
Abstract
Wnts are secreted glycoproteins that regulate stem cell self-renewal, differentiation, and cell-to-cell communication during embryonic development and in adult tissues. Bone marrow mesenchymal stem cells (BM-MSCs) have been shown to stimulate dermis repair and regeneration; however, it is unclear how BM-MSCs may modulate downstream Wnt signaling. While recent reports implicate that Wnt ligands and Wnt messenger RNAs (such as Wnt4) exist within the interior compartment of exosomes, it has been debated whether or not Wnts exist on the exterior surface of exosomes to travel in the extracellular space. To help answer this question, we utilized flow cytometry of magnetic beads coated with anti-CD63 antibodies and found, for the first time, that Wnt3a protein is detectable exteriorly on CD63+ exosomes derived from BM-MSCs over-secreting Wnt3a into serum-free conditioned media (Wnt3a CM). Our data suggest that CD63+ exosomes significantly help transport exterior Wnt3a signal to recipient cells to promote fibroblast and endothelial functions. During purification of exosomes, we unexpectedly found that use of ultracentrifugation alone significantly decreased the ability to detect exteriorly bound Wnt3a on CD63+ exosomes, however, polyethylene glycol (PEG)-mediated exosome-enrichment before exosome-purification (with ultracentrifugation into a sucrose cushion) resulted in exosomes more likely to retain exterior Wnt3a detectability and downstream Wnt/beta-catenin activity. Our findings indicate the important role that purification methods may have on stem cell-derived Wnt-exosome activity in downstream assays. The ability for BM-MSC Wnt3a CM and exosomes to stimulate dermal fibroblast proliferation and migration, and endothelial angiogenesis in vitro, was significantly decreased after CD63+-exosome depletion or knockdown of Wnt coreceptor LRP6 in recipient cells, suggesting both are required for optimal Wnt-exosome activity in our system. Thus, BM-MSC-derived CD63+ exosomes are a significant carrier of exterior Wnt3a within high Wnt environments, resulting in downstream fibroblast proliferation, migration, and angiogenesis in vitro.
Introduction
T
Wnts are thought of as short-range morphogens that transfer developmental signaling between cells in close proximity [1]. Many Wnts are modified by an endoplasmic reticulum acyltransferase (Porcupine) to append a palmitic acid on Wnt, making the ligand more active with Fzd receptors; this modification simultaneously makes the ligand more lipophilic/hydrophobic and less diffusible in the hydrophilic extracellular environment. Thus, we hypothesized that extracellular vesicles (EVs) may play a significant role in long-range cell-to-cell communication via transportation of Wnts.
Cells communicate with direct cell-to-cell contact or transfer of secreted materials, including with EVs, which include (1) exosomes, (2) microvesicles, and (3) apoptotic bodies, all of which are ubiquitous in circulation and body fluids [10 –14]. Exosomes, mostly sized between 30–150 nm, are generated within the multivesicular bodies (MVBs), which are endosome compartments in which “intraluminal vesicles” develop; intraluminal vesicles are later secreted into the extracellular space as “exosomes” [13]. Microvesicles are formed by direct shedding from the plasma membrane, averaging ∼200 nm, but can range from 50 to 1,500 nm [13,15,16]. Apoptotic bodies are larger than exosomes and microvesicles, typically ranging from 1,000 to 2,000 nm [13,15,16].
These EVs are potential candidate carriers of Wnts, helping them to travel from producers (ex. bone marrow cells) to recipients (ex. fibroblasts and endothelial cells). Recently, it has been debated whether Wnts utilize exosomes interiorly for long-range travel. Furthermore, it is unknown whether Wnts ride exteriorly on exosomes before docking with receptors on target cells to stimulate Wnt/beta-catenin signaling.
We undertook the following study to examine whether Wnt3a, when overexpressed, is detectable on the exterior surface of CD63+ exosomes derived from BM-MSCs, and whether BM-MSC Wnt-exosome activity could be purified using traditional exosome purification techniques. Furthermore, because it is unclear how exosome-Wnt3a affects fibroblast proliferation and migration, and angiogenesis in vitro, we sought to characterize how BM-MSC Wnt3a CM and purified EVs derived from a “high-Wnt” environment affect these surrogate assays of dermis repair.
Materials and Methods
Human BM-MSCs
Collection of primary human donor bone marrow was under approval of University of Miami Institutional Review Board (IRB). BM-MSC “line 2” and “line 3” were obtained from healthy male and female donors, respectively, according to quality control and manufacturing established by AllCells®, and isolated according to the procedures below. All experiments were performed in accordance with relevant guidelines and regulations and complied with the Declaration of Helsinki. Informed consent was obtained for all human subjects and permission was given by all human subjects to publish results derived from the tissues and cells and, if necessary, to publish any identifying information, including images. The human donor of bone marrow (for BM-MSC “line 1”) was a 26-year-old healthy, male donor with no known medical problems.
Specifically, the donor tested negative for anti-HIV-1/HIV-2, anti-HTLV I/II, anti-HCV, HIV-1 nucleic acid test, HCV nucleic acid test, HBsAg, anti-HBc (IgG and IgM), anti-CMV, WNV nucleic acid, Trypanosoma cruzi ELISA (Chagas), RPR for syphilis, and had no clinical/history/laboratory evidence to suggest CJD. Bone marrow (∼80 mL) was aspirated from the posterior iliac crests as per standard practice of the University of Miami Bone Marrow Transplant Programs. The marrow was aspirated into heparinized syringes. The labeled syringes were transported at room temperature to the Good Manufacturing Practices (GMP) facility at the Interdisciplinary Stem Cell Institute at the University of Miami. Bone marrow (BM) was processed using Lymphocyte Separation Medium (LSM; specific gravity 1.077) to prepare the density-enriched, mononuclear cells (MNCs). The cells were diluted with Plasmalyte A or PBS buffer and layered onto LSM using conical tubes to isolate MNCs following established standard operating procedures (SOPs). The MNCs were washed with Plasmalyte A or PBS buffer containing 1% human serum albumin (HSA).
The washed cells were sampled to determine the total number of viable nucleated cells. MSCs were initially cultured in alpha MEM media supplemented with 2 mM
Plasmids
The following plasmids were purchased from Addgene (as Escherichia coli bacterial stabs: M50 Super 8x TOPflash, M51 Super 8x FOPFlash (Howard Hughes Medical Institute, University of Washington), pIS1-Actb5UTR-renilla (Howard Hughes Medical Institute, Whitehead Institute for Biomedical Research), and pcDNA-Wnt3a (University of California, Irvine). Bacteria was streaked on LB agar plates containing 100 μg/mL ampicillin and incubated in 37°C overnight. Single colonies were picked with a sterile pipette tip and transferred to sterile 2 mL falcon tubes with LB media plus 100 μg/mL ampicillin and incubated overnight.
The next day, the growing bacteria in broth was transferred to a 500 mL LB media plus 100 μg/mL ampicillin and cultured at 37°C for 16 h in shaking incubator. Bacteria was pelleted in sterile centrifuge tubes at 5,000 g for 10 min using FiberLite® rotor, model F15-8x50C (Piramoon Technologies, Inc., Santa Clara, CA) in Heraeus Multifuge 3SR+ centrifuge (Thermo Scientific, Waltham, MA). Plasmid DNA from bacterial pellets were purified using EndoFree Plasmid Maxi Kit, according to the manufacturer's instructions (Qiagen, Hilden, Germany). Final plasmid DNA concentrations were measured using NanoDrop 2000 (Thermo Scientific).
Generation of BM-MSC Wnt3a-conditioned media
BM-MSCs (as prepared above) were split into 12-well plates (Corning, Corning, NY) at 50,000 cells per well in alpha-MEM (Corning) supplemented with 20% FBS hyclone (JR Scientific) (until collection of EVs, in which media were serum-free), 1% penicillin/streptomycin (Gibco, Waltham, MA), and 1%
Dual luciferase-based transcription assays
HEK293T cells were seeded at 5 × 104 cells per well into 24-well plates (n = 3 per treatment group). The next day, cells were transfected in OPTI-MEM with 500 ng of plasmid DNA per well of M50 Super 8x TOPflash or M51 Super 8x FOPflash as negative control (Howard Hughes Medical Institute, University of Washington) and 50 ng of DNA of pIS1-Actb5UTR-renilla (Howard Hughes Medical Institute, Whitehead Institute for Biomedical Research) (TransfeX:DNA complexes were mixed by pipetting briefly and incubated at RT for 15 min; complexes were then distributed to the cells by adding 100 μL of the complexes drop-wise to different areas of the well). Media was changed 24 h after transfection. EVs (prepared as described) were added at 1 μg/mL (in PBS diluted in serum-free alpha-MEM with the volume of vehicle remaining constant among all treatments), quantified by BSA protein quantification, according to manufacturer's instructions (Thermo Scientific).
EV treatments were incubated for 36 h before cell lysate collection for Dual-Luciferase® Reporter Assay System (Promega, Madison, WI). In brief, 100 μL of Passive Lysis Buffer was added to each well of 24-well plates and placed on gentle shaker for 30 min at room temperature. Lysates were pipetted up and down gently and 20 μL was transferred to each well of a white, polystyrene 96-well plate (Costar). A dual-injector GloMax® Multi+ Detection System with Instinct® Software was used to automatically inject reagents using preprogrammed “Dual Luciferase assay” protocol, with 100 μL LARII reagent (for firefly luciferase) with 10-s measurement followed by 100 μL Stop and Glo® reagent (followed by 10-s measurement), repeated in each well. Data were analyzed by measuring relative firefly luciferase values and normalized by renilla luciferase values in each well.
Proliferation assay
Human dermal fibroblasts were obtained from healthy donors under IRB approval of University of Miami. Dermal fibroblasts were seeded 5 × 103 cells per well in a 96-well plate (n = 6 per treatment group). The next day, medium was changed and treatments were applied (Day 0). Media treatment concentration was at 50% by volume and EVs treatments were at designated concentrations in PBS kept constant by volume, all diluted in serum-free media. Cell viability at Day 0 and Day 3, as measured by Vybrant® MTT Cell Proliferation Assay Kit (Molecular Probes, Eugene, OR) was performed according to manufacturer's instructions. In brief, 10 μL of 12 mM MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was added to each well of 96-well plate (n = 6 wells per treatment) and incubated at 37°C for 4 h. All except 25 μL of medium was removed from the wells and 50 μL DMSO was added to each well and mixed thoroughly with a pipette to dissolve the formazan and incubated at 37°C for 10 min. Each sample was mixed again thoroughly before reading absorbance at 540 nm in microplate reader VersaMax (Molecular Devices, Sunnyvale, CA).
Migration assay
Dermal fibroblasts were seeded in 24-well plates at 2.0 × 105 cells per well and incubated in 37°C in 5% CO2 until forming a confluent monolayer. To inhibit cell proliferation, media was replaced with serum-free MEM containing 10 μg/mL mitomycin for 4 h. The confluent layer was scratched with 1 mL sterile pipette tip. Culture medium was replaced with fresh serum-free MEM. Images (n = 3 per treatment) from the center of the wells were taken with IX81 Olympus microscope with ORCA-AG Hamamatsu digital camera and time 0 and 24 h after scratching. Scratch area closure was determined as analysis of change in pixels due to migration front, as measured using Image J software.
Gene expression analysis
Dermal fibroblasts (1 × 105) were seeded into 12-well plates and incubated at 37°C in 5% CO2. The following day, cells were switched to serum-free MEM for 24 h before treatment applications. After 24 h of treatment, RNA was extracted with RNeasy Kit (Qiagen) following the manufacturer's instructions. Purified RNA (300 ng) samples were converted to cDNA with qScript™ XLT cDNA SuperMix (Quanta BioSciences, Beverly, MA) according to manufacturer's instructions, using BioRad CFX96 Real-Time PCR Detection System (BioRad Laboratories, Hercules, CA) (5 min at 25°C, 60 min at 42°C, and 5 min at 85°C, hold at 4°C).
Real-time quantitative polymerase chain reaction was performed using 15 ng cDNA per reaction with PerfeCTa™ SYBR® Green FastMix™, Low ROX™ (Quanta Biosciences) using Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA) (95°C, 30 s, followed by 40 cycles of 95°C, 5 s; 55°C, 15 s; and 70°C, 10 s). Human β-actin gene expression was used as a reference gene for calculations of comparative Ct method (2ΔΔCT threshold cycle method). Primers were designed via Primer designing tool (NCBI) and generated (Integrated DNA Technologies, Coralville, IA) as listed:
human fibroblast growth factor 20:
Forward: 5′-AGAGGAGTAACCGGGCCTTA-3′
Reverse: 5′-GCCATCTCTTGGAGTTCCGT-3′
human fibronectin
Forward: 5′-ACAAGCATGTCTCTCTGCCA-3′
Reverse: 5′-TTTGCATCTTGGTTGGCTGC-3′
human β-actin
Forward: 5′-CTCGCCTTTGCCGATCC-3′
Reverse: 5′-GGGGTACTTCAGGGTGAGGA-3′
Angiogenesis assay and analysis
Human umbilical vein endothelial cells (HUVEC; Gibco) were seeded at 2.5 × 104 viable cells per well in 24-well plates on 100 μL Geltrex® Reduced Growth Factor Basement Membrane medium that was cured in 37°C for 30 min before HUVEC seeding. CM or exosome-rich EVs were applied at the time of HUVEC seeding. For media studies, 50% conditioned media (CM or Wnt3a CM) was mixed with Medium 200 with Large Vessel Endothelial Supplement (LVES) (Life Technologies). For EV studies, vehicle concentration was kept constant across wells containing 1 μg/mL of EVs in all treatment conditions (<10% by volume) diluted in Medium 200 with LVES. After 24 h, tube formation occurred, and ten images were taken per well of triplicates per condition at 4× in different parts of well using IX81 Olympus microscope with ORCA-AG Hamamatsu digital camera. Images were uploaded into Image J and analyzed with Angiogenesis Analyzer.txt Macro using algorithms to identify total length, nodes, and junctions per image.
Flow cytometry
Exosome-rich EVs in CM or purified exosome-rich EVs (as described below) were bound to Dynabeads® coated with anti-human CD63 antibodies according to manufacturer's instructions (Invitrogen™, Life Technologies™). In brief, EV samples were prepared as above and incubated with the anti-CD63 bead complexes, mixing overnight in 4°C (1 μg purified EVs added to 20 μL anti-CD63 beads solution). After a series of washes with 0.1% BSA in PBS, anti-CD63-beads-exosome complexes (from CM or purified EVs) were stained with PE-conjugated anti-human Wnt3a antibody (R&D Systems), applied as 10 μL (of 25 μg/mL stock solutions) per reaction to label exosomes in polypropylene tubes in and protected from light for 60 min on a shaker at 1,000 rpm.
Samples were washed with 300 μL 0.1% BSA in PBS, pulled down with Easy-Sep™ (StemCell Technologies) separator, and washed once more. Samples were resuspended in 200 μL of PBS and detected with CytoFlex S flow cytometer (Beckman Coulter). Data were analyzed with FlowJo software (FlowJo LLC, Ashland, OR). To test the effects of CD63 depletion from the media or EVs, the supernatant was saved for testing.
EV purification
Exosome-rich EVs were harvested using differential centrifugation or modifications as described below for enrichment. All purification steps were performed in 4°C cold room or centrifuge kept at 4°C. BM-MSC CM was centrifuged for 5 min at 500 g to remove cells and large debris; supernatant was transferred to new tubes and centrifuged 2,000 g for 10 min to remove small cell debris and larger vesicles; supernatant was switched to 14 × 95 mm sterile polypropylene tubes (Beckman Coulter, Brea, CA). Media were centrifuged at 10,000 g for 30 min to deplete larger microvesicles using SW 40 Ti swinging bucket rotor (Beckman Coulter) in Optima™ L-100 XP Ultracentrifuge (Beckman Coulter). Resulting supernatant was divided into two equal parts by volume to be mixed with 16% (2 × ) PEG6000 at ratio 1:1 for a final concentration of 8% (1 × ) PEG, which has been shown to be an optimal method to enrich EVs [17]. The aforementioned 16% (2 × ) PEG solution was prepared with PEG with Mn (number of average molecular weight) of 6,000 (Sigma, St. Louis, MO) combined with filtered, pure distilled water and sodium chloride (1 M) to make a 2× concentrated stock solution (final concentrations of PEG when added to CM was 8% and sodium chloride was 0.5 M, kept constant in all mixtures). Once combined, CM+PEG was incubated overnight at 4°C on rocker in sterile centrifuge tubes. Next day, the CM+PEG mixture was transferred to high-performance 50 mL centrifuge tubes centrifuged at 10,000 g at 4°C for 60 min using FiberLite fixed-angle rotor, model F15-8 x 50C (Piramon Technologies, Inc.) in Heraeus Multifuge 3SR+ centrifuge (Thermo Scientific). The canonical tubes were decanted and allowed to drain for 5 min.
The pellet was resuspended in sterile PBS and vortexed for 5 s. PEG-enriched EVs in PBS were processed along with non-PEG-treated CM for further purification (starting volumes of all groups kept equal). Resulting EV-containing solutions were transferred to new 14 × 95 mm sterile polypropylene tubes and centrifuged at 100,000 g into 4 mL of 30% sucrose cushion/D2O (as described [17,18]) per tube for 180 min using the SW 40 Ti swinging bucket rotor in Optima L-100 XP Ultracentrifuge (Beckman Coulter). An 18-guage needle was used to puncture the tube near the bottom to remove 3.5 mL of the sucrose cushion (per tube) containing total population of floating EVs. Cushions were divided into three equal volumes (∼1.16 mL) into new 14 × 95 mm sterile polypropylene tubes and diluted with 11 mL of sterile PBS and centrifuged again at 100,000 g in 4°C for 120 min using the SW 40 Ti swinging bucket rotor in Optima L-100 XP Ultracentrifuge.
The final pellets were resuspended in 100 μL per tube, vortexed for 5 s, and allowed to resuspend in sterile PBS overnight in 4°C. The next day, pellets were vortexed for 5 s and combined in sterile microcentrifuge tubes for further use and testing.
Transmission electron microscopy of exosomes
Exosomes were loaded as 5–10 μL of sample onto a Formar/carbon-coated copper grid for 30 min and cover was applied. The grid was rinsed by floating it on top of a drop of 0.1 M PO4 buffer for 5 min, 2% glutaraldehyde fixative for 5 min, 0.1 M PO4 buffer for 5 min, and double-distilled water for 5 min and repeated. The grid was then floated on top of a drop of 4% uranyl acetate in double-distilled water for 5 min and covered for darkness. The grid was drained by gently touching the side on a piece of filter paper to wick off the excess stain and let dry, covered. Once grid was completely dry, it was transferred to a grid box. Images were acquired with JEOL JEM-1400 Transmission Electron Microscope (Peabody, MA) equipped with a Gatan Orius SC 200D digital camera. University of Miami TEM imaging core acquired all TEM images of EVs.
Nanoparticle tracking analysis of EVs
Purified exosome-rich EVs in PBS were diluted 1:1,000 in 1,000 μL PBS before testing in the NanoSight NS300 (Malvern Instruments, Malvern, United Kingdom). Videos of nanoparticles were captured via NanoSight hardware and visualized via NanoSight software. Sample images were captured from video files. NanoSight software generated particle histograms and intensity plots. Particle concentration, particle size, and particle mode was measured for each sample with NanoSight software. Particle concentrations were correlated with sample protein concentration as measured by Micro BCA™ Protein Assay Kit (Pierce Biotechnology, Thermo Scientific).
Fluorescence staining of EVs
Purified EVs were incubated with Vybrant DiO (Molecular Probes) (1 μL of DiO dye to 100 μL of EVs in PBS) and incubated in 37°C for 20 min. Solution was transferred to new 14 × 95 mm sterile polypropylene tubes and diluted with 11 mL of sterile PBS and centrifuged at 4°C for 120 min using the SW 40 Ti swinging bucket rotor in Optima L-100 XP Ultracentrifuge. Stained EVs were resuspended in 100 μL PBS and added to fibroblasts or endothelial cells for 4 h. Cell nuclei were visualized via NucBlue (Thermo Fisher).
Small interfering RNA
SignalSilence® siControl or siLRP6 (Cell Signaling Technology, Danvers, MA) was used according to manufacturer's recommendations, at concentration of 100 nM with Lipofectamine® RNAiMAX Transfection Reagent, according to manufacturer's guidelines (Thermo Scientific, Waltham, MA), 36–48 h before treatment with CM or EVs, in the assays above.
Immunoblotting
Samples were lysed with 1× RIPA buffer and were loaded as 1 μg total protein (or otherwise indicated) in Laemmli buffer with 5% β-mercaptoethanol (Sigma). EV samples were lysed with 1× RIPA and total proteins were diluted 1:10 in PBS, quantified by Micro BCA kit according to manufacturer's instructions (Thermo Scientific). Gels were run in 10% SDS-PAGE gels (BioRad). Proteins were transferred to nitrocellulose membranes (BioRad). Membranes were blocked with 5% milk in TBST. Primary antibodies (1:1,000 dilution in 5% milk in TBST) included the following: polyclonal rabbit anti-Wnt3a, LRP6, nonphospho-betacatenin, and actin (Cell Signaling Technology); monoclonal rat anti-human Wnt3a (R&D) anti-CD63 (System Biosciences) and developed with secondary antibodies (HRP-conjugated goat anti-rabbit at 1:10,000 in 5% milk in TBST). Images were acquired with SuperSignal (Thermo Scientific) according to manufacturer's instructions and imaged with Amersham Imager (GE Sciences, Pittsburgh, PA).
Statistics
Statistical analysis was performed using Graph Pad Prism® Software Version 7, using Student's t-test (two groups) or ANOVA (three or more groups) with post hoc comparisons among treatment groups, with significance noted if P < 0.05.
Results
BM-MSC Wnt3a CM stimulates dermal fibroblast proliferation, migration, and angiogenesis in vitro
BM-MSCs were harvested from a healthy male donor (Fig. 1a for schematic and methods). Two separate BM-MSC cell lines were obtained commercially for testing (see Materials and Methods section). We introduced an expression vector for Wnt3a using a method in which typically 50%–60% of BM-MSCs can be transiently transfected. Immunoblotting revealed higher levels of Wnt3a in serum-free BM-MSC Wnt3a CM versus control BM-MSC CM (Supplementary Fig. S1a; Supplementary Data are available online at
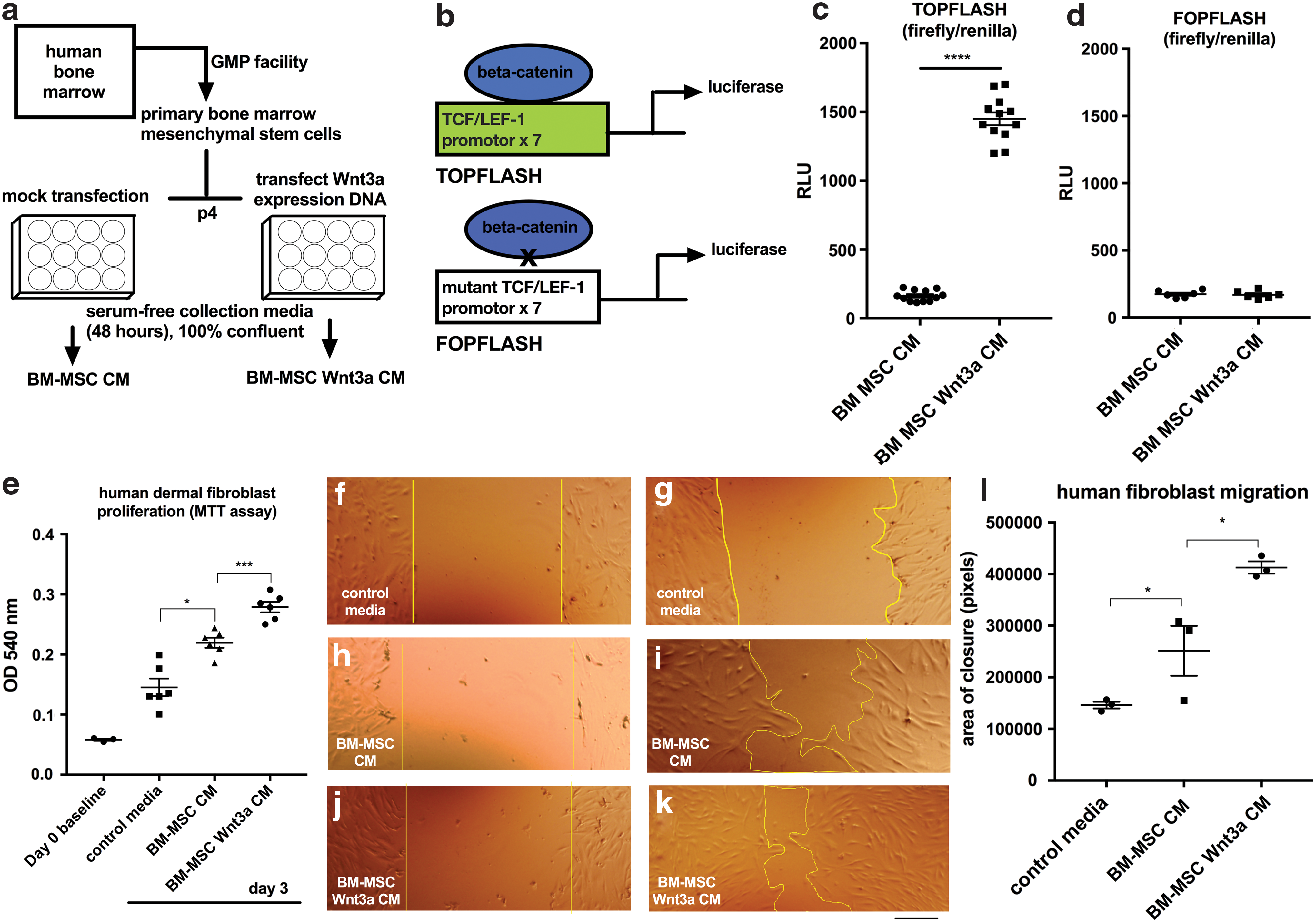
BM-MSC Wnt3a-conditioned medium stimulated dermal fibroblast proliferation and migration.
Compared to BM-MSC CM in serum-free media as control, BM-MSC Wnt3a CM increased human dermal fibroblast proliferation after 72 h (Fig. 1e). In a scratch assay (in which cell proliferation was inhibited with mitomycin in serum-free media), BM-MSC Wnt3a CM stimulated dermal fibroblast migration (Fig. 1f–k). Dermal fibroblast treatment with BM-MSC Wnt3a CM was correlated with increased fibroblast growth factor 20 and fibronectin expression, which are direct TCF/LEF-driven target genes (Supplementary Fig. S1b). We next studied the effects of BM-MSC Wnt3a CM versus control BM-MSC CM on endothelial network formation in vitro. BM-MSC Wnt3a CM significantly stimulated tube length formation, and formation of branch points among the network of endothelial cells (nodes = 2 segments joining; junctions = 3 segments joining) after 24 h of treatment (Fig. 2a–i).
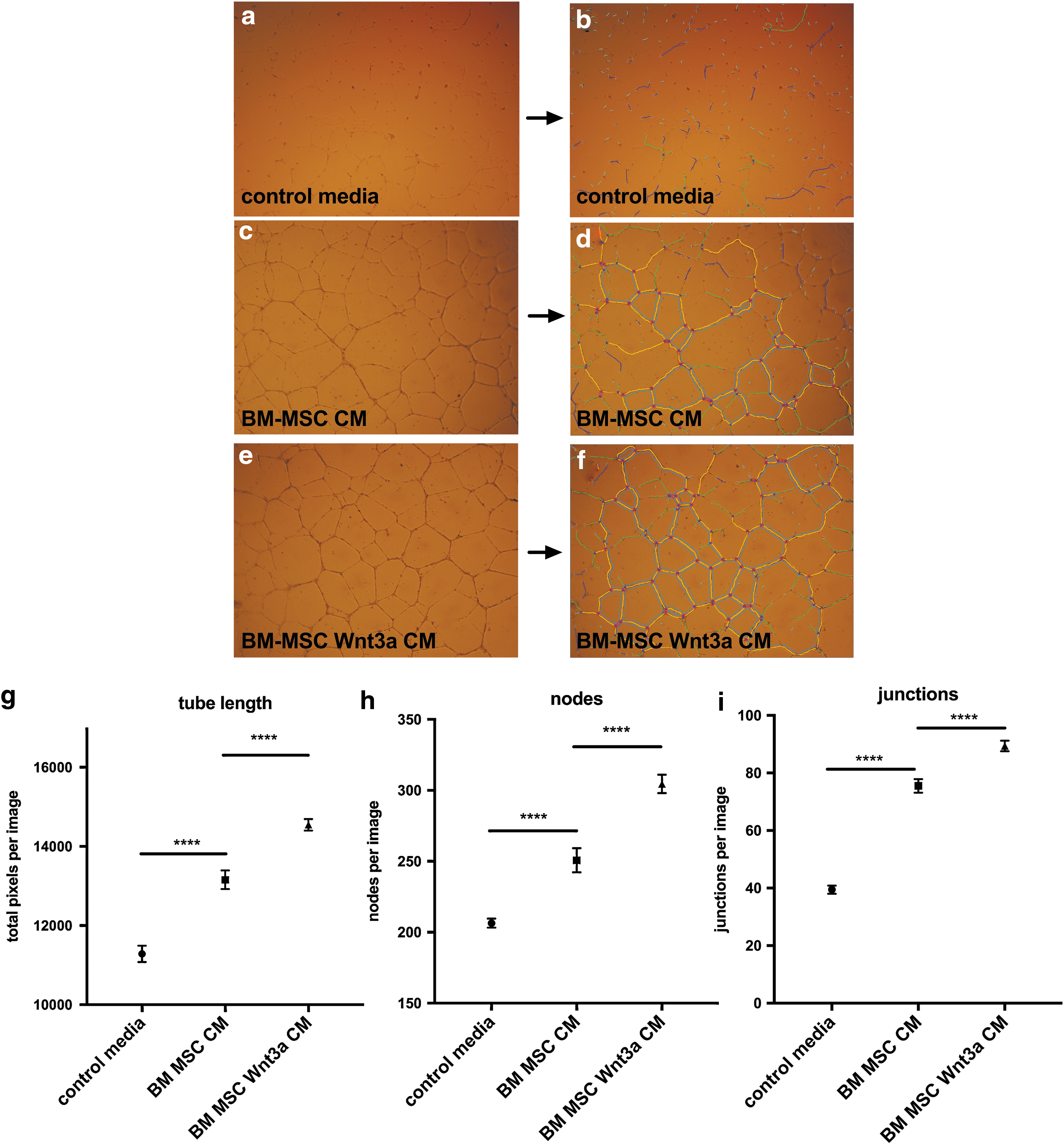
BM-MSC Wnt3a-conditioned medium increases formation of endothelial cell tube length, nodes, and junctions.
Wnt3a is detected on bead-CD63+-exosome complexes
It was specifically unknown whether any Wnt3a travels exteriorly on CD63+ exosomes of BM-MSCs. Due to the hydrophobicity of Wnt3a, we hypothesized that some of Wnt3a overexpressed by BM-MSC in CM might be associated with bilayered lipid-based exosomes exteriorly. Tetraspanin CD63 is the most commonly associated marker of exosome EVs. It was recently shown that Wnts can be detected on exosomes of drosophila cells and HEK293T cells using immunogold staining [19]. It was unclear whether BM-MSC Wnt3a is associated with BM-MSC CD63+ EVs and whether it was accessible on the exterior of the EV for antibody binding for detection in flow cytometry assays. Thus, we targeted tetraspanin CD63-EVs with magnetic beads linked to anti-CD63 antibodies known to bind to exosomes. We detected Wnt3a using PE-anti-human-Wnt3a as fluorescent signals in flow cytometry. Compared to control unstained beads and BM-MSC CM, BM-MSC Wnt3a CM contained beads highly fluorescent with PE-anti-Wnt3a signals in both bead complexes 1 (bead singlets) and 2 (bead doublets) (Fig. 3 and Supplementary Fig. S2a–d). These data suggest that some of the Wnt3a overexpressed by BM-MSCs in CM was bound exteriorly to CD63+ exosomes.
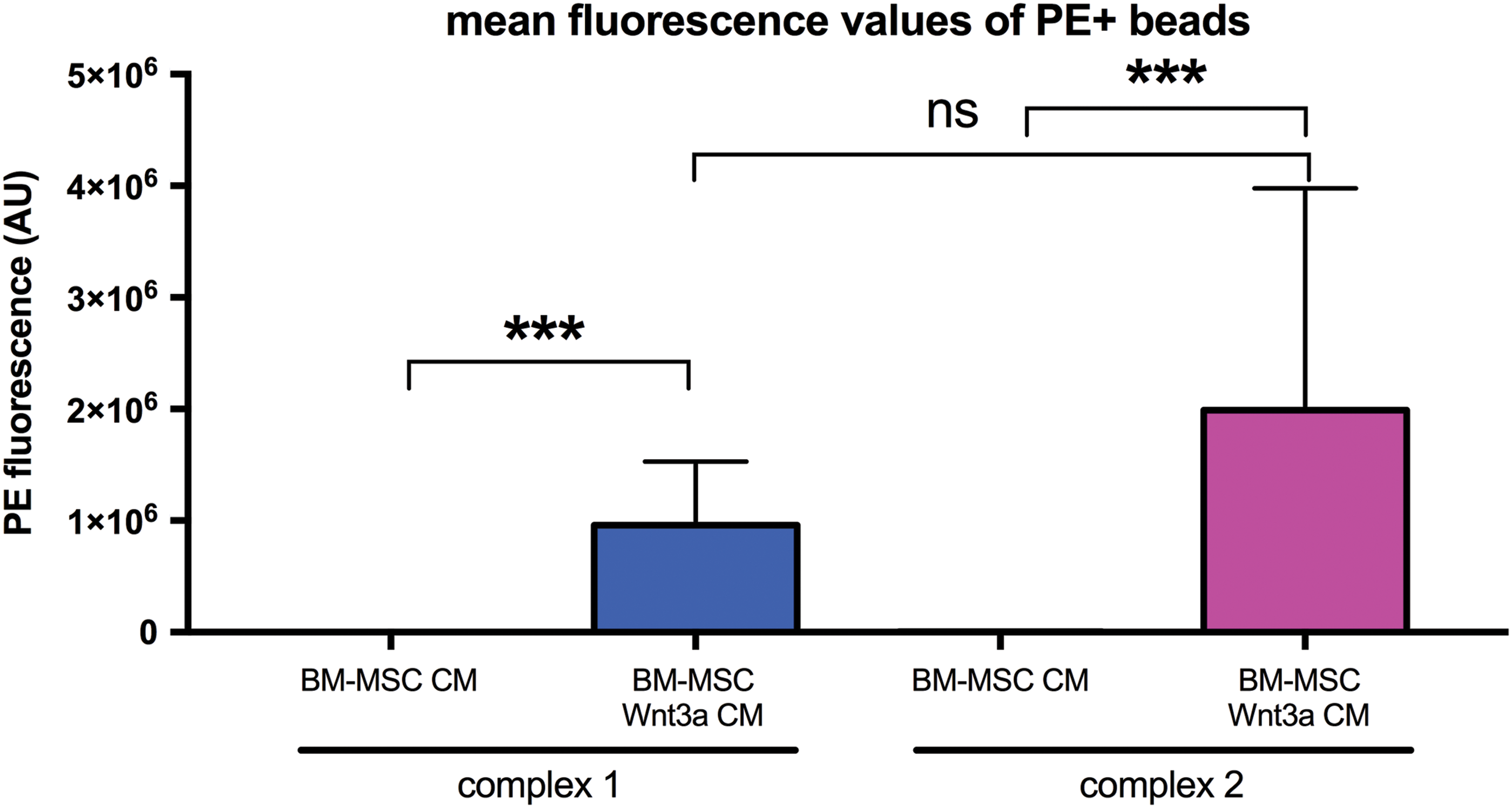
BM-MSC Wnt3a CM incubated with anti-CD63 bead-exosome complexes contained higher PE-anti-Wnt3a fluorescence signal. Mean fluorescence values of PE (AU = arbitrary units) on anti-CD63-antibody-coated magnetic beads stained with PE-anti-human Wnt3a antibody. Mean ± SEM. ***P < 0.001, ns = not significant. Flow cytometry data representative of triplicates per group. Color images available online at
Isolation and characterization of BM-MSC-secreted EVs
We sought to determine whether EV-Wnt3a from BM-MSCs could be purified with activity. EV purification methods can differentially affect EV content and downstream activity. It was recently shown PEG can enrich EVs [17], but its effect on harvesting Wnt activity was unknown until our study. We examined whether using PEG6000 to enrich EVs before purification into a sucrose cushion harvested more Wnt-EV activity per unit mass compared to traditional ultracentrifugation alone. We isolated EVs from BM-MSC CM and BM-MSC Wnt3a CM using differential ultracentrifugation combined with purification of vesicles using a sucrose cushion (UC-S) (Supplementary Fig. S3a). Additionally, using the same samples and equal starting volumes, we simultaneously purified EVs from BM-MSC CM and BM-MSC Wnt3a CM with polyethylene glycol (PEG6000) [17] to help precipitate and separate vesicles from aqueous media before ultracentrifugation. We chose 8% PEG6000 as this was the optimal PEG to enrich for exosomal markers in a previous study. The PEG-EV pellets were resuspended in PBS and further purified using the sucrose cushion method (PEG-UC-S) (Supplementary Fig. S3b, methods). In the various EV populations, there were no consistent differences in particle morphology as visualized by TEM, with most particles appearing spherical or ovoid, sometimes with a cup-shaped center, and with thin membrane bilayers with diameters under 100 nm (Supplementary Fig. S3c–j), verifying that most of the EVs in the populations were exosome-sized. Larger EVs, which were not numerous in sucrose cushion EV preparations, could be visualized using lipid staining fluorescent dye and microscopy, but most of the smaller particles were less visible with dyes. NanoSight nanoparticle tracking analysis revealed sphere-shaped vesicles (Supplementary Fig. S3k–n). We examined particle concentrations and size distributions across samples. Particle distribution curves were similar among preparations, (Supplementary Fig. 3o–r), and there were no significant differences in variations among particles per population when normalized for mass (per μg) (Fig. 4a), mean particle sizes (Fig. 4b), or particle modes (Fig. 4c) among samples and purification methods. We estimated that 100–200 particles were secreted into the serum-free media per cell, per hour (data not shown). All preparations contained the tetraspanin CD63 and no differences in CD63 content among populations were observable (Supplementary Fig. S4).
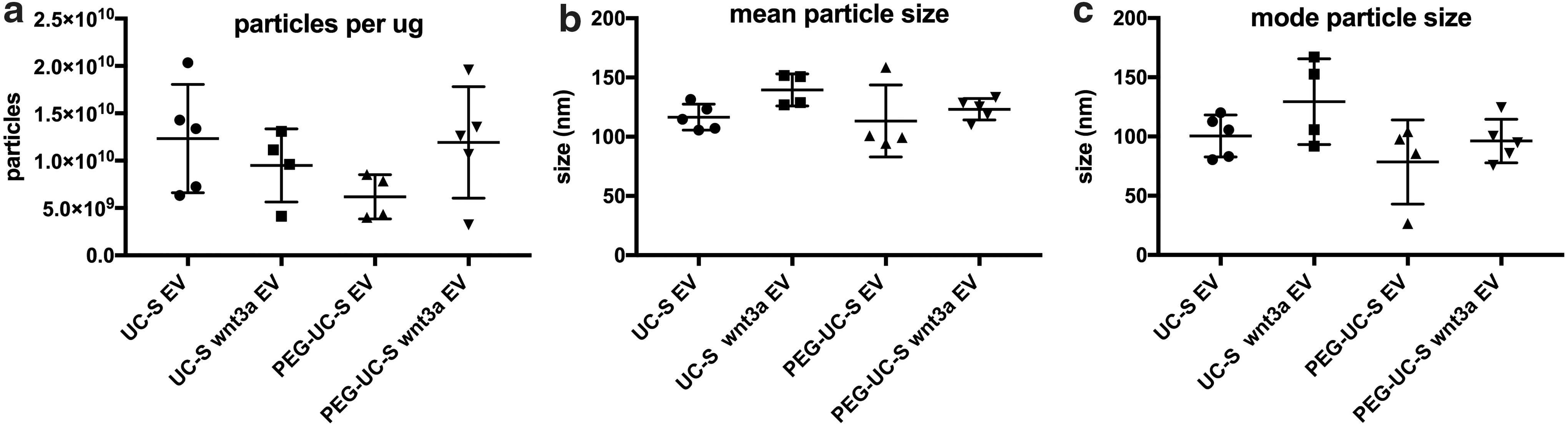
Exosome-rich EVs purified from BM-MSC CM and BM-MSC Wnt3a CM had similar particle morphologies and size distributions.
We next harvested the exosome-rich populations to test in Wnt-TCF/LEF-beta-catenin activity assays to determine the lowest concentration necessary to activate Wnt signaling. PEG-mediated exosome enrichment before ultracentrifugation resulted in a higher ratio of detectable Wnt3a ligand relative to detected CD63, as detected in immunoblotting with both monoclonal (Supplementary Fig. S4a) and polyclonal (Supplementary Fig. S4b) antibodies against human Wnt3a. BM-MSC Wnt3a CM EVs (purified through sucrose cushion) at 1.0 μg/mL resulted in a higher probability to activate TCF/LEF-driven luciferase activity (TOPFLASH), ranging up to nearly eight-fold activation in HEK293T-based transcription assays at the dose of 1.0 μg/mL, compared with vehicle alone, while 0.1 μg/mL doses and 1.0 μg/mL of other EV preparations were not as likely to induce TOPFLASH activity compared to vehicle (Fig. 5a). All EV preparations did not stimulate the negative control for TCF/LEF-driven luciferase activity (FOPFLASH) (Fig. 5b). It has been established that exosomes can interact with cells via direct fusion, endocytosis, or macropinocytosis [20]. We recently showed fibroblasts and endothelial cells can uptake exosomes via immunofluorescence [21]. Here, we visualized vesicle-membrane co-localization and confirmed recipient cells contain increased CD63 content after exosome treatment (Supplementary Fig. S5a–d).
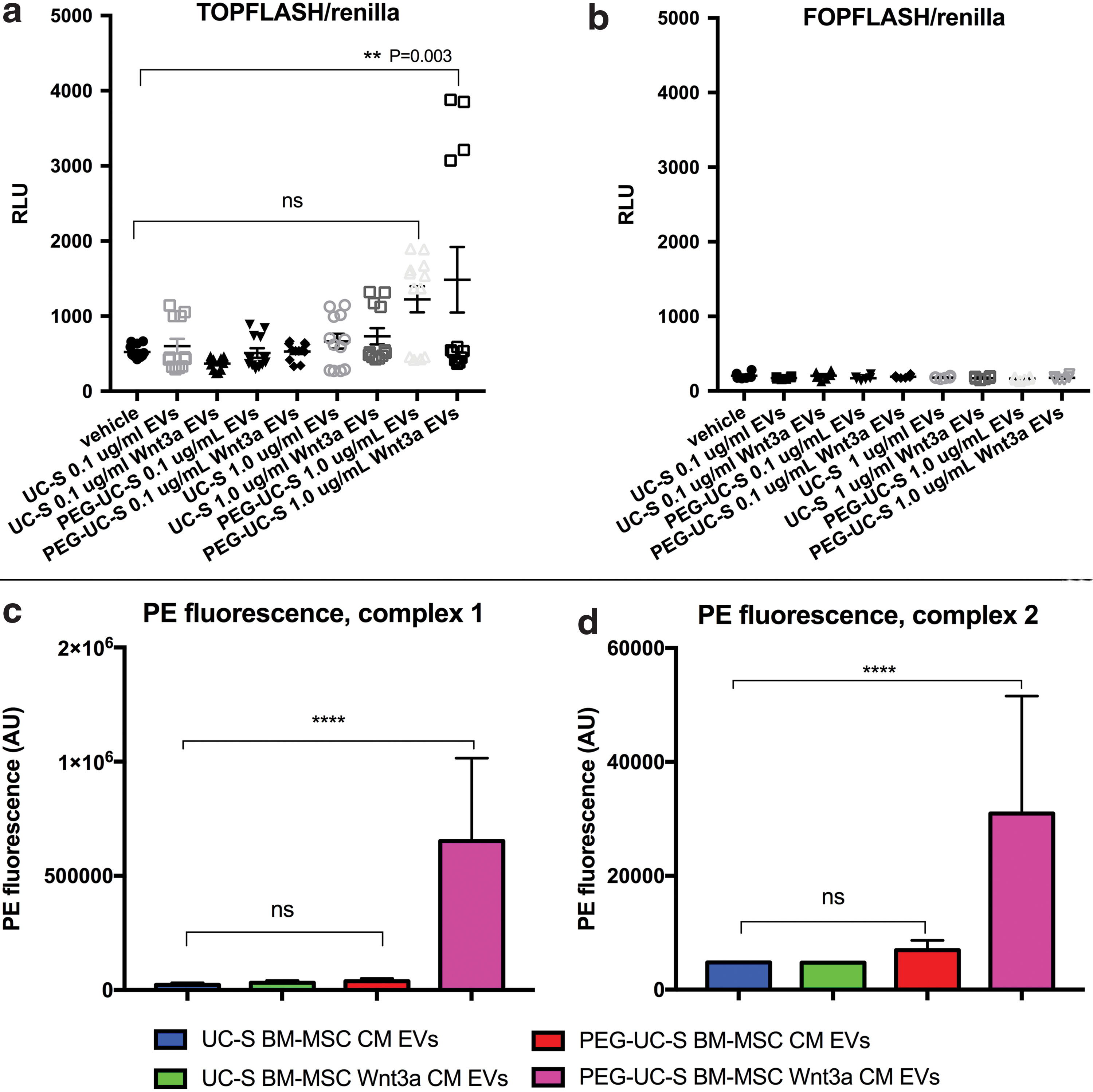
Detection of Wnt signaling activity on exosome-rich EV populations purified from BM-MSC media.
In flow cytometry assays, PE-anti-Wnt3a was detected at higher average fluorescence levels on magnetic beads targeting human CD63 mixed with exosome-rich populations from BM-MSC Wnt3a CM purified using PEG-EV-enrichment purified through a sucrose cushion (PEG-UC-S) (Fig. 5c, d and Supplementary Fig. S6a–c; quantification of additional BM-MSC lines in Supplementary Fig. S6d, e). Interestingly, UC-S alone did not result in EVs with significant Wnt detection from the BM-MSC Wnt3a CM. Thus, because sucrose cushion requires floating vesicles, our results suggest purification of EVs from BM-MSC Wnt3a CM using PEG-UC-S method was more likely to harvest EVs with Wnt3a-activity physically tethered to the EVs. We did not detect an increase in internalized Wnt3a after treatment in recipient lysates, suggesting any internalized Wnt3a in the nonenriched cell lysates were below the level of detectability on western blot (data not shown). Although less sensitive than the TOPFLASH assay in our system, we detected an increase in nonphospho-beta-catenin (active beta-catenin) after treatment with Wnt3a-enriched EVs (Supplementary Fig. S7a, b). Thus, Wnt3a-enriched EVs are associated with increased ability to induce Wnt/TCF/LEF-beta-catenin activity in recipient cells.
BM-MSC Wnt3a CM EVs containing the most Wnt3a exteriorly stimulated dermal fibroblast proliferation, migration, and endothelial angiogenesis, dependent on Wnt signaling co-receptor LRP6 in recipient cells and CD63+ exosomes in CM and EV populations
Once we identified the exosome-rich population that contained the most detectable exterior Wnt3a (PEG-UC-S), we tested them in the previous surrogate assays of dermis repair. Compared to vehicle, EVs purified from BM-MSC Wnt3a CM using PEG-UC-S significantly stimulated dermal fibroblast proliferation (Fig. 6a) and migration (Fig. 6b), while the other EV preparations at the same dose did not stimulate dermal fibroblast proliferation and migration to the same extent over 72 h (Fig. 6a, b). In angiogenesis assays, compared to vehicle, all EV fractions significantly stimulated tube length formation, however, treatment with the PEG-UC-S Wnt3a EV population exhibited the largest differences versus vehicle in all three parameters of angiogenesis in vitro (Fig. 6c–f). Thus, compared to vehicle and other preparations, the PEG-UC-S Wnt3a-exosome-rich EV population demonstrated the most activity in surrogate assays of dermis repair.
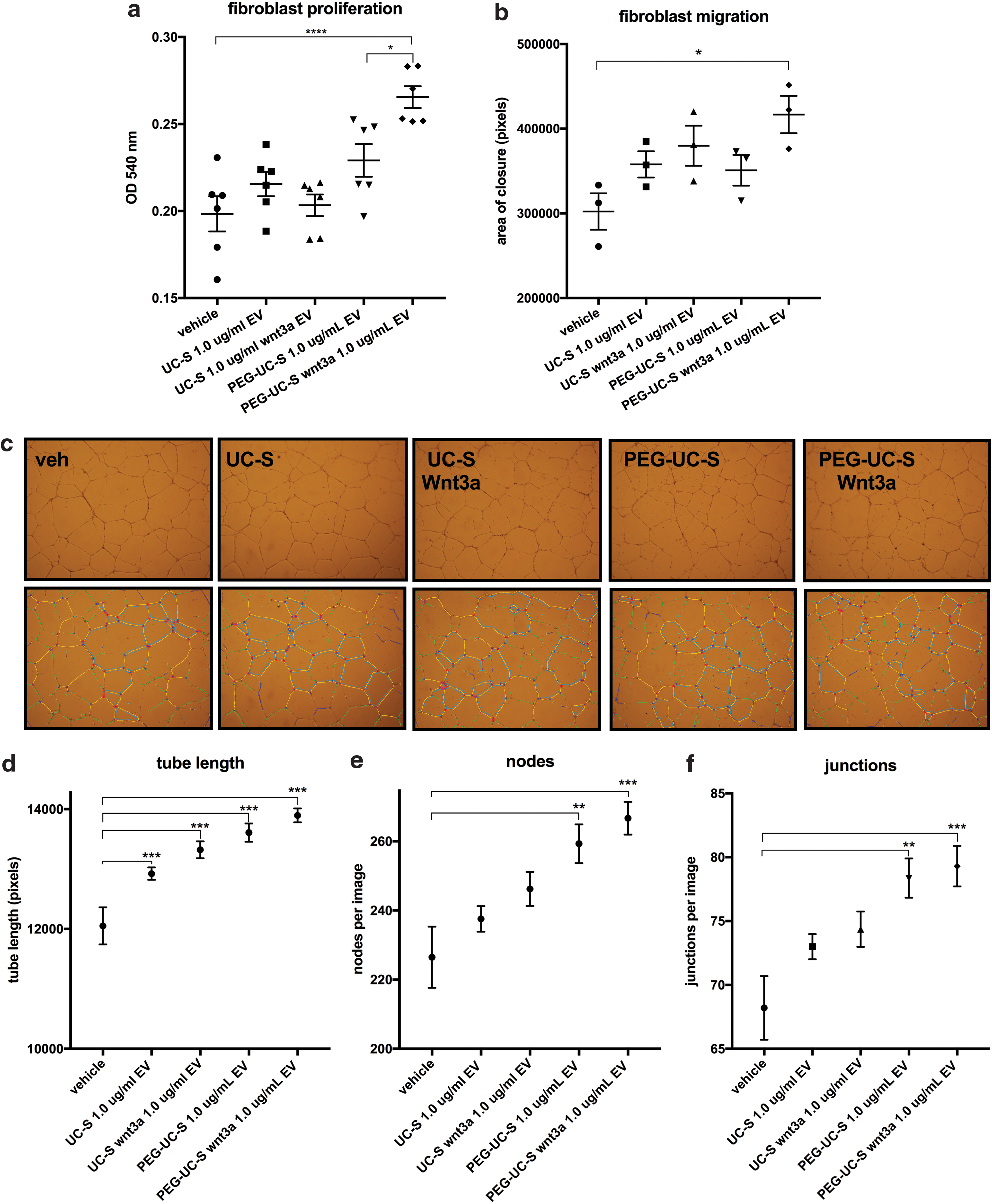
Treatment of dermal fibroblasts and endothelial cells with PEG-isolated exosome-rich EVs from BM-MSC Wnt3a CM was associated with higher fibroblast proliferation, migration, and angiogenesis in vitro.
These vesicles were highly likely to stimulate these assays through canonical Wnt signaling, because when LRP6 was knocked down in recipient cells (Supplementary Fig. S8b, c) or when CD63+ vesicles were depleted from the BM-MSC media or EVs (using the magnetic beads coated with anti-CD63 antibodies to remove the exosomes), the ability of both the BM-MSC Wnt3a CM and PEG-UC-S Wnt3a EVs to stimulate TOPFLASH activity (Fig. 7a–d and Supplementary Fig. S8d–g), dermal fibroblast proliferation (Fig. 7e–h and Supplementary Fig. S8 h–k), migration (Fig. 7i–l and Supplementary Fig. S8l–p), and angiogenesis in vitro (nodes, junctions, and tube length, Fig. 8a–e and Supplementary Fig. S9a–l) was significantly attenuated or completely blocked. Thus, CD63+ exosomes from donor BM-MSCs and LRP6 in recipient cells appear to be required for optimal canonical Wnt activation and functional effects in recipient cells.
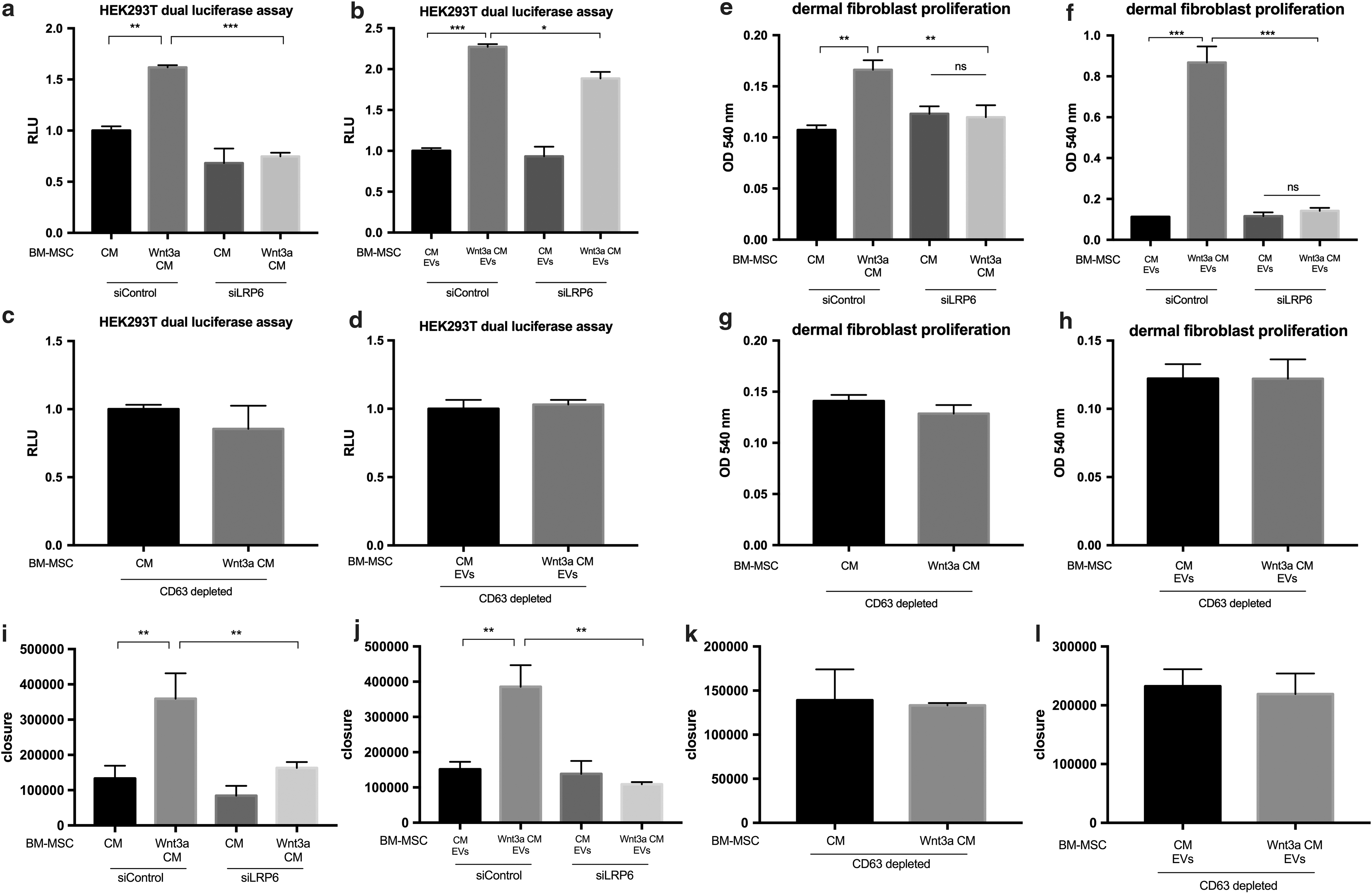
Knockdown of Wnt co-receptor LRP6 in recipient cells and depletion of CD63+ exosomes from BM-MSC CM and EV populations separately attenuate dermal fibroblast proliferation and migration induced by BM-MSC Wnt3a CM and Wnt3a-enriched-EVs.

Knockdown of Wnt co-receptor LRP6 in recipient cells and depletion of CD63+ exosomes from BM-MSC CM and EV populations attenuate endothelial angiogenesis in vitro induced by BM-MSC Wnt3a CM and EVs.
Discussion
The effects of the various Wnt ligands on functions of different skin cell types are incompletely understood. Furthermore, it is unclear how such lipophilic Wnt ligands travel from donor stem cells to recipient skin cells. We undertook this study to determine whether BM-MSC Wnt3a CM has stimulatory effects on human fibroblasts and endothelial cells and whether Wnt3a is associated exteriorly with CD63+ exosomes, at least, in serum-free conditioned media. Exterior positioning of Wnt3a would allow for easier access with Frizzled receptors and Wnt co-receptors on the surface of recipient cells upon vesicle-membrane fusion.
Taken together, our results suggest Wnt3a-loaded exosomes stimulate fibroblast proliferation, migration, and angiogenesis in vitro and that some Wnt3a is physically tethered exteriorly to BM-MSC CD63+ exosomes after purification of floating vesicles from a sucrose cushion, emphasizing an under-recognized mechanism for Wnt signaling between BM-MSCs and dermal fibroblasts and endothelial cells. Because Wnt3a is lipophilic due to its posttranslational modification of palmitoylation in the endoplasmic reticulum of cells, we hypothesized that some Wnt3a might associate with EVs manufactured by BM-MSCs, resulting in Wnt3a stability, delivery, and activity. We provide evidence that CD63+ exosomes, at least, represent one of the major EV populations on which Wnts can travel from BM-MSCs to fibroblasts and endothelial cells.
There are 19 separate Wnt ligands, all of which may have different functions in repair and regeneration and are implicated as important factors secreted by stem cells for both self-renewal and differentiation [22]. Wnt members should be studied individually (ex. Wnt3a, vs. Wnt4, vs. Wnt5a) to determine their effects on various cell types. “Canonical” Wnt/β-catenin signaling has been implicated in modulating adult cells in normal physiology and disease. In mammals, only certain tissues with high cell turnover have maintained Wnt/β-catenin pathway activation, such as the epidermis, intestinal epithelium, bone marrow, and stem cell compartments. During development, the Wnt pathway may promote fibroblast cell proliferation [23] and, during wound healing, endothelial angiogenesis [5]. Furthermore, the Wnt pathway may stimulate cutaneous differentiation and epithelialization [5,24,25]. Prolonged and unchecked Wnt signaling might promote a scarring phenotype if fibroblasts over-proliferate with excessive collagen I production [26]. Furthermore, lithium chloride, an activator of β-catenin stabilization via inhibition of GSK-3β, stimulates angiogenesis in the wound bed during the proliferation phase and can promote cutaneous wound healing [5,27], but can also inhibit wound closure of keratinocytes directly or if cells over-proliferate so much as to prevent keratinocyte coverage of the underlying wound bed [28]. There is likely a delicate balance with Wnt signaling that must be struck for optimal wound healing in various cell types. Wnt3a-loaded exosomes have potential to stimulate pro-repair functions in dermal fibroblasts and endothelial cells.
Although Wnt ligands have recently been suggested as possible EV cargo members interiorly, this concept has not been settled [19,29 –34]. This study was the first to demonstrate that primary human BM-MSCs can secrete Wnt3a that associated exteriorly with exosomes. Unlike most extracellular signaling proteins, Wnts are modified by the addition of a lipid (palmitic acid) during biosynthesis by the acyltransferase porcupine, affecting Wnt protein solubility and diffusion in the extracellular space [31].
Previously, it was only thought that Wnt proteins associate with Wntless/Evi carrier protein to be escorted to plasma membrane to be secreted freely [35]. However, Gross et al. reported the concept of active Wnts traveling on exosomes of HEK293T cells overexpressing Wnt in Drosophila cells; since then, both positive and negative results have been reported while looking for Wnt activity associated with EVs from various cell types, resulting in confusion as to whether they exist on or in EVs [31,36 –38]. Here, we present evidence that Wnt3a exists on CD63+ exosomes exteriorly, and purification methods can affect Wnt-exosome activity. Specifically, our study demonstrated that the chosen method of purification could affect whether or not exterior Wnt3a detection and activity was significantly retained. The bond between Wnt3a and the exosome may be compromised during the ultracentrifugation force of 100,000 g alone; however, PEG enrichment may increase the chance of isolating Wnt3a-bound exosomes due to increased Wnt3a-exosome membrane stability or increased numbers of Wnt3a-loaded EVs before ultracentrifugation, however, this hypothesis remains for future in-depth study.
Gross et al. estimated that ∼20%–40% of active Wnts/Wingless may be associated with EVs [19,30], however, it is unclear whether, and to what extent, exosome-dependent and free (exosome independent) Wnts are functionally different. Tools to perturb exosome formation and secretion (including interference with MVB formation or RAB and SNARE function) affects cellular health and other signaling pathways [31], thus, making it difficult to determine, in that way, to what extent Wnts depend on exosomes for beneficial effects of BM-MSC Wnt3a CM in the surrogate assays presented. Our study found CD63+ exosome depletion significantly attenuates the activity of BM-MSC Wnt3a CM. Thus, the functional activity of BM-MSC Wnt3a CM relied heavily on CD63+ vesicles. Furthermore, in the presence of siLRP6, BM-MSC Wnt3a CM significantly lost its stimulatory effects, suggesting the Wnt co-receptor LRP6 is an important hub for signaling between Wnt3a-exosomes and the recipient cells.
In summary, our study highlights the important concept that Wnt3a can exist exteriorly on exosomes of BM-MSCs in conditioned media, and that methods of exosome purification can affect the Wnt3a content of exteriorly loaded exosomes, affecting downstream applications. We found that utilization of PEG-mediated exosome enrichment can enhance the probability of extracting Wnt-loaded exosomes with activity in transcription assays and surrogate assays of dermis repair. Detection via flow cytometry, activity in transcription and functional assays is retained during purification of exosomes into a sucrose cushion via ultracentrifugation, suggesting that at least some Wnt3a is tethered to the exosomes exteriorly during purification. It is likely that more studies are needed to determine the optimal purification methods of Wnt-loaded exosome populations from different cell types to determine future applications in modulation of growth signals in the skin.
Footnotes
Acknowledgments
We would like to thank Dr. Camillo Ricordi and the Diabetes Research Institute for the use of NanoSight nanoparticle tracking analysis. We would like to thanks Dr. Joshua Hare's laboratory and Irene Margitech for use of the ultracentrifuge and BM-MSC lines, Dr. Marc Lippman and Dr. Philip Miller for use of the luminometer, Oliver Umland of the Flow Cytometry Core facility in the University of Miami Diabetic Research Institute, and Vania Almeida for assistance with transmission electron microscopy image acquisition at the University of Miami TEM Core. We thank Dr. Marian Waterman and Addgene for Wnt3a expression vector and Dr. Randall Moon and Addgene for the TOPflash and FOPflash vectors.
Author Disclosure Statement
The authors state no conflicts of interest or competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.