
Abstract
The secretome of human amniotic fluid stem cells (AFSCs) has great potential as a therapeutic agent in regenerative medicine. However, it must be produced in a clinically compliant manner before it can be used in humans. In this study, we developed a means of producing a biologically active secretome from AFSCs that is free of all exogenous molecules. We demonstrate that the full secretome is capable of promoting stem cell proliferation, migration, and protection of cells against senescence. Furthermore, it has significant anti-inflammatory properties. Most importantly, we show that it promotes tissue regeneration in a model of muscle damage. We then demonstrate that the secretome contains extracellular vesicles (EVs) that harbor much, but not all, of the biological activity of the whole secretome. Proteomic characterization of the EV and free secretome fraction shows the presence of numerous molecules specific to each fraction that could be key regulators of tissue regeneration. Intriguingly, we show that the EVs only contain miRNA and not mRNA. This suggests that tissue regeneration in the host is mediated by the action of EVs modifying existing, rather than imposing new, signaling pathways. The EVs harbor significant anti-inflammatory activity as well as promote angiogenesis, the latter may be the mechanistic explanation for their ability to promote muscle regeneration after cardiotoxin injury.
Introduction
S
The notion of a paracrine effect is supported by many studies showing that stem cells secrete a spectrum of proteins, small RNAs, and mRNAs that have biological activity [8 –10]. In this scenario, secreted stem cell factors modulate host cellular signaling pathways, for example, promoting an anti-inflammatory response or by protecting against senescence [4,11,12]. A number of studies have shown that factors released by stem cells are packaged in a variety of phospholipid membrane-bound extracellular vesicles (EVs), including microvesicles and exosomes [10,13]. Several species of EVs, such as exosomes, are shed constitutively, but more so during cellular stress [14]. Release of EVs from the cell is believed to enable effective long-range signal transduction by protecting the cargo against degradation. Furthermore, it is proposed that molecules on the surface of EVs permit homing to specific tissues, thereby facilitating a degree of target specificity [15].
Numerous studies have focused on identifying an optimal population for stem cell therapies by taking into account differentiation repertoire, ease of isolation, longevity, and any associated ethical issues. One particularly attractive source is amniotic fluid-derived stem cells (AFSCs). These cells support the regeneration of bone [2], lung [16], kidney [17,18], intestine [4], heart [19], and skeletal muscle [20,21]. They are easily isolated from human amniotic fluid that is routinely collected during clinical prenatal diagnostic procedures, such as amniocentesis, not associated with any ethical issues. Furthermore, human AFSCs have been shown to be capable of ∼250 population doublings [22]. However, in many reports, regeneration takes place without significant AFSC engraftment, again pointing to a paracrine mechanism of action [4,9].
In this study, we determined whether a human AFSC secretome collected under conditions concordant with clinical translation would support tissue regeneration. Specifically, we wanted to obviate contamination with nonhuman proteins/molecules, including those found in fetal calf serum. We developed conditions that allowed the collection of a secretome that was not only serum free but also induced cellular stress without promoting cell death. In this study, we report that the human AFSC secretome produced in nutrient-free and hypoxic conditions contains a spectrum of proteins and miRNAs. The whole secretome protected against senescence and promoted stem cell proliferation and migration.
Using reporter cell lines, we demonstrate significant anti-inflammatory potential of the human AFSC-conditioned medium (AF-CM). In vivo experiments show that the AF-CM promoted skeletal muscle regeneration. We then isolated and characterized the EV fraction [amniotic fluid stem cell extracellular vesicle (AF-EV)] from AF-CM and showed that it too had anti-inflammatory properties and protects against senescence. We profiled the protein and miRNA cargo of both the EV and non-EV fractions and found that each contained specific entities. Importantly, we show that AF-EVs were able to promote adult mouse muscle regeneration.
Methods
Ethical approval
Human amniotic fluid samples were collected from women undergoing amniocentesis for prenatal diagnosis for therapeutic purposes in the Fetal Medicine Unit of University College London Hospital (UCLH). All patients gave informed written consent to an approved researcher under ethical approval (UCL/UCLH Joint Committee for the Ethics of Human Research, REC Reference: 08/0304). Invasive procedures were performed under ultrasound guidance using sterile equipment. In vivo animal experiments were performed under a project license from the United Kingdom Home Office in agreement with the Animals (Scientific Procedures) Act 1986.
Animal maintenance
Male wild-type C57BL/6 mice (2–3 months old) were maintained in accordance with the Animals (Scientific Procedures) Act 1986 (United Kingdom). Mice were housed under standard environmental conditions (20–22°C, 12-h light–12-h dark cycle) and were provided with food and water ad libitum.
Cell culture
Human AFSCs were harvested from a single amniotic fluid sample based on C-kit expression using MACS (Miltenyi Biotec) following one passage. C-kit+ AFSCs were cultured on nontissue culture-treated plastic in alpha-minimum essential medium (α-MEM) (Gibco, Invitrogen) containing 15% fetal bovine serum (FBS), 1% glutamine, and 1% penicillin/streptomycin (Gibco), supplemented with 18% Chang B and 2% Chang C (Irvine Scientific).
U251 cells were cultured in antibiotic-free Dulbecco's modified Eagle's medium (DMEM; Gibco), 10% FBS, and 1% glutamine.
Human fetal IMR-90 lung fibroblast cells (IMR-90) were a gift from Dr. Debacq-Chainiaux (University of Namur, Belgium). Cells were used at 27–30 population doublings. IMR-90 cells were cultured in α-MEM containing 10% FBS, 1% glutamine, and 1% penicillin/streptomycin.
StemPro® human adipose-derived mesenchymal stem cells (hADMSCs; Life Technologies; lot 2117), derived from a 49-year-old female donor, were from StemPro (lot no. 2117). hADMSCs were cultured in MesenPro RS™ (Gibco), 1%
All cells were cultured at 37°C with 5% CO2 and passaged upon reaching 70% confluency unless otherwise stated.
CM generation and EV isolation
At passage 10, AFSCs were pelleted at 300 g in 1.5-mL microfuge tubes at a density of 1 × 106 cells/tube. Cells were incubated in 400 μL of sterile phosphate-buffered saline (PBS) for 24 h. Following incubation, the cells were pelleted at 300 g for 5 min, and the supernatant was aspirated, pooled, and centrifuged at 2,000 g for 20 min. The supernatant (AF-CM) was collected and stored at −20°C. AF-EVs were isolated using sequential ultracentrifugation of AF-CM at 200,000 g using a 70.1Ti fixed-angle rotor (Beckman Coulter) and resuspending EVs in ultracentrifuged PBS.
Flow cytometry
Expression of surface markers was assessed using monoclonal antibodies against the cluster of differentiation (CD) antigens: anti-human CD44/Alexa Fluor® 647 (Millipore); anti-human CD73/PE (Biolegend; 1:20); anti-human CD90/PE (Millipore; 1:20); anti-human CD34/FITC (Millipore; 1:10); and anti-human CD45/FITC (Biolegend; 1:20).
Post-CM generation, AFSCs were fixed in 4% paraformaldehyde (PFA; Sigma-Aldrich) for 20 min and nonspecific binding blocked with 10% FBS (Gibco) at room temperature (RT). Antibodies were diluted in PBS and incubated with the AFSCs for 1 h at 4°C. Ten thousand events were captured and recorded using the BD Accuri C6 flow cytometer C-sampler. Data were analyzed using FlowJo, v10, analysis software (FlowJo LLC).
Adipogenic differentiation
AFSCs were seeded at 4,000 cells per cm2 and grown to 95% confluency. Growth media were then removed; cells were washed once with PBS and cultured for 21 days with StemXVivo Adipogenic Base Media (R&D Systems) supplemented with StemXVivo Adipogenic Supplement (100 × ) according to the supplier's instructions. Cells were stained for 30 min with Oil Red O and washed with ddH2O.
Osteogenic differentiation
AFSCs were seeded at 4,000 cells per cm2 and grown to 95% confluency. Growth media were then removed; cells were washed once with PBS and cultured for 21 days with StemXVivo Osteogenic Base Media (R&D Systems) supplemented with StemPro Osteocyte/Chondrocyte basal medium (Life Technologies) according to the supplier's instructions. Calcium staining was carried out for 30 min using Alizarin Red S and washed with ddH2O before capturing images.
Chondrogenic differentiation
For chondrogenic differentiation, 1 × 106 AFSCs were pelleted at 300 g. Cell pellets were cultured in StemPro Osteocyte/Chondrocyte basal medium (Life Technologies) supplemented with StemPro Chondrogenesis supplement (Life Technologies) according to the supplier's instructions. After 21 days, cells were fixed for 15 min in 4% PFA/PBS and washed twice with ddH2O. Chondrogenic cultures were stained for 1 h with Alcian blue at RT. Cell pellets were washed twice for 1 h in 6:4 ethanol/acetic acid destain solution. Pellets were washed once in ddH2O before freezing and blocking in optimal cutting temperature media (OCT). Ten-micrometer cryosections were taken using a Bright OT5000 cryostat. Cells and pellet sections were imaged using a bright-field microscope.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis and silver staining
Protein concentrations in the CM were determined using the Bio-Rad protein assay (Bio-Rad). CM protein samples were denatured at 100°C then loaded onto precast NuPAGE® 4%–12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE; Novex) gels and run at 100 V for 3 h. Silver staining was carried out using the SilverXpress® silver staining kit (Invitrogen). Protein bands were imaged using Syngene G:Box and GeneSys software.
Cellular proliferation assay
hADMSCs were transferred into 96-well culture plates at a density of 400 cells/well and allowed to adhere for 24 h. Subsequently, media were removed before replacing with corresponding treatments. Cells were treated for 48 h, then washed with PBS, fixed with 4% PFA/PBS for 15 min, and then washed again in PBS. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) and counted using a fluorescence microscope (Zeiss A1).
Migration assay
hADMSCs were cultured to reach 100% confluency in 24-well plates. A single scratch was created along the diameter of the well with a p200 tip. Media were then aspirated and cells washed once with PBS before treating with their corresponding medium/CM treatments. Plates were immediately positioned onto a time-lapse microscope and images were captured at three areas of the wound. Total wound coverage was measured using ImageJ software at T = 0 h and T = 9 h.
Cellular senescence assay
IMR-90 cells were grown in 12-well plates until 70% confluency. Media were removed and replaced with media containing AF-CM and left for a further 24 h. Media were removed and cells were stressed for 2 h with 100 μM H2O2 in serum-free media for 2 h. Media were removed and cells washed twice with PBS before being incubated in 1 mL growth media for 48 h in normal culture conditions. Cells were passaged into three wells of a 24-well plate at 800 cells/well. After 24 h, cells were fixed with 0.2% glutaraldehyde/2% PFA for 5 min. Subsequently, cells were then washed twice in PBS and incubated with 2.5 mg/mL X-Gal solution for 18 h at 37°C in the dark [23]. Subsequently, wells were rinsed twice with PBS and then twice with methanol before air drying. Cells stained blue were considered senescent (Fig. S1; Supplementary Data are available online at
NF-κB p65 nuclear translocation assay
U251 cells were cultivated on cell culture-treated coverslips for 24 h. After 24-h pretreatment with CM, cells were exposed to 10 ng/mL tumor necrosis factor alpha (TNF-α) for 30 min. Cells were washed using PBS and fixed in 4% PFA (4% PFA, 15 min, RT). Permeabilization and blocking were carried out as previously described [24]. NF-κB-p65 was detected by immunostaining with p65 antibody (1:100, sc-8008; Santa Cruz Biotechnology), followed by incubation with Alexa Fluor 488 goat anti-mouse immunoglobulin G (IgG) secondary antibody (Molecular Probes; 1:200), and mounting in medium containing 2.5 μg/mL DAPI. Imaging was carried out using a Zeiss Axio Imager epifluorescence microscope and p65 nuclear pixel intensity was measured using ImageJ software.
NF-κB luciferase gene reporter assays
U251 cells (Cell Line Service, Eppelheim, Germany) were cultivated in DMEM high glucose (Sigma-Aldrich) containing 1%
In vivo cardiotoxin induced mouse tibialis anterior injury and AFSC CM/EV treatment
C57BL/6 mice (12 weeks old) were tail vein injected with 100 μL of hAFSC CM, hAFSC EV, or ultracentrifuged PBS. Thirty minutes later, mice were injected with a total of thirty microliters of 50 μM Naja pallida cardiotoxin (CTX; Latoxan, Valence France) into the tibialis anterior (TA). After 5 days, mice were sacrificed and TA muscles were collected and immediately frozen.
Histology
Frozen TA muscles were embedded in Tissue-TEK® OCT compound (PST). Twelve-micrometer cryosections were processed for immunohistochemistry [24]. The following antibodies were used: monoclonal anti-mouse NF-κB-p65 (Santa Cruz; 1:200); monoclonal anti-mouse Pax-7 (Developmental Studies Hybridoma Bank; 1:1); monoclonal anti-mouse myosin heavy chain 3 (Santa Cruz Biotechnology; 1:200); rat anti-mouse CD31 (Bio-Rad; 1:150); rat anti-mouse F4-80 (Bio-Rad; 1:200); and polyclonal rabbit anti-MyoD (Santa Cruz Biotechnology; 1:200). All primary antibodies were preblocked in wash buffer for 30 min before use. The following secondary antibodies were used: Alexa Fluor 488 goat anti-mouse IgG (Molecular Probes; 1:200), Alexa Fluor 594 goat anti-mouse IgG (Molecular Probes 1:200), and rabbit anti-rat horseradish peroxidase (Dako; 1:200).
Uptake of PKH26-stained EVs by IMR-90 cells
AF-EV staining was carried out using the PKH26 Red Fluorescent Cell Linker Kit (Sigma). IMR-90 cells were cultured until 70% confluency and treated for 3 h with stained AF-EVs. Cells were washed twice with PBS and EV uptake visualized using a Zeiss Axio Imager epifluorescence microscope.
Sample preparation for mass spectrometry
AF-EV and non-EV fractions isolated from AFSC secretome were obtained as described above. Proteins were suspended in SDS-PAGE loading buffer 1× (NuPAGE; Invitrogen, Darmstadt, Germany), denatured, and reduced with 1 mM dithiothreitol (DTT) for 10 min at 95°C, then alkylated using 5.5 mM iodoacetamide for 30 min at 25°C in the dark. Proteins were resolved by SDS-PAGE using 4%–12% Bis-Tris minigradient gels (NuPAGE; Life Technologies). Each lane was cut into 10 equal slices, and proteins therein were in-gel digested with trypsin (Promega). Peptides were extracted, purified by STAGE tips, dried to less than 5 μL, and resuspended in 15 μL of 0.5% acetic acid for mass spectrometry (MS) analysis.
MS analysis
LTQ Orbitrap XL mass spectrometers (Thermo Fisher Scientific) coupled to an Agilent 1200 nanoflow-HPLC (Agilent Technologies GmbH) were used to measure peptides derived from trypsin digestion. Samples, applied directly onto self-packed HPLC-column tips of around 20 cm, were subjected to a gradient formed by solution A [0.5% acetic acid in liquid chromatography (LC)-MS-grade water] and by increasing organic proportion of solution B (0.5% acetic acid in 80% LC-MS-grade acetonitrile [ACN] in water) within 120 min [25]. MaxQuant software, version 1.4.1.2, was used to identify proteins based on peptides and to perform label-free quantification [26]. Both MS and MaxQuant parameters were set as previously described [27]. The analysis of Molecular Function gene ontology (GO) terms (GOMF) was performed through Perseus software, version 1.5.8.5, using Fisher's exact test with a P value threshold of 0.02 [28].
miRNA array and analysis
GeneChip® miRNA 4.0 arrays were used to analyze the miRNA content of AF-EVs using FlashTag™ Biotin HSR RNA-Labeling Kits according to the manufacturer's instructions.
Probe cell intensity data from Affymetrix GeneChip® miRNA 4.0 arrays were analyzed in the Affymetrix Expression Console™ software. Normalization was performed using the robust multichip analysis + detection above background (DABG) algorithm [29]. Only miRNAs calculated as present in all three samples were declared as generally expressed after 24 h. Focus was given to the miRNAs present with the highest signal intensities (top 50). For these top 50 miRNAs, validated target mRNAs were amalgamated using miRWalk2.0 software [30]. In the next step, GO Slim classification for the biological process was performed using WebGestalt software to provide a high-level functional classification for validated target mRNAs [31]. Morpheus software was used to design the heat map showing intensity variability between biological replicas (n = 3) for the top 50.
Transmission electron microscopy
For transmission electron microscopy, EV pellets were resuspended in 40 μL of 0.1 M PBS. Drops of these suspensions were placed on parafilm. Carbon-coated copper mesh grids (Plano, Germany) were placed on the drops for 5 min for probe adsorption. After 5 min of fixation on drops of 1% glutaraldehyde (Roth, Germany), grids were washed four times for 30 s and negative contrasted using 1% uranyl acetate. Grids were air-dried and analyzed using a Zeiss 906 transmission microscope (Zeiss, Germany).
Statistics
All experiments were performed three times unless otherwise stated. Statistical analysis was performed using GraphPad Prism software. Data analyzed between multiple groups were tested using one-way analysis of variance, followed by Tukey's post hoc testing. For comparison between two groups, independent sample t-tests were completed. Significance values were always set at the 95% confidence interval. All P values were indicated as *P < 0.05, **P < 0.01, or ***P < 0.001. All data are displayed as mean ± standard error of the mean.
Results
AFSCs retained multipotency following the collection of AF-CM rich in protein and nucleic acid (Fig. 1)
AF-CM contained a high level of protein (834.25 μg/mL ±72.73) and a low level of nucleic acid (21.93 μg/mL ±2). Human AFSCs retained expression (Fig. 1A–C) of mesenchymal stem cell markers CD44 (97.5%), C73 (92.7%), and CD90 (42.7%), while being negative (Fig. 1D, E) for CD45 (0.095%) and CD34 (5.92%). Following the collection of AF-CM under nonphysiological conditions, human AFSCs not only survived but were also able to expand in normal culture conditions. Additionally, human AFSCs cultured under differentiating conditions after CM collections were capable of undergoing adipogenesis, osteogenesis, and chondrogenesis (Fig. 1F–H′), confirming their multipotent differentiation capabilities.

AF-CM content and multipotency of post-CM generated AFSCs. Flow cytometry analysis for
AF-CM is able to suppress NF-κB signaling (Fig. 2A–E)
The ability of AF-CM to modulate the inflammatory response was tested using U251 cells stimulated with TNF-α, which resulted in an increase of nuclear NF-κB p65 (Fig. 2D). Treatment with AF-CM for 24 h before and during TNF-α stimulation led to a reduction in nuclear p65 by 42.5%, from 2.28 ± 0.02 to 1.31 ± 0.11 (Fig. 2D), at levels comparable with unstressed U251 control cells. Activity of NF-κB was quantitatively demonstrated using NF-κB-luciferase/GFP lentiviral-infected U251 cells (Fig. 2E). TNF-α elevated the levels of NF-κB-dependent luciferase by 46.3%, which was reduced by 15.4% to 40 ± 1.63 with AF-CM. AF-CM treatment alone showed no change in NF-κB-dependent luciferase activity compared with non-TNF-α-stimulated control U251 cells. Therefore, AF-CM attenuates NF-κB activity, a transcription factor widely regarded to regulate proinflammatory signaling [32].
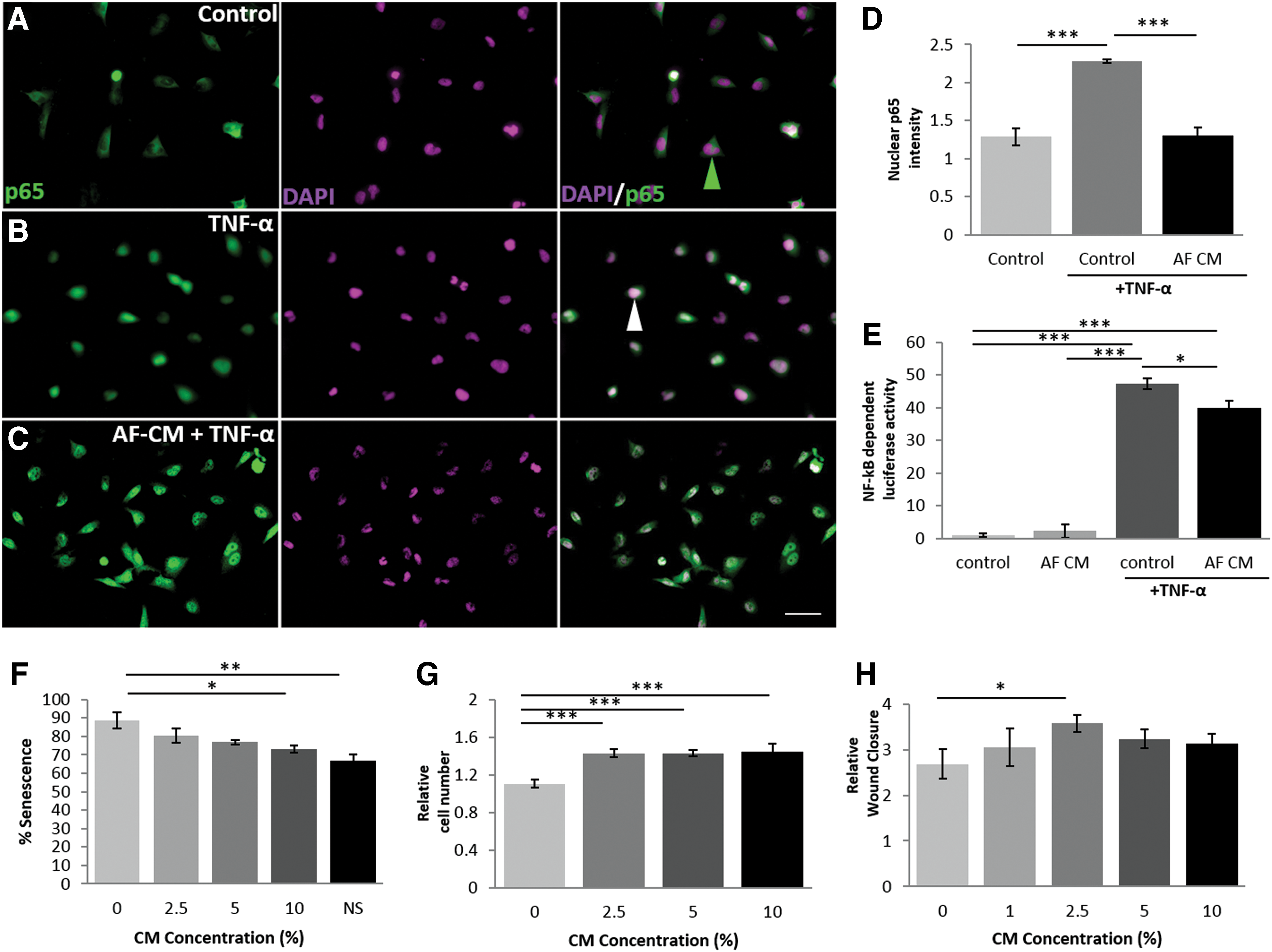
In vitro assessment of biological activity of whole AF-CM.
AF-CM modulates exogenous stem cell properties and protects against cellular stress (Fig. 2F, G)
The efficacy of AF-CM as a regulator of cellular behavior was assessed by examining three properties: (i) the ability to reduce levels of cellular senescence, (ii) to promote proliferation, and (iii) to promote migration. First, we demonstrated a reduction in H2O2-induced, senescence-associated β-galactosidase staining (Fig. 2F) following AF-CM treatment stress exposure, from 88.75% ± 4.39 to 73.25% ± 1.89. Additionally, AF-CM increased the proliferation capability of ADMSCs at a range of concentrations (Fig. 2G). The 30% increase in cell number attributed to the lowest concentration of AF-CM was similar to changes seen with higher CM concentrations, but 2.5% AF-CM induced the optimal increase in ADMSC migration in a wound closure assay (Fig. 2H). Therefore, AF-CM demonstrates an ability to protect against senescence induced through stress as well as to promote the proliferation and migration of allogeneic stem cells.
AF-CM enhances regeneration in an in vivo acute muscle injury model (Fig. 3)
To directly assess its regenerative capabilities, AF-CM was intravenously injected into mice before injecting naja mossambica mossambica CTX intramuscularly into the TA.
Expression of embryonic myosin heavy chain was used to identify newly regenerating muscle fibers (Fig. 3A). Analysis of regenerating fiber cross-sectional area (CSA) showed a dramatic 73.6% increase in fiber size (from 381.12 ± 4.43 μm2 to 661.67 ± 6.31 μm2) in the AF-CM-treated mice (Fig. 3B, C), particularly in those fibers sized 600 μm. Since CTX does not directly affect the resident satellite cell population, endogenous stem cells become activated following fiber damage, subsequently differentiate into myoblasts, and then fuse to replace lost muscle fibers. Although the levels of quiescent Pax7+/MyoD− and activated Pax7+/MyoD+ satellite cells did not significantly differ between CTX and CTX AF-CM-treated mice, the level of muscle lineage-committed Pax7−/MyoD+ progenitor cells was increased by 76.4% in AF-CM-treated mice (28.45 ± 4.66 cells to 50.20 ± 5.62 cells) compared with CTX-treated controls (Fig. 3D, E). Additionally, AF-CM-treated mice had increased numbers of CD31-positive capillaries per area of regenerating muscle (Fig. 3G). More importantly, the number of CD31+-positive vessels per fiber was increased by 75% (from 0.96 ± 0.10 to 1.68 ± 0.25) when compared with the CTX-treated controls (Fig. 3H). Further examination uncovered a larger infiltration of cells expressing the pan-macrophage marker, F4-80+, within TA muscles of AF-CM-treated mice compared with control CTX-damaged mouse TAs (Fig. 3I, J).
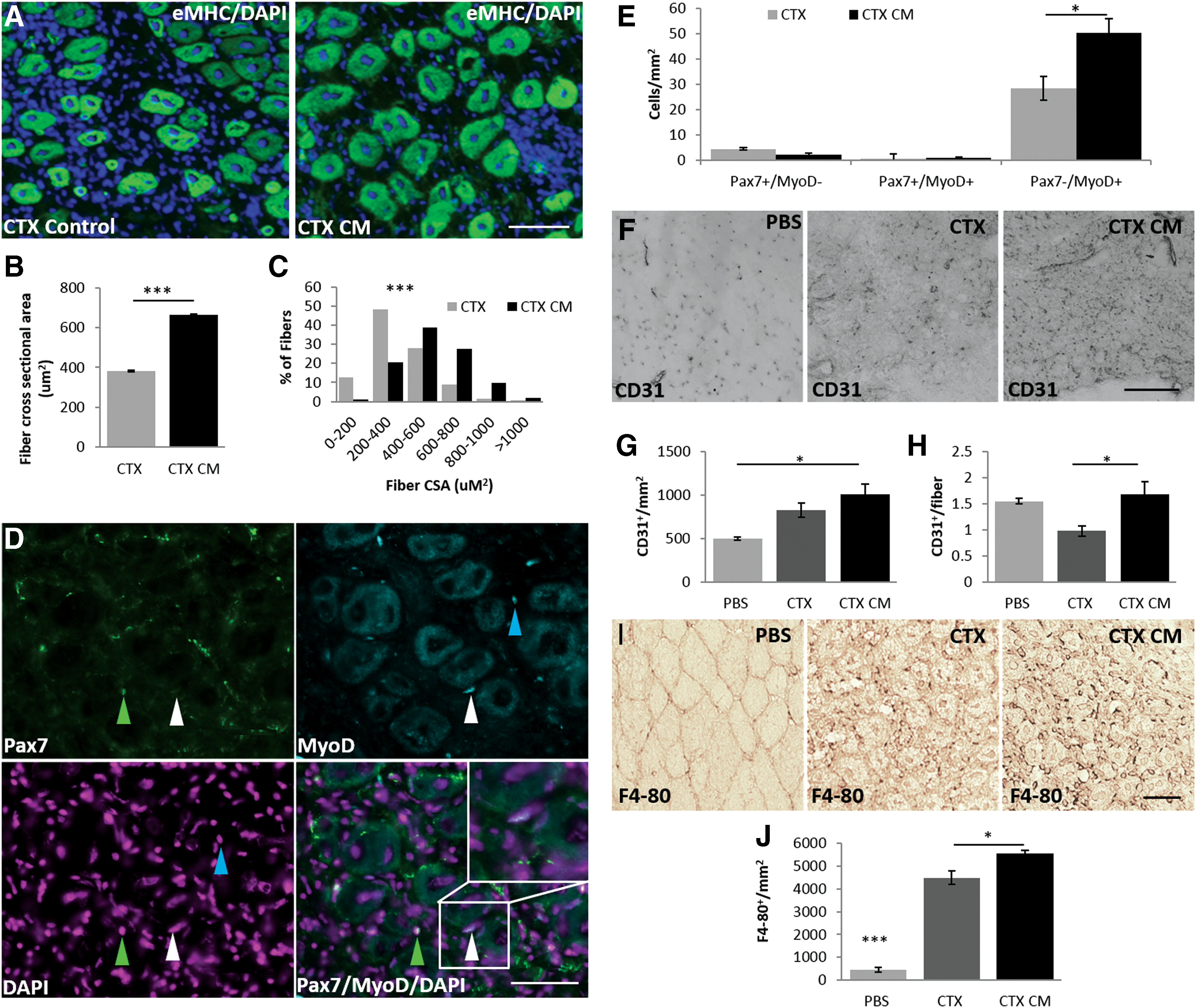
AF-CM and muscle regeneration.
AF-CM EV size is characteristic of exosomes and they are rich in miRNA (Fig. 4)
Stepwise ultracentrifugation was used to isolate EVs (AF-EV) from the AF-CM. The RNA profile showed that the AF-EV fraction was almost exclusively made up of small species [∼21–25 nucleotides in size (Fig. 4A)]. Small RNAs such as miRNA are known to be vulnerable to enzymatic breakdown and have been proposed as being contained within EVs released by cells for intercell communication [33,34]. Size analysis of vesicles was originally carried out to determine the type of vesicle being secreted by the AFSCs in AF-CM (Fig. 4B, C). Average vesicle diameter was 72.5 ± 4.66 nm, suggesting that the majority were exosomes. Levels of proteins within the AF-EV were found to be very high (595 μg/mL). Vesicular proteins in the AF-EV were compared with those found in the whole AF-CM using silver staining (Fig. 4D). Silver staining of proteins in the AF-EV revealed a higher concentration of proteins within the 40–50 kDa range compared with the wide spectrum of proteins found in the AF-CM (Fig. 4D). Fluorescent red PKH26-stained AF-EVs were taken up by IMR-90 target cells and staining was found within the cells, as opposed to the cell membrane (Fig. 4E).
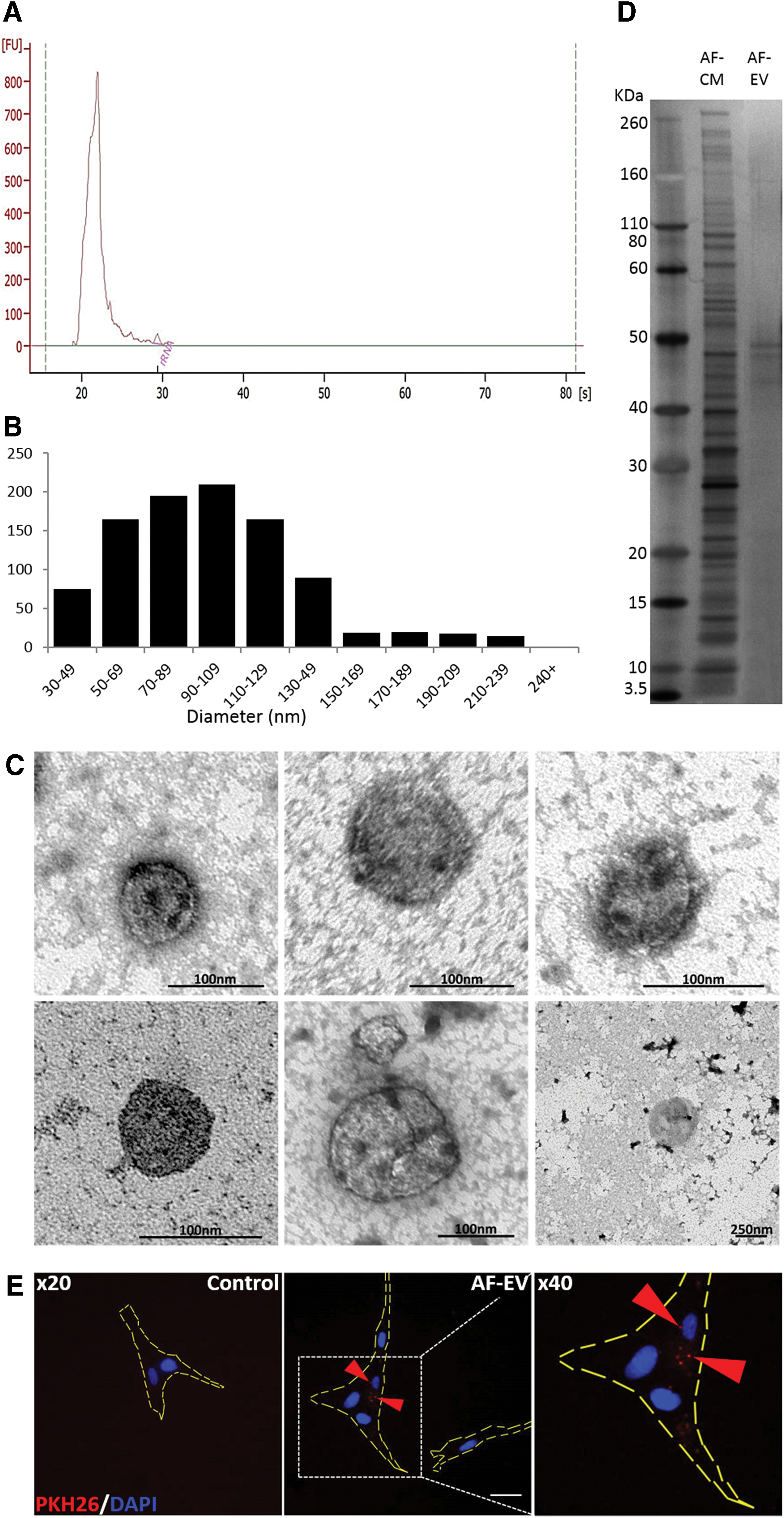
Characterization of AF-CM and AF-EV fraction.
AF-CM EVs protect against cellular stress and suppress NF-κB activation (Fig. 5)
In vitro assays testing cellular protection from senescence and ability to reduce NF-κB activity were carried out to compare the AF-EV fraction with the whole AF-CM. The p65 nuclear translocation assay (Fig. 5A–E) showed that the AF-EV provided a 34.3% greater suppressive effect on the translocation compared with AF-CM (1.47 ± 16.15 and 2.39 ± 0.12, respectively). Quantitative evaluation of AF-EV NF-κB activity through the luciferase-based assay showed no change in luciferase expression compared with the AF-CM (Fig. 5F). The AF-EV fraction induced a decrease in the levels of H2O2-induced senescence by 78% (from 35.50 ± 1.99 to 7.58 ± 0.94; Fig. 5G). However, there were no differences in senescence between the whole and AF-EV fractions.
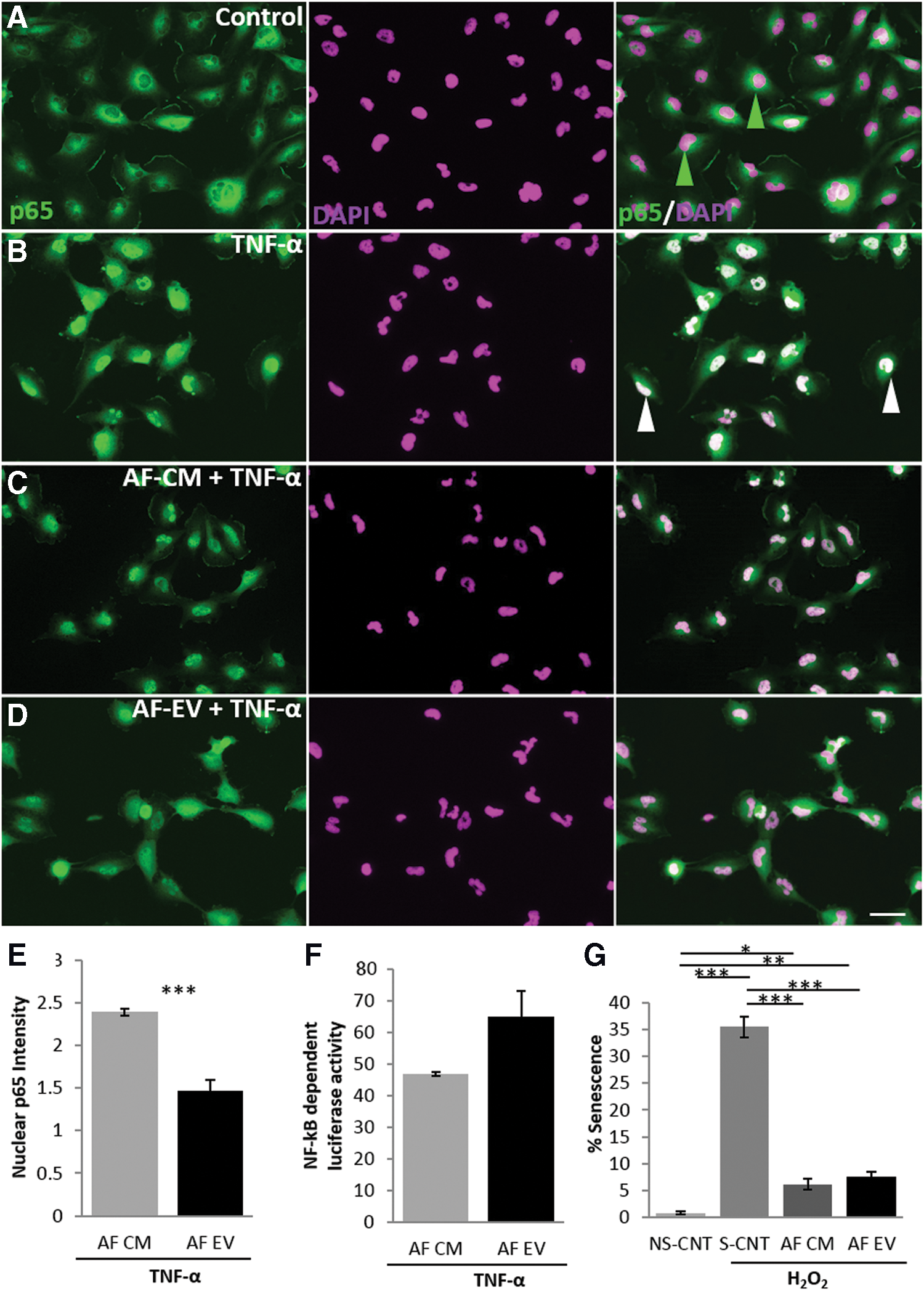
In vitro assessment of biological activity of AF-EV.
AF-EVs retain the ability to enhance regeneration within an acute muscle injury model in vivo (Fig. 6)
Regeneration of CTX-damaged TA muscle was used to determine whether the AF-EV fraction promoted muscle regeneration. CSA of newly regenerating TA fibers was increased by AF-EV treatment compared with the CTX damage control (926.66 μm2 ± 24.23 and 484.51 μm2 ± 68.27, respectively), resulting in a 91.2% increase in the size of newly formed fibers (Fig. 6A, B). Levels of CD31+ capillaries/fiber (Fig. 6C, D) were also increased by 29.7% (from 1.01 ± 0.08 to 1.31 ± 0.07 capillaries/fiber) by the AF-EV treatment compared with the vehicle treatment. AF-EV treatment likewise amplified capillary density toward that similarly seen in an undamaged control, mirroring what was observed in AF-CM. On the other hand, levels of F4-80+ cells were not altered by the AF-EV treatment (Fig. 6E, F). AF-EV treatment did not alter the levels of quiescent (Fig. 6G) and activated satellite cells (Fig. 6H) and committed progenitor cells (Fig. 6I) compared with that of the CTX damage control. In vivo analysis of AF-EV regenerative capabilities showed that treated mice presented with increased vascularization and regenerating fibers with a larger CSA following CTX damage compared with vehicle-treated controls.
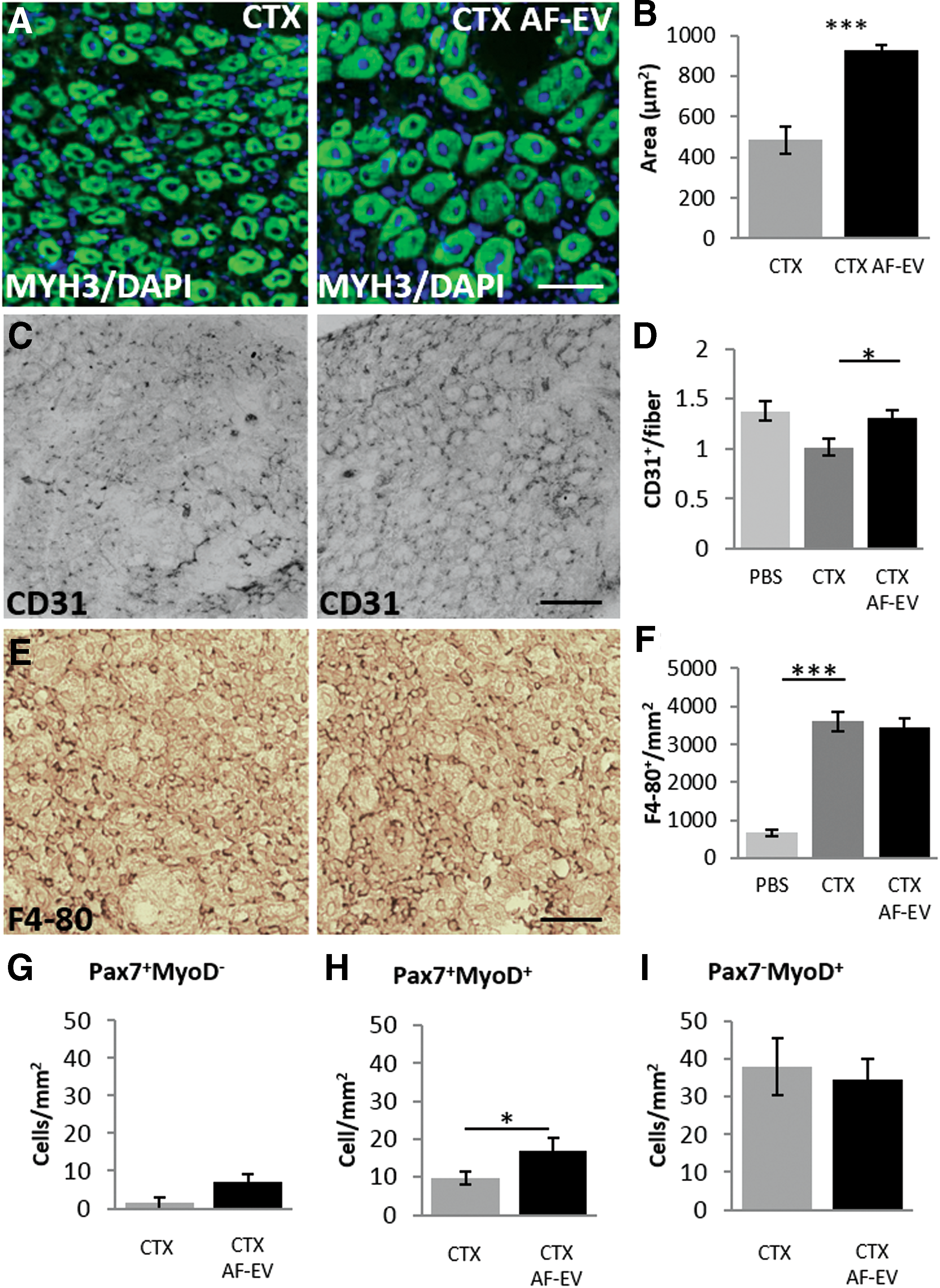
AF-EV and muscle regeneration.
AF-EVs harbor unique proteins and regeneration, promoting miRNA (Figs. 7 and 8)
MS was used to identify proteins in both the AF-EV and non-EV fractions from the human AFSC secretome (Fig. 7B). A total of 856 protein groups were identified. A variety of proteins were found to be present in both the soluble fraction and EV fraction of the AF-CM (Fig. 7A). However, a very distinctive quantitative protein profile was found in the EV fraction compared with the soluble fraction (Fig. 7B). Four hundred sixteen protein species were shared between the two fractions. However, 250 protein species were unique to the soluble fraction, whereas 190 species were exclusive to the AF-EV fraction (Fig. 7A). Proteins enriched in the different AF-CM fractions were shown to be important in shared cellular processes (Table 1), cellular processes individual to that of the soluble fraction (Table 2), and processes only found to be controlled by proteins within the AF-EV fraction (Table 3). Of interest was the heat shock family of proteins present in the AF-CM. A large number of heat shock proteins were secreted, some of which were found only within the AF-EV fraction, including HSPA (70 kDa)-4L, whereas others were found only in the soluble fraction, including the mitochondrial 60 kDa heat shock protein (HSPD-1). Finally, many were found in both fractions, for example, HSPA-6 protein. Transmembrane transporters and receptors were enriched within the AF-EV, including ABCE1, ABCF1, ABR, AP2A1, CYFIP1, CYFIP2, DNM1, DNM2, DNM3, HDAC2, HDAC1, HK1, MAPK1, NCKAP1, PPP1CB, ROCK2, RTN4, STRAP, USP9X, USP9Y, and XPO1 (Fig. 7B). A2B1 ribonucleoprotein was also found in the AF-EV fraction, while no RNA-induced silencing complex (RISC) complex-associated proteins were found in either fraction.
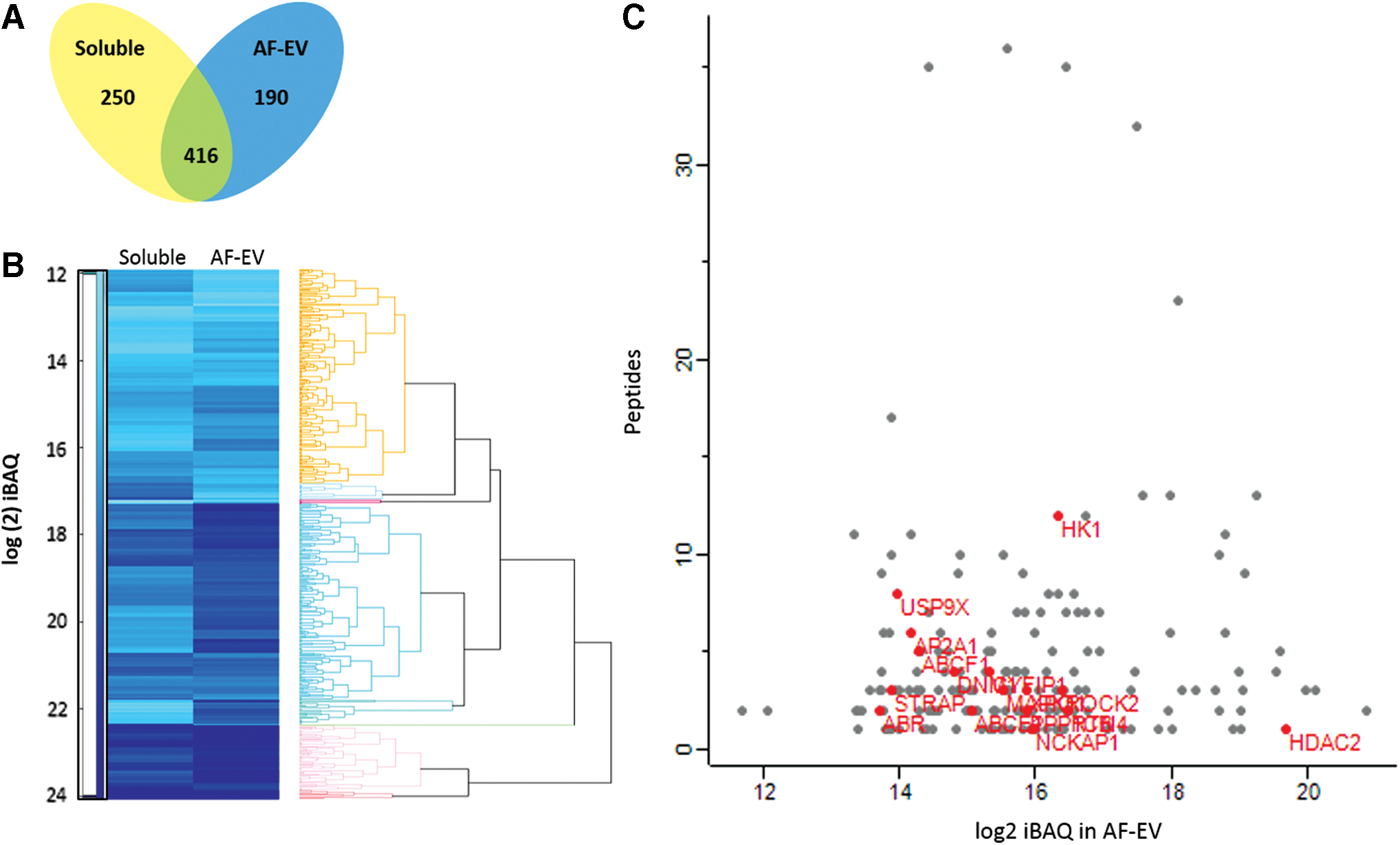
Mass spectrometry analysis and comparison between the AF-EV and AF-CM-soluble fractions.
FDR, false discovery rate; GOMF, gene ontology molecular function.
SRP, signal recognition partical; ER, endoplasmic reticulum.
Detailed analysis of the AF-EV miRNA led to the identification of 586 miRNA species and 21,399 pathway targets (Fig. 8B). The majority of miRNAs were found to target not only mRNA-controlling processes such as metabolic activity (Fig. 8B) but also more specifically responses to stress (Fig. 8C). Within the top 50 miRNAs in the AF-EV, many were postulated to promote angiogenesis, proliferation, migration, differentiation, and autophagy, such as miR382, miR24-3p, and miR3960 [35 –37]. Other species of miRNAs found potentially inhibit apoptosis, including miR3196, miR762, miR125b-5p, and miR1273-3p [38 –41], while others present such as miRNA let-7b-5p are known to also possess anti-inflammatory effects [42,43].
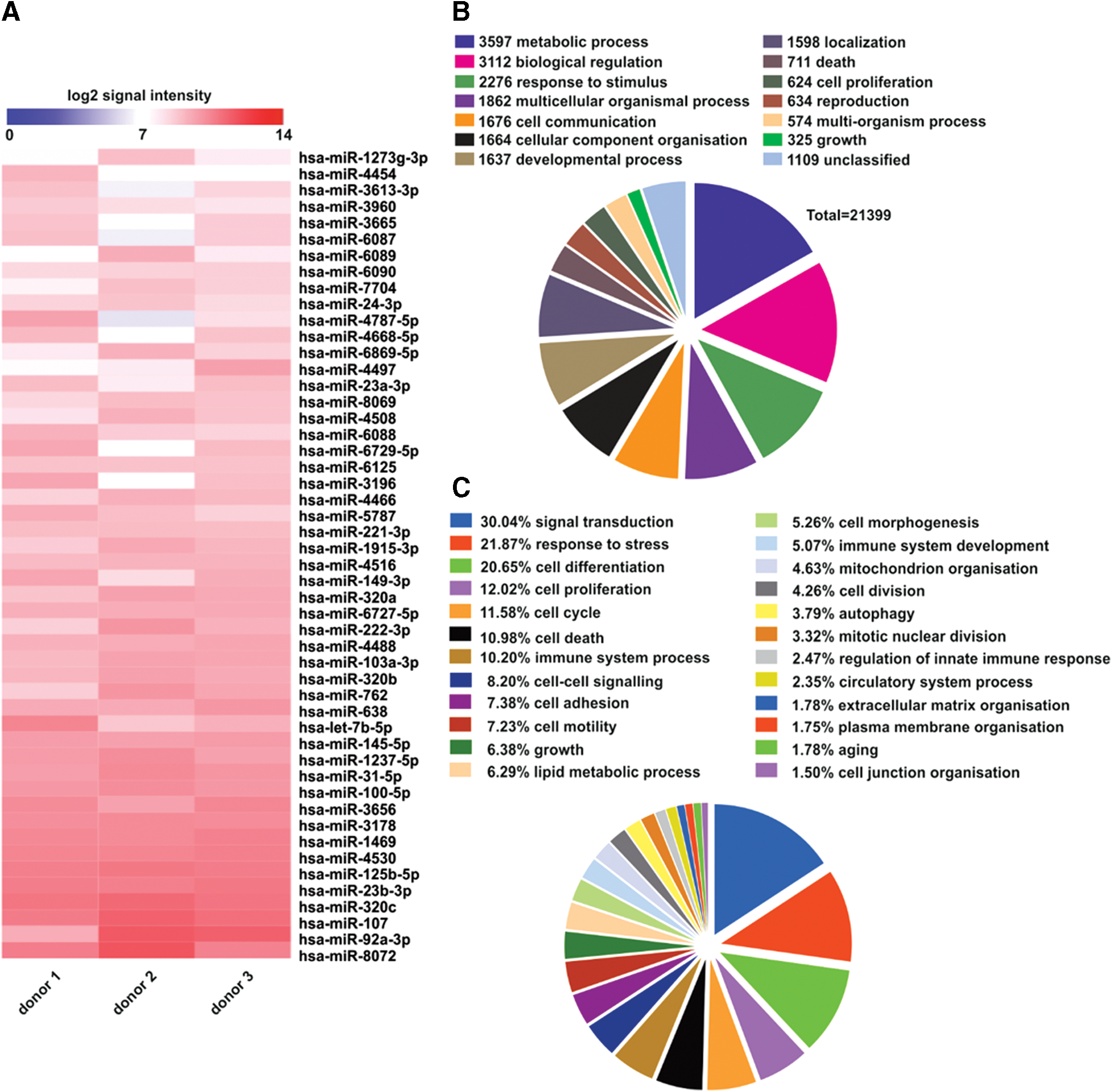
Profiling of AF-EV miRNA content (n = 3).
Discussion
Our extensive characterization of AF-CM and its AF-EV fraction created under nonphysiological conditions from human AFSCs provides crucial information about their protein and RNA cargo. We demonstrated that human AFSC-derived AF-CM and AF-EV were able to modulate allogeneic stem cell activity and inflammatory signaling. Our results showed that alterations in inflammatory signaling following treatment with AF-CM were mediated, at least in part, through NF-κB pathway repression. Furthermore, our data suggest that many paracrine factors that support regeneration were in the AF-EV. Interestingly, the RNA fraction of the secretome was made entirely of miRNAs and lacked mRNA molecules.
Much promise surrounds the clinical potential for MSCs in regenerative therapies. Fetal-derived MSCs such as AFSCs may have greater differentiation potential compared with their adult counterparts, such as ADMSCs [22]. AFSCs are multipotent and promote regeneration of an array of tissues [4,9,22]. However, the ability of AFSCs and other stem cells to regenerate tissue is often through the paracrine-acting secretome, which includes proteins and RNA molecules [4,8]. Paracrine factors are often found in membrane-bound vesicles such as exosomes and microvesicles that aid transit for cell communication and protect the cargo from degradation [34]. Low levels of basal EV secretion are often increased after stress [14]. The ability to increase EV shedding following stress was proposed as a means to produce a more potent secretome [44]. Additionally, cell-free-based therapies reduce the risk of immunological reactivity often associated with the use of whole cells. The potential for tumorigenesis using whole cells is also avoided and it is believed that cell-free therapies are a safer option for clinical use.
Previous studies have either used whole stem cells or basal medium and/or serum-derived CM collected under physiological conditions [4,10]. Collection of AF-CM in PBS at RT, as used in this study, was postulated to induce cellular stress, which would enhance the secretion of regenerative molecules [44]. It is in accordance with a recent study, which demonstrated that hypoxia promoted EV secretion from human AFSCs [45]. However, in our study, we used even more severe conditions and demonstrated that not only did the human AFSCs survive 24 h in nonoptimal nutrients, temperature, and oxygen conditions but they also expressed multipotency markers, CD90, CD73, and CD44, as well as retained the adipogenic, osteogenic, and chondrogenic potentials. These results show that human AFSCs are extremely resilient, which would be beneficial for not only surviving in a hostile cellular environment, which might exist in the uterus during gestation, but also in damaged tissue.
A wealth of evidence shows that paracrine factors released by stem cells are packaged into small membrane-bound EVs [10,34,46 –48]. We isolated vesicles from our AF-CM, exhibiting an average diameter of 72.5 nM, which is consistent with them being exosomes rather than microvesicles [34,49]. Protein analysis also provided evidence for the presence of exosomes due to the existence of known structure-specific markers such as CD63, HSP70, HSP90, ACTB, GAPDH, and 14-3-3 protein zeta/delta (YWHAZ) [50,51]. AF-EV cargo profiling identified a number of polypeptides unique to this fraction. The presence of specific proteins within the AF-EV fraction and soluble fraction implies that both may have therapeutic value and contrasts suggestions promoting the use of AF-EV fractions alone [45]. Our data show that although the AF-EV may provide the AF-CM principle effects, the use of AF-EV alone could lead to the loss of potentially important protein species for regeneration.
The library of excreted paracrine factors identified using MS suggests that they alter the cellular signaling activity in target cells. Identification of heat shock proteins was of particular interest. For example, the presence of heat shock protein 70 in the AF-CM has been previously shown to initiate a preparatory-like signaling mechanism to brace cells for incoming stress [52 –54]. Furthermore, the alarmin protein HMGB1 was present in the CM, which may further potentiate protective mechanisms [55]. A reduction in the level of H2O2-induced senescence following AF-CM treatment also showed a protective effect. The suggestion that there is a safeguarding against cellular stress following tissue necrosis could, in part, be explained by the presence of these alarmins within the AF-CM. In vitro experiments showed that AF-CM increased proliferation and migration of ADMSCs, thus showing an ability to modulate allogeneic stem cell activity in favor of regeneration. AF-CM caused no aggravation of a proinflammatory response in itself. Interestingly, the capacity to reduce NF-κB-p65 nuclear translocation and TNF-α−driven NF-κB activity demonstrated the ability of AF-CM to reduce inflammation, a key characteristic required for regeneration [56].
Our work also sheds light on the possible role of AFSCs in their natural environment, the amniotic fluid. It has been shown that there is an increase in p65 signaling toward the end of a pregnancy, which is believed to help trigger labor. It is conceivable that AFSCs not only play a role in dampening p65 signaling and therefore prevent preterm labor but also constrain a proinflammatory environment known to be damaging for the fetus [57 –59]. We suggest that this is a regulated process such that periods of stress promote the secretion of anti-inflammatory molecules by AFSCs into the amniotic fluid.
Treating IMR-90 cells with PKH26-stained AF-EVs not only showed that AF-EVs were capable of being taken up by target cells but it also provided further evidence that uptake was through an active process. The lack of stained membrane lipid around the periphery of the cell, and instead presence within the cell, suggests that the AF-EV membranes did not fuse with the target cell membrane. Instead, its uptake has been proposed to be facilitated by active mechanisms such as endocytosis, preparing cargo endocytic compartment sorting [60,61]. Membrane-bound receptors and transporters are thought to aid the facilitation of EV fusion and uptake into target cells [34]. It has been previously hypothesized that EV adsorption is cell specific, enabling cellular responses such as angiogenesis [34,48]. Endothelial progenitor-derived EV targeting to endothelial cells is believed to be facilitated by surface alpha4 and beta1 integrins [48]. In contrast, our data show a regenerative response for the AF-CM and AF-EV in a number of cell types. In keeping with this, MS profiling failed to find any specific cell surface targeting protein. These data therefore provide evidence that the AF-CM protective effects are not specific to one cell type, but afford cellular protection to a spectrum of cell types. Again, this is hypothesized to be of benefit during pregnancy to protect the fetus. This suggests that AF-CM may be therapeutically amenable for use in a number of pathological situations and tissues.
The A2B1 ribonucleoprotein is known to transport miRNA into EVs. The presence of this protein in the AF-EV fraction supports the idea that loading of the miRNA in this compartment is a regulated process [62]. miRNAs must be protected for long-range transport and signaling due to their instability and susceptibility to enzymatic degradation. Protection can be facilitated through interaction with the RISC protein complex. However, MS failed to detect RISC complex proteins. We suggest that the role of providing miRNA stability is fulfilled by EVs. Packing of miRNAs into EVs not only protects them from degradation but may also be essential for their biological action. Indeed, recent research has demonstrated that naked circulating miRNA has no biological activity. This property is imbued following their encasement in EVs [13]. One very important finding of this study was that all the RNAs in the CM were miRNAs with no evidence of mRNAs [33,48,63,64]. This signifies that AF-CM modifies endogenous programs of existing target cells rather than imposing ones that are completely foreign.
Enhanced regenerative properties of the EV fraction demonstrated here could be attributed to the presence of miR125b-5p, miR3196, and miR762, which target proapoptotic and senescence-related mRNA [40]. Suppression of keap1 mRNA by miRNA125b-5p has been shown to inhibit both FAS-induced apoptosis and APAP-induced necrosis, leading to inhibition of acute liver failure [40]. In addition, targeting of the RNF4 gene by miR762 inhibits apoptosis by inducing DNA repair mechanisms [39]. The modulation of H2AX expression by miR3196 and downregulation of PUMA/p53/p21 signaling were not only shown to inhibit apoptosis but also suggest subsequent suppression of downstream senescence [38]. Importantly, some of the identified miRNAs, notably Let-7b, have been previously shown to be anti-inflammatory [42]. A recent study by Balbi et al. profiled miRNA in an AF-CM with regenerative properties [45]. However, none of their candidates feature on the list of top 50 miRNAs found in our samples. We suggest that the types of miRNAs produced by human AFSCs depend on culture conditions and many miRNAs regulate mRNAs that control regeneration.
Suppression of NF-κB signaling by the AF-EV fraction was shown to be equal to or stronger than that of the whole AF-CM. This implies that the components subduing NF-κB activity reside mainly in the EV fraction. Our data provide direct evidence of AF-CM and AF-EV having anti-inflammatory effects through suppression of NF-κB signaling. Studies on the ability of AFSC CM to alter polarization of M1 to M2 macrophages will provide further evidence of its anti-inflammatory ability, as was shown recently with ADMSC CM [65]. Cellular protection may also derive from the vesicular portion of the CM, as evidenced by a reduction in cellular senescence, with AF-EV being equally potent as AF-CM. Regulation of NF-κB signaling homeosis is important in determination of not only the inflammatory response but also many other directly and indirectly related pathways such as cellular growth, proliferation, and the stress response [66,67]. These results suggest that the AF-CM, including its EV fraction, may drive signaling toward a normal equilibrium-like state.
Our results show that both the CM and EV from AF cells promote muscle regeneration. However, we believe that the subtle differences in results from these two experiments offer not only mechanistic insights into how this is occurring but also the relative importance of differing factors in the process. Whole CM experiment showed an increase in the number of committed muscle cells (Pax7+/MyoD+), an increase in capillary density, and an increase in the number of macrophages. Capillary density, but not macrophage infiltration, was changed in the EV experiment compared with controls. Furthermore, in the EV experiment, the number of activated precursor muscle cells was increased. These results suggest that the suprabasal infiltration of macrophages is not a key determinant in this study for the enhanced fiber regeneration effect of AF-CM or AF-EV. AF-CM and AF-EV treatments both increased the capillary density. A highly vascularized tissue is documented to support regeneration [68]. Indeed, recent work shows that an enhancement in vascularization can overcome even a deficit in muscle stem cell numbers to promote muscle regeneration [68]. We suggest that the impressive enhancement in muscle regeneration seen after AF-EV treatment is due to a combined effect of not only increased capillary density but also an increase in the number of Pax7+/MyoD+ cells. A number of studies have shown that these cells undergo more rapid division than committed Pax7−/MyoD+ cells [69]. We suggest that constant supply of precursors in a highly vascularized environment, promoted by AF-EV, results in superior regeneration compared with AF-CM treatment.
Notably, our in vitro results were in general accordance with the data obtained in the in vivo model of muscle regeneration. Our data showed that both AF-CM and AF-EVs accelerated muscle regeneration after CTX damage. The most obvious sign of regeneration was the enhanced CSA of newly regenerating fibers 5 days after damage. Both the AF-CM and AF-EV doubled the size of regenerating fibers. Our data directly showed significant evidence of increased levels of capillary density following AF-CM (Fig. 3F–H) and AF-EV (Fig. 8C, D) [45]. The level of angiogenesis during regeneration has been suggested as being more important than the number of satellite cells present [68]. Let7b has been linked to function as a proangiogenic miRNA as well as miR382, which was also found to be present and previously demonstrated to promote angiogenesis through phosphatase and tensin protein (PTEN) repression [35,43].
The increase in F4-80+ cells following AF-CM treatment could be due to the time frame in which muscles were collected following damage. Hypothetically, the increase in macrophage numbers is due to a surge in anti-inflammatory M2 macrophages. However, unlike the AF-EV, the AF-CM significantly elevated the number of Pax7−/MyoD+ myogenic-committed progenitor cells. A greater level of committed cells in the CM-treated mice along with the increased exogenous stem cell migration and proliferation in vitro evokes the possibility that AF-CM enhances satellite cell proliferation and migration. This would lead to increased levels of myogenic-committed cells seen 5 days later upon dissection. More myogenic progenitor cells would also account for an increased fiber size due to elevated myoblast fusion events.
To summarize, the whole AF-CM had an ability to increase regenerating fiber size, increase the number of capillaries/fiber, and increase the level of committed muscle progenitors available. On the other hand, the AF-EV was shown only to retain the ability to increase the regenerating fiber size and the number of capillaries/fiber. Although the AF-EV fraction is able to promote regeneration, these results imply that the use of whole AF-CM may be more beneficial therapeutically than AF-EV fraction alone because it mediated wider regenerative effects. The array of factors in both the soluble and AF-EV fractions could provide more means of regeneration compared with that of the AF-EV alone. On the other hand, in the case of miRNA, there is strong evidence suggesting that it can only significantly alter the signaling of other cells when packaged, secreted, and intercepted within EVs [13].
In summary, we show that AF-CM produced from human AFSCs under conditions compliant with clinical translation contained a spectrum of proteins and miRNA as the predominant molecular species. The secretome was shown to regulate key stem cell activities as well as promote skeletal muscle regeneration. AF-CM contained exosomes that carried a unique cargo, which in itself was able to promote muscle regeneration. Proteomic and molecular profiling of the cargo identified molecules capable of modulating inflammation and angiogenesis, which could mechanistically underpin tissue regeneration. Future investigations will focus on specific cargo molecules for their ability to promote the repair of tissue.
Footnotes
Acknowledgments
This research was funded by BBSRC, National Institute for Health Research Great Ormond Street Biomedical Research Centre, and Rosetrees Trust. A.L.D. is funded by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. P.D.C. is funded by the National Institute for Health Research and Great Ormond Street Hospital Children's Charity. T.B.H. was supported by the DFG (CRC1140, CRC 992, HU 1016/8-1), by the BMBF (01GM1518C), by the European Research Council-ERC grant 616891, and by the H2020-IMI2 consortium BEAt-DKD. D.W. was supported by a grant from the DFG (WI4318/2-1).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.