluorescence was performed using anti-BrdU antibody (BioLegend; 1:300), as described above.
Flow cytometry analysis and fluorescence-activated cell sorting of outgrowth cells
Cells were harvested by trypsinization (0.1% trypsin and 0.04% EDTA; Sigma-Aldrich), washed in PBS, and stained with anti-PDGFRα-APC (R&D; 1:10), anti-c-Kit-PE (BD Biosciences; 1:20), anti-CD31-PE (R&D; 1:20), anti-CD90-FITC (BioLegend; 1:20), anti-PDGFRβ-PE (BioLegend; 1:20), and anti-CD34-AF488 (BioLegend; 1:20) for 60 min in the dark. For pluripotency proteins, permeabilized cells were stained with KLF4 (Cell Signaling; 1:200), MYC (Cell Signaling; 1:1,000) and NANOG (Cell Signaling; 1:200) antibodies for 1 h followed by secondary antibody (Invitrogen; AF488, 1:500). Isotype controls were performed concurrently. The fluorescence intensity of the cells was analyzed by flow cytometry (BD FACSCanto II; BD Biosciences). Cells were sorted for four populations (1) PDGFRα+/CD90+/CD31−, (2) PDGFRα+/CD90−/CD31−, (3) PDGFRα−/CD90+/CD31−, and (4) PDGFRα−/CD90−/CD31− using Influx machine (BD Biosciences). To examine the effect of c-Kit, cells were sorted for (1) PDGFRα+/CD90+/CD31−/c-Kit+, (2) PDGFRα+/CD90+/CD31−/c-Kit−, (3) PDGFRα−/CD90+/CD31−/c-Kit+, and (4) PDGFRα−/CD90+/CD31−/c-Kit−.
Clonogenicity assays
Single cells at passage 2 (P2) and passage 12 (P12) were sorted and plated into 96-well plates using a fluorescence-activated cell sorting (FACS) influx machine (BD Biosciences). Two 96-well plates were used for each sample (N = 3–4 patient samples). Single cells were cultured in complete MEMα containing 20% FBS and monitored every day for cell growth. On day 10, the number of clones generated in each plate was counted and the percentage of single cells plated per plate that have generated clonal colonies was calculated. After quantification of clonogenic efficiency, colonies were trypsinized and expanded for subsequent clonal analysis experiments.
Quantitative real-time polymerase chain reaction analysis
Total RNA was extracted using the Isolate II RNA Mini Kit (Bioline) according to the manufacturer's protocol. cDNA was synthesized from 1 μg RNA using Reverse Transcription System with random primers (Promega) following the manufacturer's instructions. mRNA expression of specific genes was carried out using SensiFAST SYBR No-ROX Mix (Bioline) using gene-specific primers as follows: PDGFRα (F: 5′-AACCGTGTATAAGTCAGGGGA-3′, R: 5′-ATTTCTTCCAGCATTGTGAT-3′), CD90 (F: CGGAAGACCCCAGTCCA-3′, R: 5′-ACGAAGGCTCTGGTCCACTA-3′), SOX4 (F: 5′-TCGCTGTCGGGTCTCTAGTT-3′, R: 5′-AATGTATGTTTCCCCCTCCC-3′), MYC (F: 5′-TTTCGGGTAGTGGAAAACCA-3′, R: 5′-CACCGAGTCGTAGTCGAGGT-3′), NANOG (F: 5′-GATTTGTGGGCCTGAAGAAA-3′, R: 5′-CAGGGCTGTCCTGAATAAGC-3′), KLF4 (F: 5′-GAGTTCCCATCTCAAGGCAC-3′, R: 5′-CCCCGTGTGTTTACGGTAGT-3′), c-KIT (F: 5′-GGGATTTTCTCTGCGTTCTG-3′, R: 5′-GATGGATGGATGGTGGAGAC-3′), and GAPDH (housekeeping gene, F: 5′-ACCCACTCCTCCACCTTTG-3′, R: 5′-CTCTTGTGCTCTTGCTGGG-3′). Relative gene expression was calculated using the 2−ΔΔCt method, which normalized against housekeeping gene.
Western blot
Protein extraction, SDS-PAGE, electrophoretic transfer of proteins to nitrocellulose membrane, and immunoblotting were performed as previously described [17]. The primary antibodies for Klf4 (1:1,000), Myc (1:1,000), and Nanog (1:2,000) (Cell Signaling) were used. Bound antibodies were detected with IRDye 800CW-conjugated secondary antibody (1:5,000; LICOR Biosciences). Membranes were scanned using an Odyssey Infrared Imaging system (LICOR Biosciences).
In vitro vascular cell and myocyte differentiation assays
To determine the differentiation potential, 2,000 cells were seeded onto gelatin-coated glass coverslips in 24-well plates and cultured in complete MEMα media containing 20% FBS. At 70%–80% confluency, the basal medium was removed and specific differentiation medium (detailed below) was added for 14 days with changes every 3–4 days. Cells were fixed after 14 days of differentiation. Immunofluorescence staining was performed as described above. Samples were analyzed using an Olympus FV 1000 Confocal Laser Scanning microscope with FV10-ASW 1.7c software (Olympus, Japan). Ten fields per sample were randomly selected for quantification.
CM differentiation with neonatal rat ventricular cardiomyocytes
Neonatal rat ventricular myocytes (NRVMs) were isolated from 2- to 3-day-old neonatal Sprague Dawley rats. Hearts were digested in trypsin (Sigma-Aldrich; 1 mg/mL) overnight at 4°C followed by digestion in collagenase (Worthington Biochemical; 1 mg/mL) for 45 min at 37°C. Cells were preplated in M199 medium (Invitrogen) containing 10% FBS to remove fibroblasts. Nonadherent cells were then plated in 24-well plates and cultured overnight at 37°C. For coculture, explant-derived CPCs were transduced with lentivirus encoding enhanced green fluorescent protein (eGFP) to allow identification of human cells. They were then plated in the 24-well plates that contain NRVMs. Cocultures were maintained with M199 medium containing 2% FBS. Media were changed every 2–3 days and cells fixed after 14 days of differentiation. They were then stained for cTnT (DSHB; 1:800) and α-actinin (Sigma-Aldrich; 1:800).
EC differentiation
To assess the endothelial differentiation potential, cells were cultured in IMDM (Invitrogen) supplemented with 10 ng/mL VEGF (R&D Systems), 10 ng/mL bFGF and 2% FBS for 14 days [7]. Medium was changed every 2–3 days. The cells were then stained for von Willebrand Factor (vWF, Dako; 1:1,000) and examined under the fluorescence microscope.
Smooth muscle cell differentiation
Cells were cultured in DMEM-HG supplemented with 50 ng/mL PDGF-BB (R&D Systems) and 2% FBS for 14 days [7]. The cells were then stained for smooth muscle myosin heavy chain 11 (MYH11, Abcam; 1:300).
Colony-forming unit fibroblast assay
Five hundred cells were plated into six-well plates (triplicates for each sample). The cells were cultured in completed MEMα containing 20% FBS for 12 days. The colonies were then fixed with 4% (wt/vol) PFA and stained with 0.05% (wt/vol) Crystal Violet. The number of colonies was quantified according to their size: large (>2 mm), small (<2 mm), and micro (∼25–50 cells) colonies [11].
Long-term growth studies
Long-term growth was performed as previously described [11]. Outgrowth of CPCs was expanded in bulk culture after plating 20,000 cells per T75 flask. Resulting cells were counted and passaged every 10 days or when cells reached 80%–90% confluence. Cumulative cell numbers were calculated and plotted (log10 scale).
Analysis of telomerase activity and telomere length
Telomerase activity
The polymerase chain reaction (PCR)-based telomeric repeat amplification protocol assay was used to detect telomerase activity [18]. Cell lysates were prepared from 1,000,000 cells using CHAPS lysis buffer. Telomerase extension products were amplified using the M2 (5′-AATCCGTCGAGCAGAGTT-3′) and ACX primers (5′-GCGCGGCTTACCCTTACCCTTACCCTAACC-3′), separated in a 10% polyacrylamide gel, and visualized with SYBR Gold on a Typhoon FLA 9500.
Telomere length by terminal restriction fragment analysis
Terminal restriction fragments (TRFs) were prepared from genomic DNA by digestion with HinfI and RsaI, and separated by pulsed field gel electrophoresis in a 1% agarose gel at 14°C, run at 6 V/cm with an initial switch time of one and a final switch time of six for 14 h. Gels were dried, denatured, and subject to in-gel hybridization in Church buffer with a telomere-specific oligonucleotide probe. Gels were washed and visualized by Phosphorimage analysis on a Typhoon FLA 9500.
Telomere length analysis by quantitative PCR
Telomere qPCR was carried out using the Rotor-Gene SYBR Green PCR Kit (Qiagen) using forward (5′-CGGTTTGTTTGGGTTTGGGTTTGGGTTTGGGTTTGGGTT-3′) and reverse (5′-GGCTTGCCTTACCCTTACCCTTACCCTTACCCTTACCCT-3′) telomere primers, and compared with the single-copy gene 36B4 using forward (5′-CAGCAAGTGGGAAGGTGTAATCC-3′) and reverse (5′-CCCATTCTATCATCAACGGGTACAA-3′) primers. PCR reactions were performed in the Rotor-Gene Q (Qiagen) real-time cycler for 95°C for 15 min, followed by 30 cycles of 95°C for 7 s and 58°C for 10 s (telomeric primers), and for 95°C for 5 min, followed by 35 cycles of 95°C for 15 s and 58°C for 30 s (36B4 primers). Serially diluted DNA standards were used to generate standard curves and Rotor-Gene Q series software was used to analyze data.
Statistical analysis
Data are presented as Mean ± Standard Errors of the Mean (SEM) or number (percent). All data were analyzed with SigmaPlot 12.5 software. Statistical comparisons were performed by unpaired Student's t-test. P < 0.05 was considered statistically significant.
Results
Cardiac explant-derived cells express MSC and cardiac progenitor markers
We cultured multiple small (100–200 mg) pieces of previously cryopreserved human LV tissue (Fig. 1A) from eight different donors of varying age and gender (Table 1). Initial cell outgrowth appeared within 3–4 days and displayed a fibroblastic spindle-shaped morphology. Cultures reached full confluence in 14–21 days (Fig. 1B). These times and morphological characteristics did not vary with the age of the heart at original cryopreservation or whether the hearts were from female, male, young, or old hearts (Fig. 1C).
CPCs have previously been isolated by other explant-based methods and these produce a mixed population of mesenchymal and progenitor cells [8,19]. We characterized the phenotype of outgrowth cells from our cryopreserved human cardiac samples using qPCR, immunofluorescence and flow cytometry. As expected, our analysis revealed heterogeneity within this population. There was high expression of CD90 and PDGFRα transcript (Fig. 2A) suggesting subpopulations with a fibroblast and mesenchymal cell phenotype. There was also robust expression of known pluripotency genes (SOX4, NANOG, MYC, KLF4) (Fig. 2A) suggesting a progenitor/stem cell phenotype. These findings were validated by flow cytometric analysis of the relevant proteins (Fig. 2B, C). Interestingly, the vast majority of cells expressed PDGFRα (88.3% ± 3.6%) and almost half expressed CD90 (43.6% ± 9.8%), which we have previously shown to mark a population of cardiac MSC-like cells isolated from murine and human hearts [7,11].
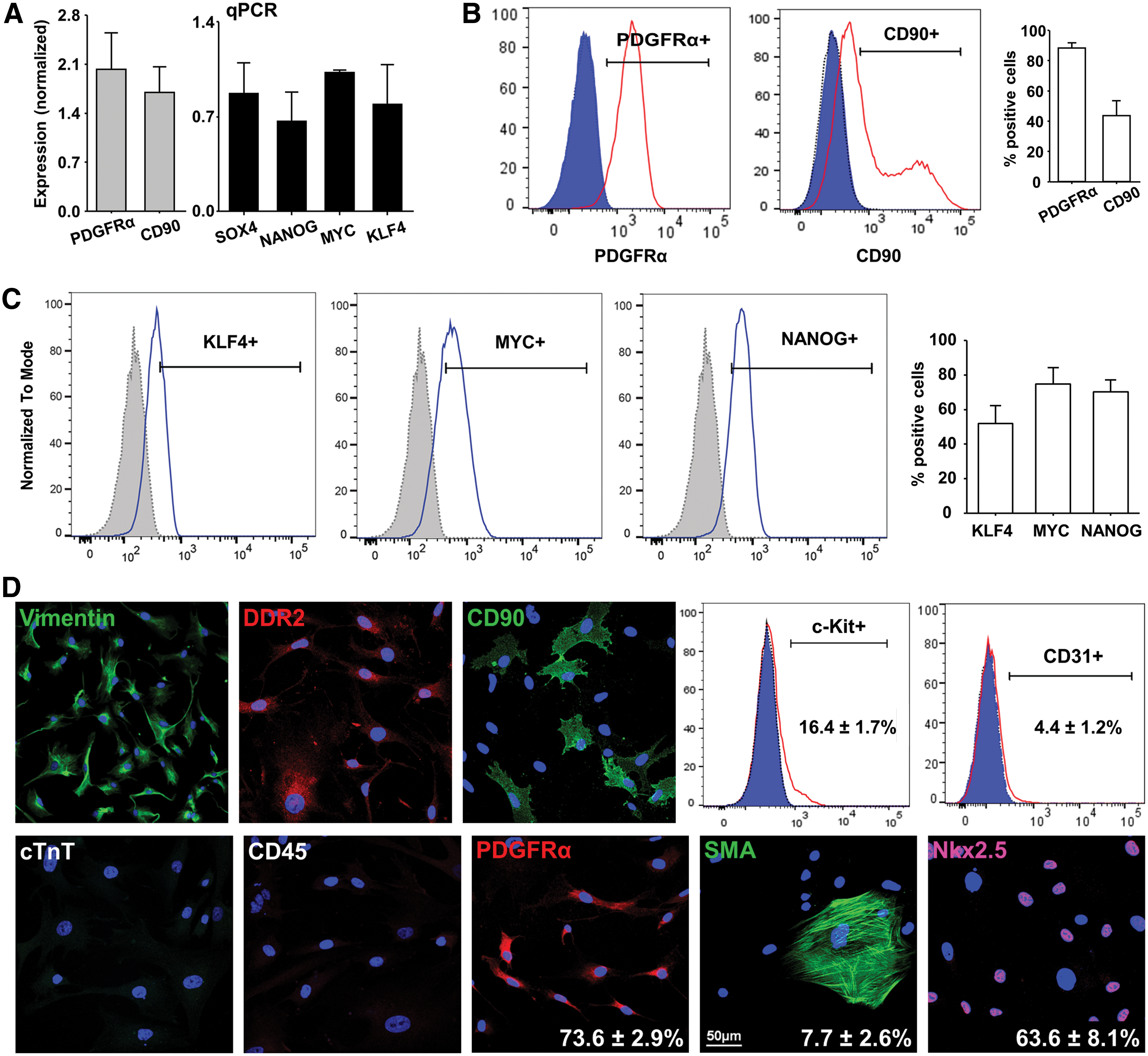
Phenotypic characterization of outgrowth cells.
We then performed immunohistochemical and flow cytometry analysis to further characterize the phenotype of outgrowth cells (Fig. 2D). Virtually all of the explant-derived cells expressed Vimentin and DDR2. While these markers have traditionally been associated with a fibroblast phenotype, recent reports have highlighted the ambiguity of the cardiac fibroblast population and demonstrated that many cardiac fibroblasts express MSC and cardiac progenitor markers [20]. Consistent with this, the majority of outgrowth cells from our explant cultures expressed the cardiogenic transcription factor Nkx2.5 (63.6% ± 8.1%). There was low expression of the EC marker CD31 (4.4% ± 1.2%) and the smooth muscle cell and myofibroblast marker SMA (7.7% ± 2.6%). All cells were negative for cTnT (a myocyte-specific marker) and CD45 (a pan-hematopoietic cell marker).
To confirm the karyotypes of outgrowth-derived cells we analyzed the chromosomal status using the GTG-banding method. This showed maintenance of normal karyotype (Supplementary Fig. S1A; Supplementary Data are available online at
Cardiac explant-derived cells demonstrate clonogenicity, self-renewal, and vascular cell and myocyte differentiation potential
To explore the multipotent potential of progenitor cells from cryopreserved cardiac explants we subjected outgrowth cells to in vitro myogenic and vascular differentiation assays. Cells grown in medium, without specific differentiation reagents served as controls. Consistent with our previous studies on developing and diseased hearts [7], PDGF-BB induced smooth muscle differentiation (MYH11+, 13% ± 3% vs. 2% ± 1% in the controls, P < 0.05) (Fig. 3A) and VEGF induced EC differentiation (vWF+, 78% ± 5% vs. 45% ± 2% [controls], P < 0.05) (Fig. 3B). Although explant-derived outgrowth cells express high numbers of the cardiogenic transcription factor Nkx2.5 (63.64%), there was no expression of either cardiac troponin T (cTnT) or sarcomeric α-actinin when cultured in basal medium alone (without NRVMs) (Fig. 3C). In contrast, 2 weeks after cardiomyocyte differentiation by coculture with NRVMs, we observed differentiation into cardiomyocyte-like cells as indicated by the expression of cTnT and α-actinin (Fig. 3C). The number of cTnT- and α-actinin-positive cells was very low (<1%) and although there was some myofibrillar organization, there was no clear cross striation as evident in surrounding NRVMs (Fig. 3C).
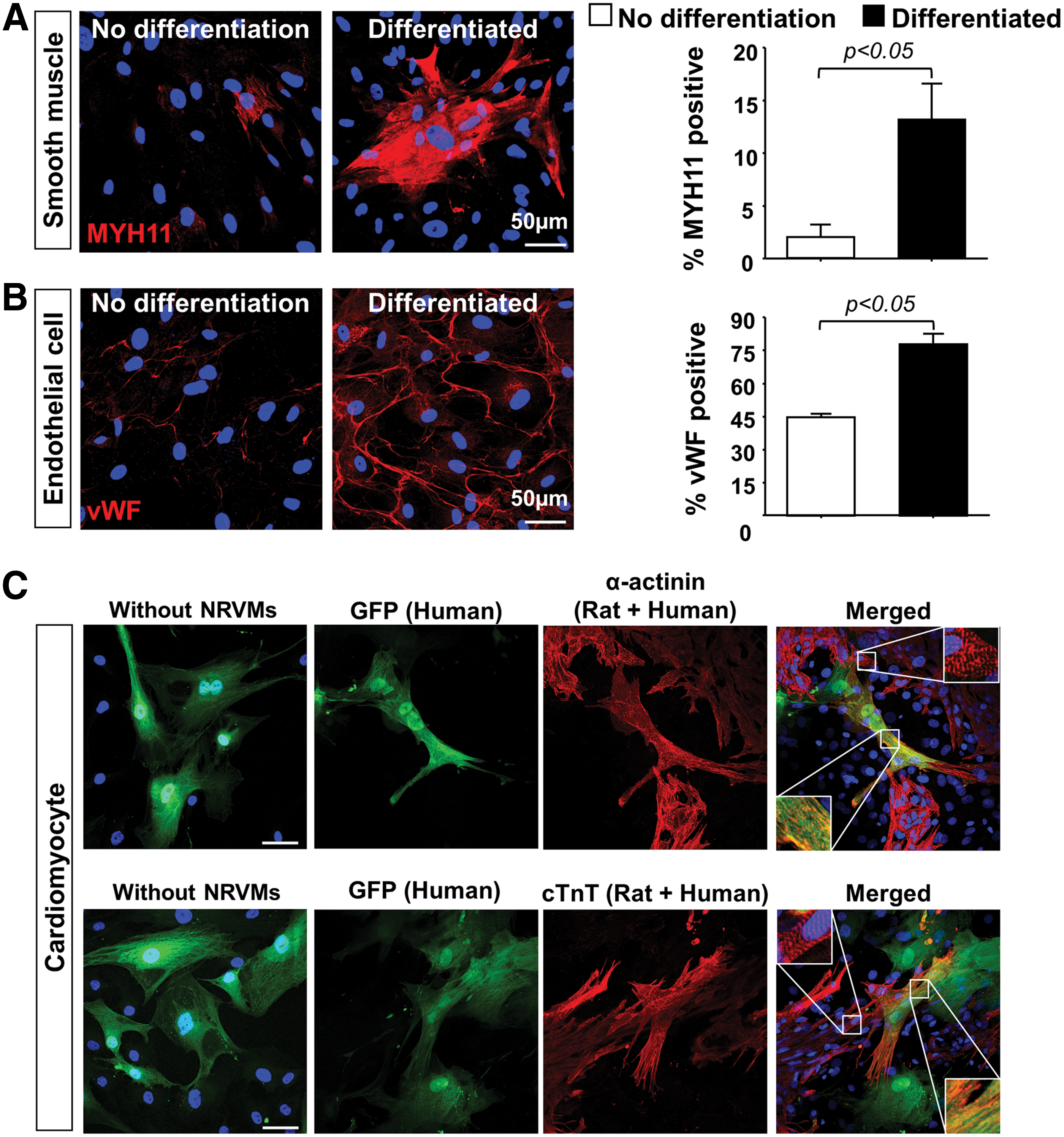
Explant-derived cells are capable of in vitro differentiation to vascular and cardiomyocyte lineages.
Other than differentiation potential, key stem/progenitor cell characteristics include clonogenicity and extended self-renewing capacity. Colony-forming assays were first used to characterize BM-MSCs [21] and later to define cardiac MSC-like populations from the adult murine heart [11,22]. We previously reported that colonies with the largest diameter are founded by cells with the greatest stem/progenitor capabilities whereas other colonies result from more differentiated founding cells [11]. Total colony numbers and distribution of sizes resulting from explant-derived cells were similar to previously described results (large >2 mm, 12 ± 4 colonies, small <2 mm, 9 ± 3 colonies, micro 25–50 cells, 5 ± 1 colonies, Fig. 4A).
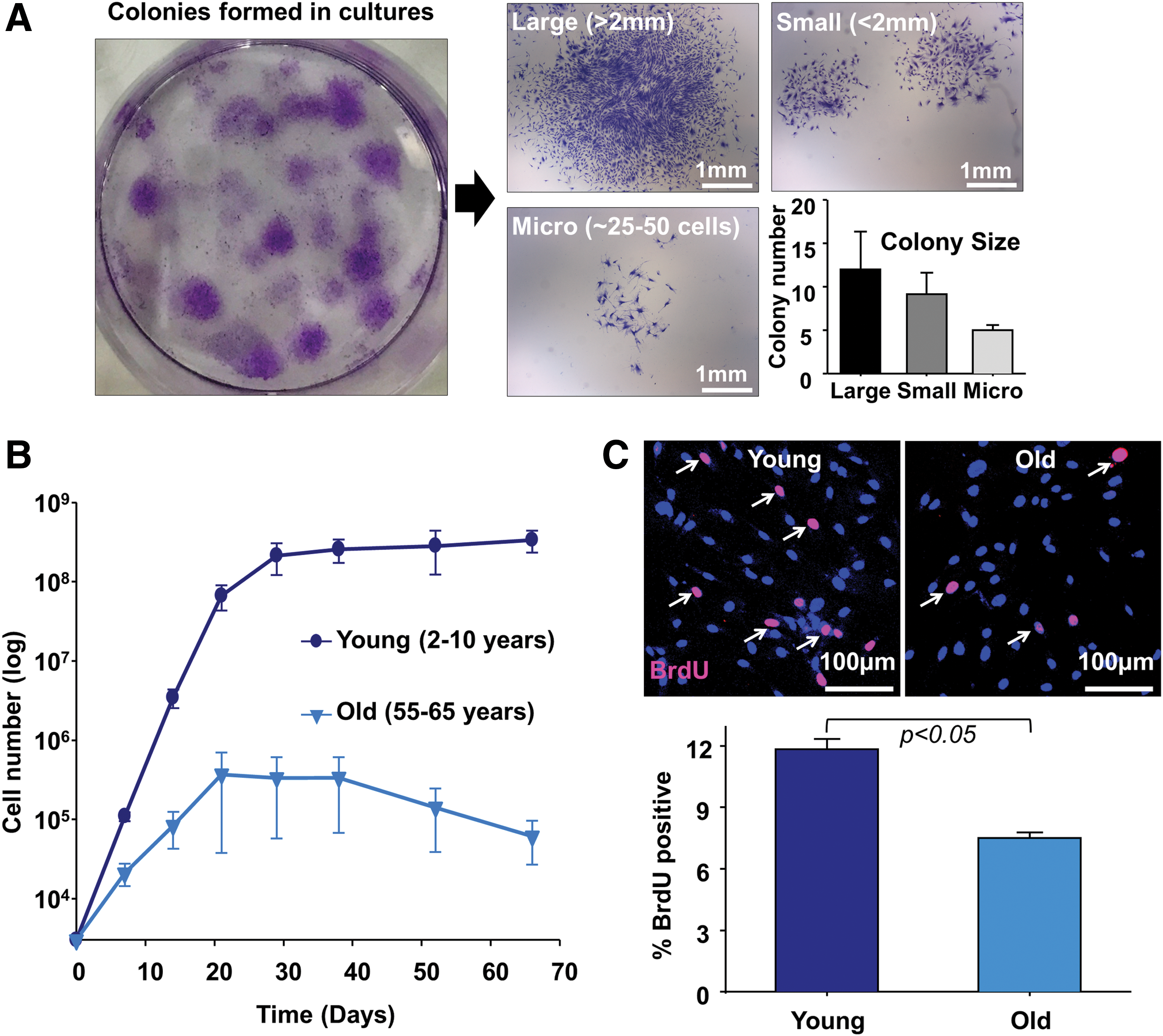
Colony-forming assays and growth characteristics of explant-derived cells show clonogenicity and long-term self-renewal.
We then used serial passaging and growth curve analysis to demonstrate the self-renewing capacity of explant-derived outgrowth cells from young and old hearts. At 80%–90% confluence, the cells were dissociated and repassaged. Initial and cumulative cell numbers were counted at each passage. Outgrowth cells expanded over successive passages. Cells grew for at least 2 months, but this was significantly reduced in aged hearts (cumulative cell number, young = 3.4 ± 1.0 ( × 109) cells vs. old = 6.1 ± 3.4 ( × 105) cells, P < 0.05) (Fig. 4B). The cells proliferated rapidly at early passages, but after 30 days this proliferative potential declined (Fig. 4B). The proliferative activity of outgrowth cells was assessed by the incorporation of BrdU, a synthetic analog of the nucleoside thymidine. It is incorporated into replicating DNA during the S phase in dividing cells and is subsequently detected by anti-BrdU antibodies [23]. Consistent with reduced growth in aged hearts (Fig. 4B), significantly fewer BrdU-positive cells were observed from old (>55 year old, 7.5% ± 0.3%, P < 0.05) compared with young (<10 year old, 11.8% ± 0.5%) hearts (Fig. 4C). Taken together these results indicate that outgrowth cells from cryopreserved cardiac explants contain a subpopulation of clonogenic and self-renewing CPCs with similar characteristics and differentiation potential to PDGFRα+ MSC-like cells reported in our previous studies [7,11].
Cardiac explant-derived cells lack telomerase and have decreasing telomere lengths with age
Telomeres cap the ends of chromosomes and progressively shorten with each cell division until reaching cellular senescence [24,25]. The enzyme telomerase is necessary for telomere repeat synthesis and maintenance of telomere length required for sustained cell proliferation and growth. Although pluripotent stem cells are well known to robustly express telomerase and maintain short telomere lengths [26], the literature on telomerase expression in adult stem/progenitor cells is inconsistent [27,28]. Therefore, we analyzed telomere length and telomerase activity in cardiac explant-derived cells. TRF analysis (Fig. 5A) and qPCR (Fig. 5B) identified significantly shorter telomere lengths in outgrowth cells from old (T/S ratio = 0.49 ± 0.06) compared with young heart samples (T/S ratio = 0.78 ± 0.02, N = 3 patients/group, P < 0.05). This correlated with the reduced proliferative capacity observed in cells isolated from old hearts (Fig. 4B). No telomerase activity was detected in cells derived from any of the hearts (Fig. 5C). The lack of telomerase activity may explain reduced growth potential of the cells in our study and represent an opportunity to enhance stem/progenitor cell function in future studies.
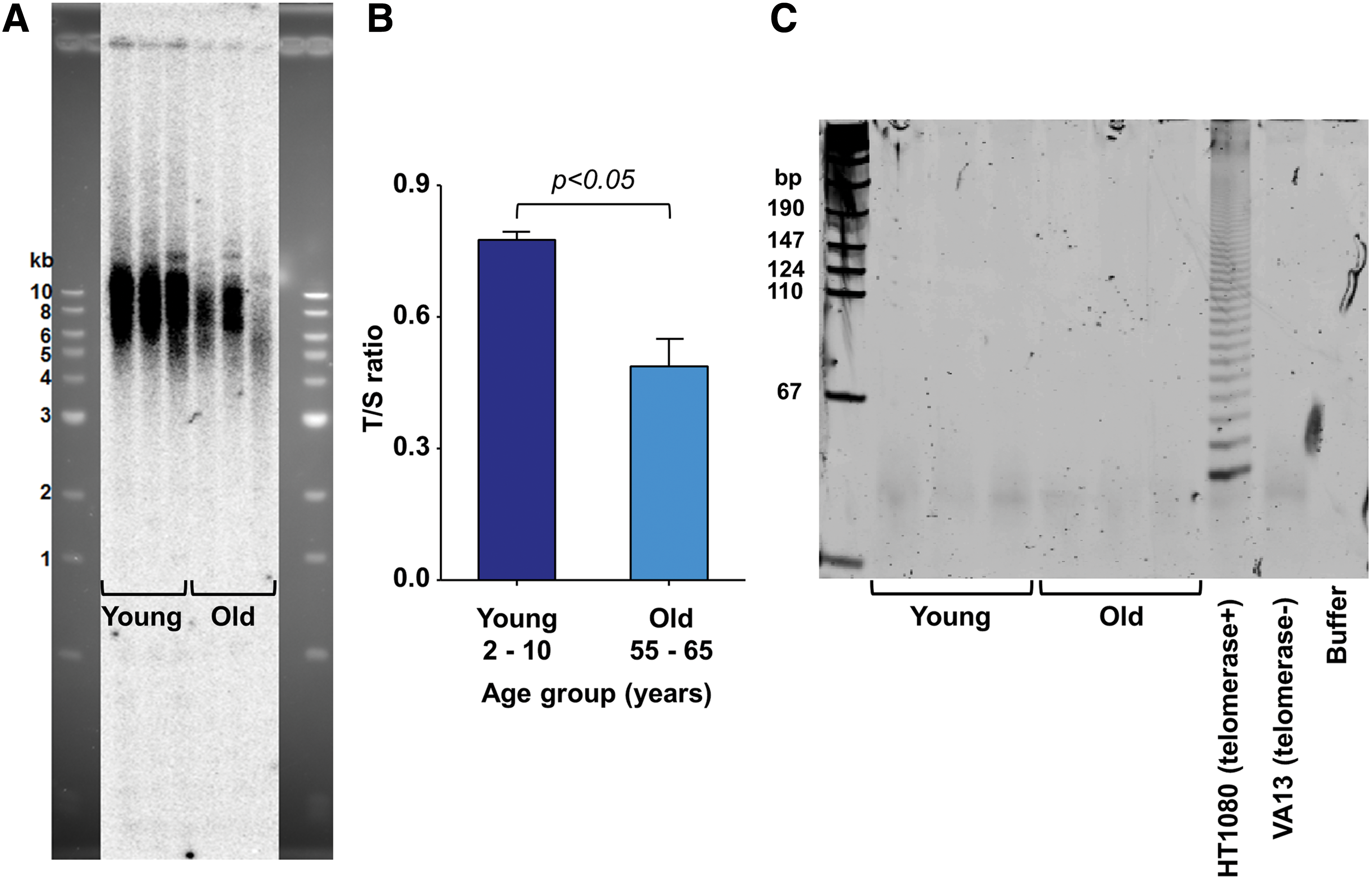
Telomere length and telomerase activity analysis of explant-derived cells from young and old hearts. Telomere length analysis by
CPCs are enriched within the PDGFRα+/CD90+/CD31− cell fraction
Although the results above demonstrate that outgrowth cells contain CPCs, development of a method to enrich CPCs from the heterogeneous population of outgrowth cells would be beneficial. Our previous murine studies demonstrate that FACS for the Sca-1+/PDGFRα+/CD31− fraction highly enriches for CPCs [11]. However, there is no known human ortholog for rodent Sca1. Since CD90 is a mesenchymal cell marker that enriches for cardiomyogenic cells within other CPC populations [29], we used PDGFRα, CD90, and CD31 markers to construct a suitably enriching surface marker signature. Four populations were sorted using the gating strategy in Fig. 6A, including PDGFRα+/CD90+/CD31−, PDGFRα+/CD90−/CD31−, PDGFRα−/CD90+/CD31−, and PDGFRα−/CD90−/CD31− fractions with colony-forming assays subsequently performed.

Cell sorting for PDGFRα+/CD90+/CD31− cells enriches explant-derived cells for progenitors.
As discussed above, colony-forming assays can be used to estimate progenitor/stem cell proportions within specific cell fractions with large (>2 mm diameter) colonies correlating with the highest CPC enrichment [11] and small (<2 mm) or micro (∼25–50 cells) colonies deriving from more differentiated founder cells. The PDGFRα+/CD90+/CD31− fraction had the highest number of large colonies compared with the other cell fractions (Fig. 6B) suggesting significant enrichment for CPCs. To validate this result we performed growth curve analysis on cells from the PDGFRα+/CD90+/CD31− fraction compared with cells that were not sorted. PDGFRα+/CD90+/CD31− cells demonstrated greater self-renewing capacity (Fig. 6C). The expression of pluripotency genes (KLF4, MYC, NANOG) in purified PDGFRα+/CD90+/CD31− population was further confirmed by western blot (Fig. 6D).
Although, c-Kit has been widely used to identify resident cardiac progenitor populations [6,30], our previous studies in mice suggest that c-Kit expressing CPCs are a distinct population from PDGFRα+/CD31− CPCs. Therefore, we investigated the impact of c-Kit+ expression on CPC enrichment. Colony-forming assays demonstrated no significant difference in the number of large (>2 mm) colonies from the PDGFRα+/CD90+/CD31−/c-Kit+ compared with the PDGFRα+/CD90+/CD31−/c-Kit− cell fractions (Supplementary Fig. S2A). Interestingly, there were significantly lower colony numbers in PDGFRα− cell fractions (Supplementary Fig. S2A), suggesting that PDGFRα has a predominant role in the enrichment of this CPC population rather than c-Kit. The expression of c-KIT transcript in outgrowth cells from the four FACS-sorted populations was also analyzed by qPCR (Supplementary Fig. S2B). This confirmed low overall c-Kit expression, but three- to four-fold increased enrichment of c-Kit in the c-Kit+-sorted populations. Based on these results (Fig. 6 and Supplementary Fig. S2), FACS sorting for the PDGFRα+/CD90+/CD31− fraction was used to enrich for human CPCs in subsequent experiments.
PDGFRα+/CD90+/CD31− CPCs maintain stem/progenitor cell phenotype after long-term propagation
To confirm the stem/progenitor cell phenotype of PDGFRα+/CD90+/CD31− CPCs, we sorted and cultured single cells at early (P2) and late (P12) passages. PDGFRα+/CD90+/CD31− CPCs at P2 and P12 maintained the ability to generate single cell-derived colonies (Fig. 7A). No significant difference in clonogenic efficiency was observed from P2 (14.7% ± 7.7%) compared with P12 (11.3% ± 4.7%, P = 0.54) (Fig. 7A). Since pluripotency genes are implicated in maintaining self-renewing capacity and multipotent potential, we compared pluripotency gene expression and cardiomyocyte differentiation potential of these clonal-derived cells at P2 and P12. Single cell-derived colonies from both populations (P2 & P12) maintained expression of CPC markers (PDGFRα, CD90) as well as pluripotency genes (SOX4, NANOG, MYC, and KLF4) (Fig. 7B). Similarly, no difference in the cardiomyocyte differentiation potential was observed between the cells isolated at P2 and P12 (Fig. 7C, D). Furthermore, chromosome karyotype analysis of P2 and P12 PDGFRα+/CD90+/CD31− CPCs showed maintenance of normal karyotype (Supplementary Fig. S1B, C). These results demonstrate that clonogenic PDGFRα+/CD90+/CD31− CPCs can be isolated and propagated from previously cryopreserved cardiac tissue samples.

Clonogenicity, self-renewing capacity, and differentiation potential of PDGFRα+ CPCs at different passages.
Tissue cryopreservation does not influence isolation or phenotype of explant-derived human CPCs
To determine whether cryopreservation affects explant-derived CPC properties, we isolated and characterized cells from fresh versus frozen samples. Atrial appendage biopsies from patients at the time of cardiothoracic surgical procedures were collected (Table 2). Each tissue specimen was divided into two sections for fresh cell isolation (fresh tissue) and tissue cryopreservation (frozen). One of the frozen samples was stored for 2.5 years and others for 3–16 days. Cryopreservation did not influence the cell morphology and overall cell yields from plated tissue (Fig. 8A, B). Furthermore, no significant difference in clonogenic efficiency was observed between cells from fresh (8.6% ± 1.1%) and cryopreserved tissue (7.7% ± 0.9%) (Fig. 8C). Flow cytometry of cells derived from fresh and cryopreserved tissue also showed no significant difference in PDGFRα (43% [fresh] vs. 46% [frozen]), PDGFRβ (41% [fresh] vs. 43% [frozen]), CD90 (76% [fresh] vs. 79% [frozen]), c-Kit (1.5% [fresh] vs. 2.2% [frozen]), CD31 (12% [fresh] vs. 9% [frozen]), and CD34 (1.1% [fresh] vs. 1.4% [frozen]) expression (Fig. 8D). Taken together, these results demonstrate that cryopreservation does not alter the phenotypic signature of outgrowth cells from which PDGFRα+/CD90+/CD31− CPC are derived.
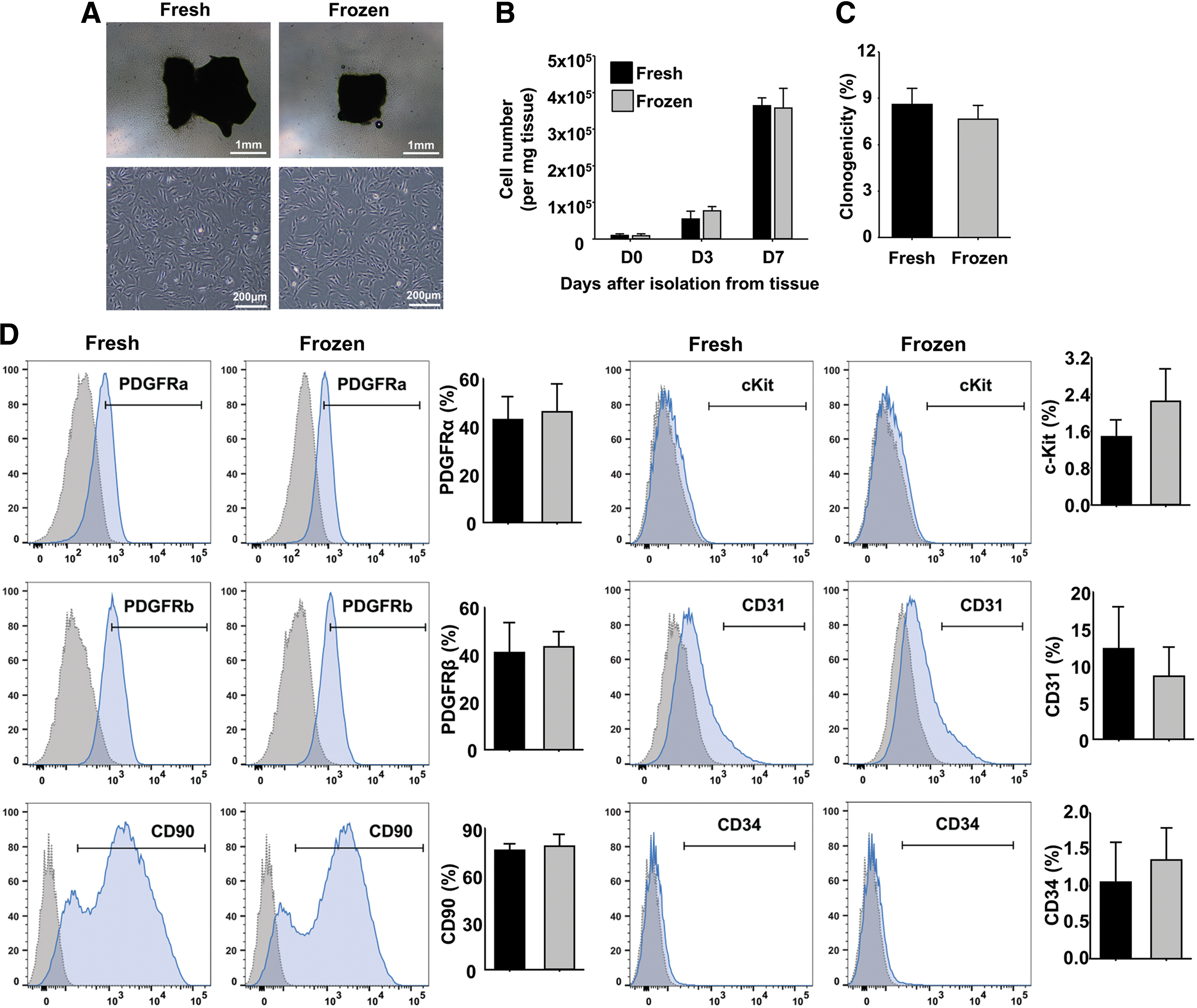
Cryopreservation does not impact phenotype or isolation of explant-derived CPCs.
Discussion
CPCs have recently been used in cardiac regeneration clinical trials [30 –32] and remain the focus of ongoing investigation. These have usually required ex vivo expansion of autologous CPCs in a time-consuming manner that prevents early administration of CPCs after cardiac injury. Another alternative is to use allogeneic CPC transplantation (ALLSTAR NCT02293603, DYNAMIC NCT01458405). However, the harvesting of CPCs requires invasive cardiac biopsy, which is not without risk and may often only be procured from individuals who have pre-existing cardiac disease. A better solution could be to use previously cryopreserved cardiac tissue for harvest of allogeneic CPCs.
In this study, we developed an explant-based cell culture assay to cultivate outgrowth cells with progenitor cell characteristics from healthy human hearts previously cryopreserved by the SHB. This provides a “proof-of-concept” for utilization of frozen tissue to expand allogeneic CPCs for potential subsequent clinical transplantation. Although many different CPC populations have been reported [33], we focused our efforts on a PDGFRα+ cardiac MSC-like population previously described [7,11]. We show that these CPCs can be isolated and expanded from previously cryopreserved cardiac tissue without loss of their progenitor cell phenotype. In particular, differentiation potential, clonogenicity, and prolonged self-renewal potential are retained from early to late cell culture passage.
Furthermore, we show that FACS can be used to enrich CPCs that lie within the PDGFRα+/CD90+/CD31− cell fraction and that there are minimal effects of cryopreservation on the recovery of CPCs when directly compared with isolation from noncryopreserved fresh tissue. These data support two recent studies that also find cryopreservation does not adversely affect the function of c-Kit expressing [34] or explant-derived [35] cardiac stem/progenitor cell populations. Altogether, these studies support a strategy for the formation of tissue biobanks that could provide a rapidly accessible source of CPCs for therapeutic use. Heart samples can be obtained by tissue donation after death from noncardiac causes, thereby ensuring nondiseased status and potentially young age. This avoids potential problems of age- and disease-associated reduction of CPC function and utility [36,37].
Moreover, the efficiency of derivation in our study was very good. Outgrowth cells were successfully isolated from all fresh and frozen cardiac samples (Tables 1 and 2). However, potential impediments to this method include improper tissue sample collection, storage, and processing. The samples used in this study were collected at the time of surgical organ harvest for initially intended cardiac transplantation and were isolated with clinically sterile conditions followed by careful cryopreservation at −196°C to prevent tissue degradation.
A surprising finding in our characterization of outgrowth cells from cryopreserved cardiac samples was the high expression of fibroblast proteins together with genes/proteins usually associated with stem/progenitor cells. The cardiac homeobox protein Nkx2.5, which is essential for heart development and plays a critical role in transcriptional regulation during cardiogenesis [38,39], was strongly expressed together with pluripotency gene transcripts (SOX4, NANOG, MYC, KLF4) that are important for the maintenance of self-renewing capacity and multipotent state of progenitor cells. These findings suggest that the culture conditions used are permissive for the isolation and propagation of CPCs may be otherwise labeled loosely as “cardiac fibroblasts.” Indeed, there is increasing understanding that the poorly defined cardiac fibroblast population contains a progenitor pool of cells whose physiological function currently remains to be determined [40].
Although MSCs from adult BM or adipose tissue [41,42] are capable of long-term self-renewal, they ultimately reach replicative senescence during long-term expansion. Furthermore, the number and regenerative potential of tissue resident stem cells decline with advancing age [36,37,43]. We also observed a similar replicative senescence and reduction in PDGFRα+ CPC growth, which was more pronounced when aged rather than young hearts were used for cell isolation (Fig. 4B). This decline in CPC proliferation has also been reported in other CPCs [36,37]. These findings suggest that using juvenile hearts to procure CPCs may be preferable for future clinical therapies compared with diseased or older donors. Importantly, the isolation of CPCs from young healthy donors might only be possible using banks of previously cryopreserved tissue. However, yet another strategy may be to rejuvenate the CPCs from aged hearts.
The progressive shortening of telomeres is associated with cellular aging, cell growth, and senescence [25,44,45]. Telomeres are structures that contain the repeated sequence TTAGGG, and are responsible for maintaining chromosomal stability and integrity [46,47]. Consistent with our growth curve analysis (Fig. 4B) and similar reports by others [48], outgrowth cells from old hearts have significantly shorter telomeres than younger hearts. Telomeres are maintained by telomerase, a ribonucleoprotein complex expressed in cells with extended replicative ability, such as embryonic stem cells. In mice and humans, telomerase is silenced after birth, leading to progressive telomere shortening throughout their lifespan [48 –52]. We found no telomerase activity in progenitor cells derived from adult hearts (Fig. 5A). Without this activity, telomere lengths cannot be replenished resulting in the cellular senescence observed earlier in CPCs from our older heart samples. In contrast, pluripotent stem cells and germline cells express high levels of the catalytic subunit of telomerase (human telomerase reverse transcriptase or hTERT) that maintains telomerase activity and full replication of telomeric DNA [53 –55]. As suggested in other reports of TERT overexpression [56 –58], it is tempting to speculate that TERT manipulation in CPCs could rejuvenate an aged or diseased phenotype and enhance cardiac regenerative capabilities.
Conclusions
We have successfully isolated PDGFRα-expressing CPCs from cryogenically frozen samples of healthy human hearts. The characteristics of these CPCs were similar to those isolated from fresh (never frozen) tissue described in our previous studies [7,11]. Isolation of CPCs from previously frozen cardiac tissue supports the creation of heart tissue banks that could increase the ease of CPC production for future clinical applications. Further investigation on telomere/telomerase roles in CPC function and enhancement of CPC function by telomerase manipulation may be important to improve clinical translational goals and are ongoing.
Footnotes
Acknowledgments
This study was supported by the National Health and Medical Research Council (APP100046). James Chong was supported by a Future Leader Fellowship (ID 100463) from the National Heart Foundation of Australia and Sydney Medical School Foundation Fellowship. Funding bodies had no role in study design, data collection, and analysis, decision to publish, or preparation of the article.
Flow cytometry was performed in the Flow Cytometry Core Facility that is supported by Westmead Institute, Westmead Research Hub, Cancer Institute New South Wales, and National Health and Medical Research Council. Immunofluorescence image acquisition was conducted using the Olympus confocal microscope in the Cell Imaging Core Facility that is supported by Westmead Institute, Westmead Research Hub, National Health and Medical Research Council, DVCR, and South Western Area Health Service. qPCR was performed in the Westmead Institute Genomics Core Facility.
Author Disclosure Statement
The authors declare that there is no conflict of interest regarding the publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.