
Abstract
Horses are susceptible to a number of neurotropic viruses, including West Nile virus (WNV), which is a pathogen of global significance in both horses and humans. However, there are no in vitro models with which to study infectious neuropathic diseases in the horse. In an effort to redress this, we have generated neurons from equine induced pluripotent stem cells (equiPSCs) that express a range of cortical neuron-specific markers, in addition to the membrane-bound ligand ephrin B3, which plays an important role in axon guidance as well as functioning as the receptor through which henipaviruses, such as Hendra virus, enter mammalian neurons. EquiPSC-derived neurons spontaneously depolarize with waves of depolarization conducted unidirectionally to adjacent neurons. We sought to confirm that equiPSC-derived neurons are a possible in vitro model for viral neuropathic diseases in the horse by examining their susceptibility to infection with flaviviruses that are known to be neurotropic in horses, including WNV and Murray Valley encephalitis virus (MVEV), and to compare these to nonpathogenic flaviviruses such as Fitzroy River virus (FRV) and Bamaga virus (BgV). All three strains of WNV tested in this study grew to high titres in the equiPSC-derived neurons, inducing a strong cytopathic effect (cpe), as did MVEV. In contrast, FRV showed restricted replication, and no cpe, which is consistent with the observation that FRV infects, but does not cause disease, in horses. BgV, which is thought to infect only marsupials, did not replicate in the equiPSC-derived neurons. Hence, our equiPSC-derived neurons display virus-specific differences in terms of viral titre and cpe that are similar to observations made in vivo, thus supporting their use as an in vitro model for neurotropic viral infection in horses.
Introduction
T
WNV and MVEV are mosquito-borne pathogenic flaviviruses that can cause severe, potentially fatal, encephalitis in humans and horses [7]. The geographic distribution of WNV is continuing to expand with outbreaks now identified in Europe, Asia, North, Central and South America, the Middle East, and Australia, making it a pathogen of global significance. Since its first incursion into the United States in 1999, WNV has been responsible for more than 16,000 human cases of neurological disease and over 1,500 deaths [8]. In 2011, Australia experienced an unprecedented outbreak of equine encephalitis caused by a virulent strain of WNV (WNVNSW2011) that affected more than 1,000 horses, with a mortality rate of 10%–15% [9,10]. Curiously, while highly virulent in horses, WNVNSW2011 caused only a single case of mild disease in the human population [9,10].
MVEV is endemic to Australasia and, although uncommon, has also caused fatal encephalitis in both horses and humans [11,12]. Australia is also host to a large number of apparently nonpathogenic flaviviruses, including two recently discovered viruses belonging to the yellow fever virus group: Fitzroy River virus (FRV) and Bamaga virus (BgV). FRV is widespread in northern Australia where it infects humans and domestic animals, including horses, although to date there is no indication that it causes disease in either horses or humans [13]. In contrast, BgV appears to have a more restricted host range that may include marsupials but unlikely other mammals [14].
Materials and Methods
Maintenance of equiPSCs
Two previously characterized clones of equiPSCs (A and K) [6] were cultured on feeder layers of irradiated mouse embryonic fibroblasts (MEFs) at a density of 20,000 MEFs/cm2. Maintenance medium consisted of KnockOut DMEM (Gibco), 15% (v/v) embryonic stem cell-qualified fetal bovine serum (ESC-FBS) (Hyclone), 0.1 mM nonessential amino acids (NEAAs) (Gibco), 2 mM
EquiPSCs were maintained on feeder layers for 7 days with maintenance medium before undergoing neuronal differentiation (see next section). Colonies were manually dissected and ∼9 individual sections were passaged onto MEF-seeded organ culture dishes (Costar, Corning Life Sciences).
Neuronal differentiation of equiPSCs
After 7 days, the maintenance medium was replaced with neuronal medium consisting of 50% (v/v) Neurobasal medium (Gibco), 50% (v/v) KnockOut DMEM, 0.5 × N2 Supplement (Gibco), 0.5 × B27 (Gibco), 2.5 μg/mL human insulin (Invitrogen), 2 mM
EquiPSCs were cultured in the neuronal differentiation medium for 8 days before being manually dissected and segments seeded onto the Matrigel-coated (Sigma-Aldrich) wells of a six-well plate. Passaged neuronal cultures were maintained in neuronal differentiation medium for an additional 4–6 days (total time in neuronal differentiation medium 12–14 days) before dissociation into single cells with Accutase (Invitrogen) and reseeded onto Matrigel-coated six-well plates for maintenance, or chamber slides (Nunc) for viral infections and immunocytochemistry.
Immunocytochemistry
Samples were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) (Invitrogen) at room temperature (RT) for 15 min, before blocking and permeabilization with 10% goat serum (Invitrogen) and 0.1% Triton X-100 (Ajax Finechem) in PBS for 1 h at RT. Primary antibodies were diluted in 10% goat serum in PBS and incubated overnight at 4°C. Primary antibodies and their dilutions are as follows: TUJ1 (1:500; Merck), Ephrin B3 (1:200; Abcam), SOX2 (1:150; Cell Signaling Technology), BRN2 (1:150; Cell Signaling Technology), and TBR2 (1:150; Abcam). Negative control samples were incubated with 10% goat serum in PBS without primary antibody. Samples were then incubated with isotype- and species-matched Alexa-Fluor conjugated secondary antibodies (1:1,000; Invitrogen) for 1 h at RT. Nuclei were visualized with Hoechst33342 (1:10,000; Invitrogen) or DAPI (1:10,000; Invitrogen).
RNA isolation, cDNA synthesis, and quantitative polymerase chain reaction
Total RNA was isolated with TRIzol using the Ambion PureLink RNA Mini Kit (Ambion) according to the manufacturer's instructions. Complementary DNA was synthesized with the iScript DNA Synthesis Kit (Bio-Rad Laboratories). Quantitative polymerase chain reactions (qPCRs) were performed with SsoFast EvaGreen Supermix (Bio-Rad Laboratories). All target genes were normalized to endogenous equine GAPDH. Primer pairs used are listed in Table 1. Three replicate qPCRs were performed for each transcript, and the relative changes in expression of each gene were determined via the 2−ΔΔCT method [15].
Fluo-4 AM calcium dye staining and imaging
Fluo-4 AM (F14201; Life Technologies) was reconstituted to a stock solution of 2 mM in DMSO. Loading solution was prepared by diluting Fluo-4 AM to a final concentration of 10 μM in Tyrode's solution (5 mM KCl, 129 mM NaCl, 1 mM MgCl2, 2 mM CaCl2, 30 mM glucose and 25 mM Hepes). Five week-old neuronal cultures (three each from both A and K clones) were incubated in loading solution for 40 min at 37°C followed by two washes with PBS. Cultures were then incubated in Tyrode's solution for 30 min at 37°C to allow deesterification of intracellular AM esters. Neuronal cultures were excited using a 488 nm filter and imaged using a Nikon DS-Qi2 camera mounted on an Olympus IX51 microscope. Acquired time-lapse images were processed using ImageJ to calculate mean intensities for specified regions of interest where spontaneous activity was observed. Normalized fluorescence (ΔF/F0) was determined by calculating the average of baseline fluorescence (F0) before a spontaneous calcium transient, followed by dividing the measured mean fluorescence intensities (ΔF) by the average baseline fluorescence (F0). These results were plotted as line plots with the normalized fluorescence on the y-axis and time in seconds on the x-axis.
Viruses and virus infection cultures
The following flaviviruses were assessed for replication in equiPSC-derived neurons: WNV strains NSW2011 [9,16], K68967 [16], and Gu0631 [16]; MVEV strain 51, BgV [14], and FRV [13]. EquiPSC-derived neurons were seeded into eight-well chamber slides (Nunc). Four chambers per slide were mock-infected, while the other four chambers each received 4 × 103 infectious units of one of the flaviviruses listed. Due to the dense colony growth pattern of the cells, an exact multiplicity of infection (MOI) could not be determined. The chamber slides were incubated at 37°C in humidified 5% CO2 air. At 24, 72, and 120 h postinfection (pi) one slide per virus was terminated. The culture supernatants were collected and stored at −80°C until tested (see Virus titration section), while the cells were immediately fixed in 4% neutral-buffered formalin for 15 min followed by air-drying. The slides were stored dry, in the dark at RT until subjected to immunocytochemistry.
For comparison, primary equine fibroblasts, established from the joint capsule of an adult stockhorse, and used in passage 6–7, were infected with WNVNSW2011, MVEV, BgV, and FRV at a MOI of 2, 1, and 0.5 in duplicate wells of 96-well tissue culture plates. After 5 days, the cultures were examined visually for cytopathic effect (cpe), fixed in 20% acetone overnight at 4°C and air dried. The cells were subsequently immunolabeled with a flavivirus envelope protein-specific mouse monoclonal antibody (mAb) 4G2 [17] as described in the section below.
Immunocytochemistry for virus antigen
Detection of virus replication in cells was achieved by immunolabeling for the nonstructural protein-1 (NS1) using the flavivirus NS1-specific mAb 4G4 (all WNV strains and MVEV) or the flavivirus envelope protein-specific mAb 4G2 (BgV and FRV) using a protocol adapted from Prow et al. [18]. In brief, the formaldehyde-fixed cells were subjected to antigen retrieval by heating in EDTA, pH 9 (DAKO Antigen Retrieval solution) for 20 min at 96°C, followed by cooling to RT for 25 min. This was followed by three blocking steps with 0.3% H2O2 in water for 10 min, 0.15 M glycine in PBS for 10 min, and 30 min with DAKO antibody dilution buffer, with brief rinses with Tris-buffered saline with 0.1% Tween-20 (TBST) between steps. This was followed by 2 h incubation at RT with either blocking buffer (two infected and two uninfected wells) or with the mAb (neat hybridoma spent medium; two infected and two uninfected wells), followed by multiple TBST rinses over a 10–15 min period and detection of bound antibody using the DAKO Envision Kit and AEC substrate. The slides were then counterstained with Meyer's hematoxylin and mounted with DAKO Faramount medium. For each batch, a positive (tissue from a mouse experimentally infected with WNV or WNV infected C6/36 insect cells) and negative (uninfected mouse tissue or uninfected C6/36 cells) antigen control was included.
To investigate whether viral infection was specific to neurons or also occurred in neural progenitor cells (NPCs) and/or glial cells, we performed double-immunolabeling with the neuron-specific TUJ1 antibody and mAb 4G4 as described in [19]. In brief, the TUJ1 antibody (rabbit) was diluted directly in the 4G4 mAb, and the cells were incubated as described above. This was followed by sequential incubation with anti-mouse (DAKO Envision Kit with horseradish peroxidase conjugate) and anti-rabbit-IgG (Vector AB-kit with alkaline phosphatase conjugate) antibodies, followed by sequential incubation with the chromogens for the two enzymes. Controls were mouse brains as described above and wells that only received one of the primary antibodies or no primary antibody.
Virus titration
Infectious virus released into the culture supernatant was quantitated by a 50% tissue culture infective dose (TCID50) assay as described [20]. In brief, spent medium from the equiPSC-derived neuron cultures was diluted in DMEM/2% FBS in a 10-fold dilution series with 8 wells of a 96-well plate per dilution. African green monkey (Vero) cells were used for titration of all WNV strains, MVEV and the mock controls, while baby hamster kidney-derived (BSR) cells were used for FRV titration. Supernatants from BgV-infected cultures were tested on C6/36 insect cells, as this virus does not replicate in Vero cells and only to a limited extent in BSR cells [14]. Cells were added at 2 × 104 cells per well to a final volume of 200 μL per well and the culture plates incubated at 37°C in humidified 5% CO2 air for 5 days. At the end of incubation, the wells were examined visually for cpe, and the cell monolayers were then fixed in 20% acetone with 0.1% BSA by overnight incubation at 4°C. This was followed by fixed-cell ELISA using the mAb 4G2 as described [17]. The final titre was calculated using the method of Reed and Muench [21].
Results
Rapid neurogenesis, cortical identity, and functionality
Within 6 days of being transferred into neuronal differentiation medium, equiPSC colonies (Fig. 1A) had formed neurospheres with numerous axonal outgrowths (Fig. 1B) that appeared to make connections with axons from adjacent neurospheres (Fig. 1C). Neurons stained positively for the neuronal marker TUJ1 (Fig. 1D, E). Neuronal cultures passaged to single cells still displayed residual clusters from the initial neurospheres; however, individual neurons could be seen to have formed connections between these clusters (Fig. 1F). Neurons also expressed the cell surface transmembrane ligand Ephrin B3, which plays a role in axon guidance (Fig. 1G).

Temporal progression of equiPSC neuronal differentiation.
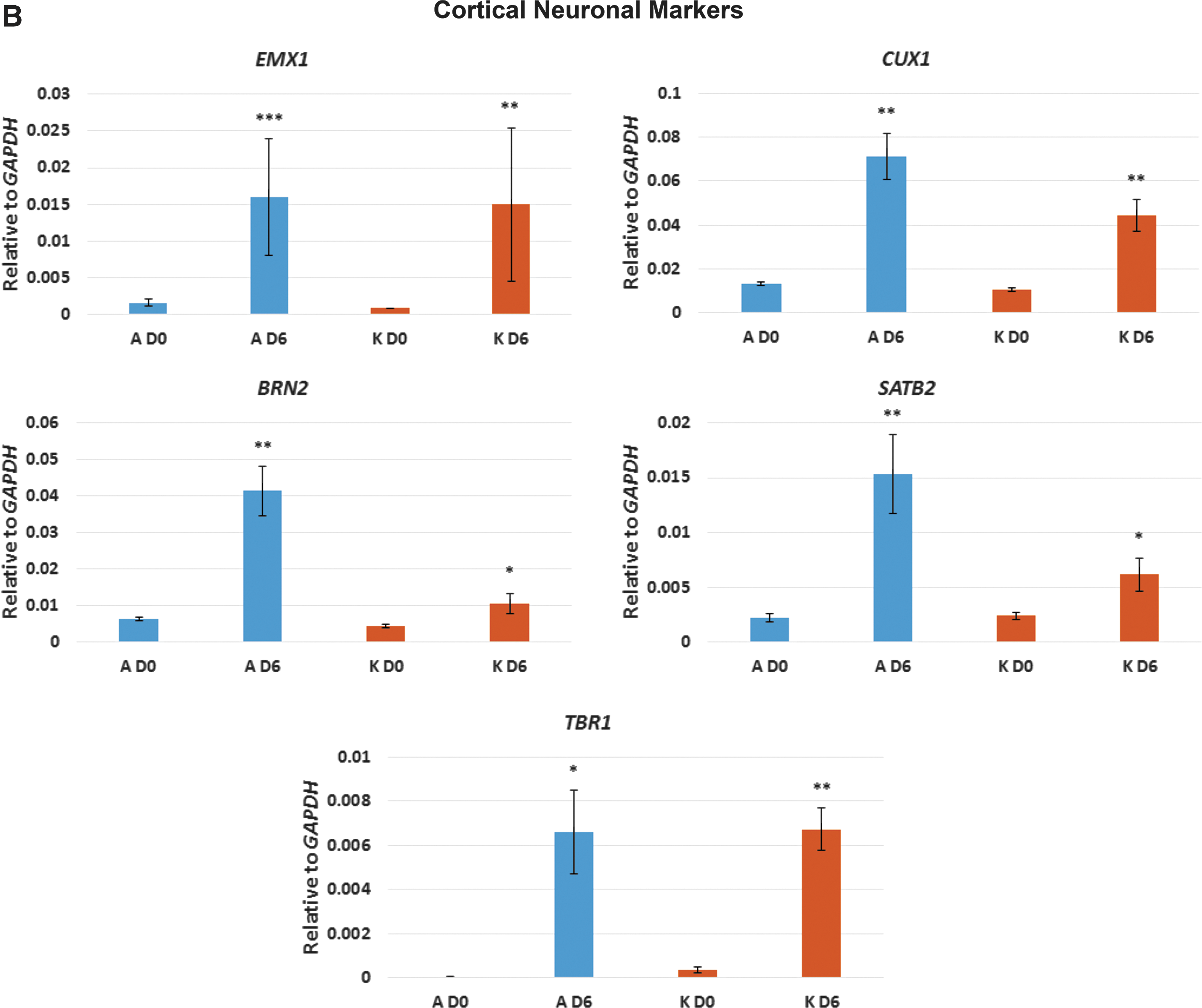
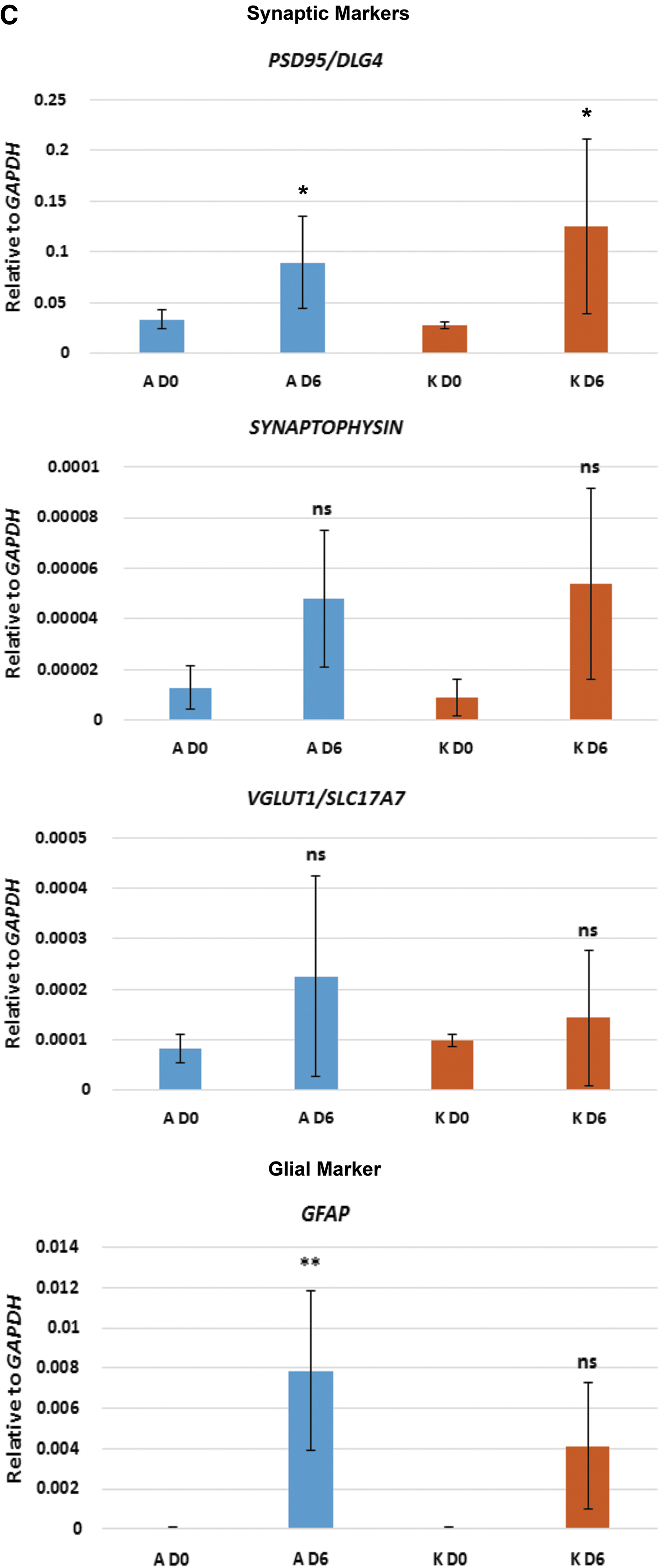
After 6 days in neuronal differentiation medium, cultures of clone A showed high expression levels of TBR2 (also known as EOMES) which, in human, is required for the transition from PAX6-expressing radial glia neural stem cells (NSCs) to intermediate neural progenitor cells (NPCs) [2,22] (Fig. 2A). In keeping with an increase in TBR2 expression, PAX6 was not expressed in clone A cultures (Fig. 2A). In contrast, clone K cultures retained low, but significant, levels of PAX6 expression, while TBR2 was not expressed (Fig. 2A). OTX1 expression is similarly associated with human NSCs [2,23] and significant levels of expression were seen in cultures of clone K, but not clone A (Fig. 2A). Expression of FOXG1 was not significantly upregulated in cultures of either clone A or K at day 6 (Fig. 2A). Thus, cultures from clone A, which express TBR2 but not PAX6 or OTX1, appear to contain a greater proportion of NPCs than cultures from clone K, which retain expression of the NSC markers PAX6 and OTX1 and do not express TBR2. Cultures from both clones expressed markers consistent with cortical neuron differentiation, including EMX1, CUX1, BRN2, SATB2, and TBR1 [2,22,23] (Fig. 2B). However, cultures from clone A, that appear to contain a greater proportion of NPCs than NSCs, consistently expressed higher levels of the cortical neuronal markers than the cultures from clone K (Fig. 2B). Interestingly, cultures from clone A also expressed the glial marker GFAP which, in the central nervous system, is expressed by astrocytes as well as radial glia NSCs [2,24] (Fig. 2C). Cultures from both clones A and K express the postsynaptic marker PSD95 (Fig. 2C); however, we did not detect significant levels of expression of SYNAPTOPHYSIN or VGLUT1/SLC17A7.
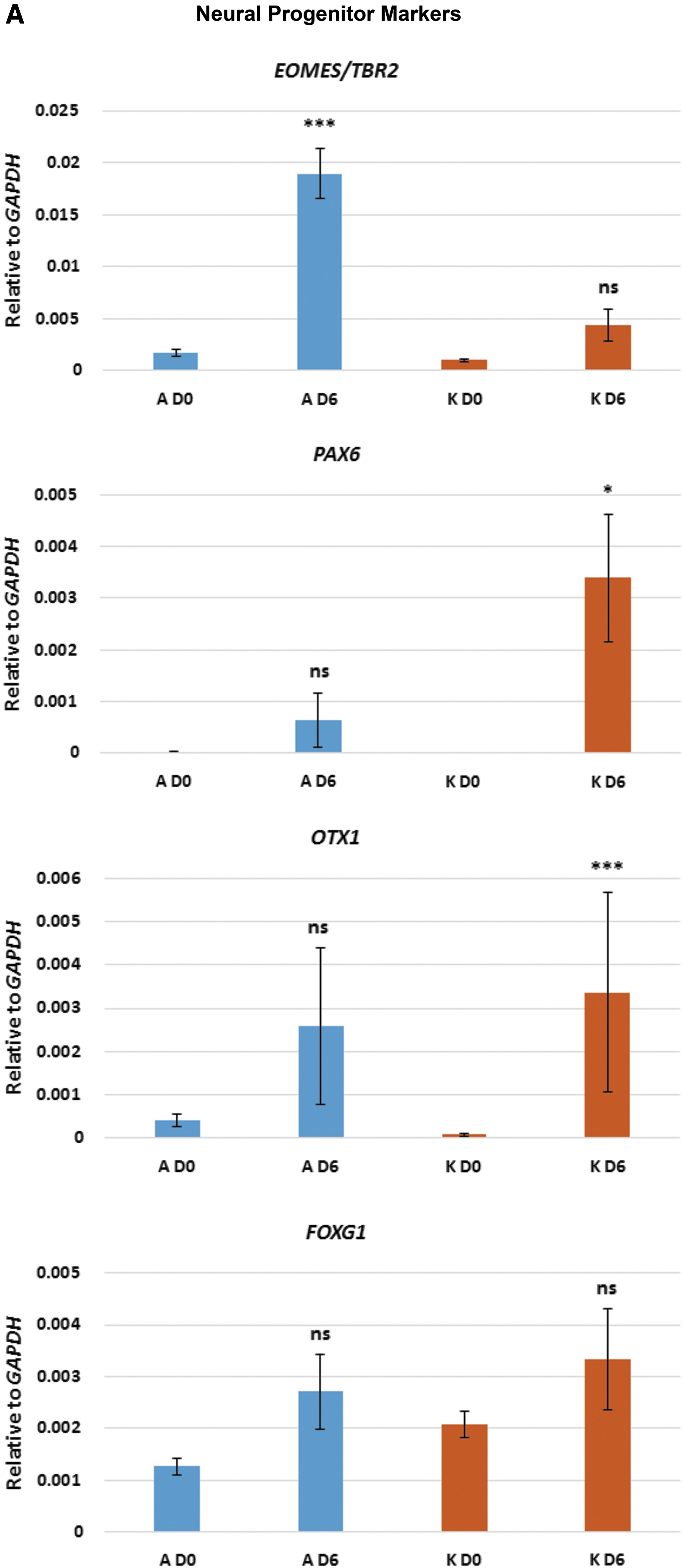
qPCR analysis of selected key markers for neural progenitors, cortical neurons, glia, and synapses. Neurons derived from both clones A and K were analyzed before neuronal induction (D0) and 6 days after neuronal induction (D6). Asterisks represent degrees of significance based on t-tests: ns = not significant (P > 0.05); *P < 0.05; **P < 0.01; ***P < 0.001.
At 2 weeks postneuronal induction, the majority of cells persisted as NPCs as shown by positive immunostaining for SOX2 within the residual neurosphere clusters and in the majority of the dissociated single cells (Fig. 3A). However, in cultures colabeled with the NPC marker TBR2 and the neuronal marker TUJ1, a proportion of cells were positive for both markers, indicating early neuronal differentiation. (Fig. 3B). Populations of developing neurons (TBR2+/TUJ1+) were observed adjacent to clusters of NPCs (TBR2+/TUJ1−) (Fig. 3B), demonstrating that neuronal differentiation is asynchronous, as has been described for neuronal differentiation of human pluripotent stem cells [25].
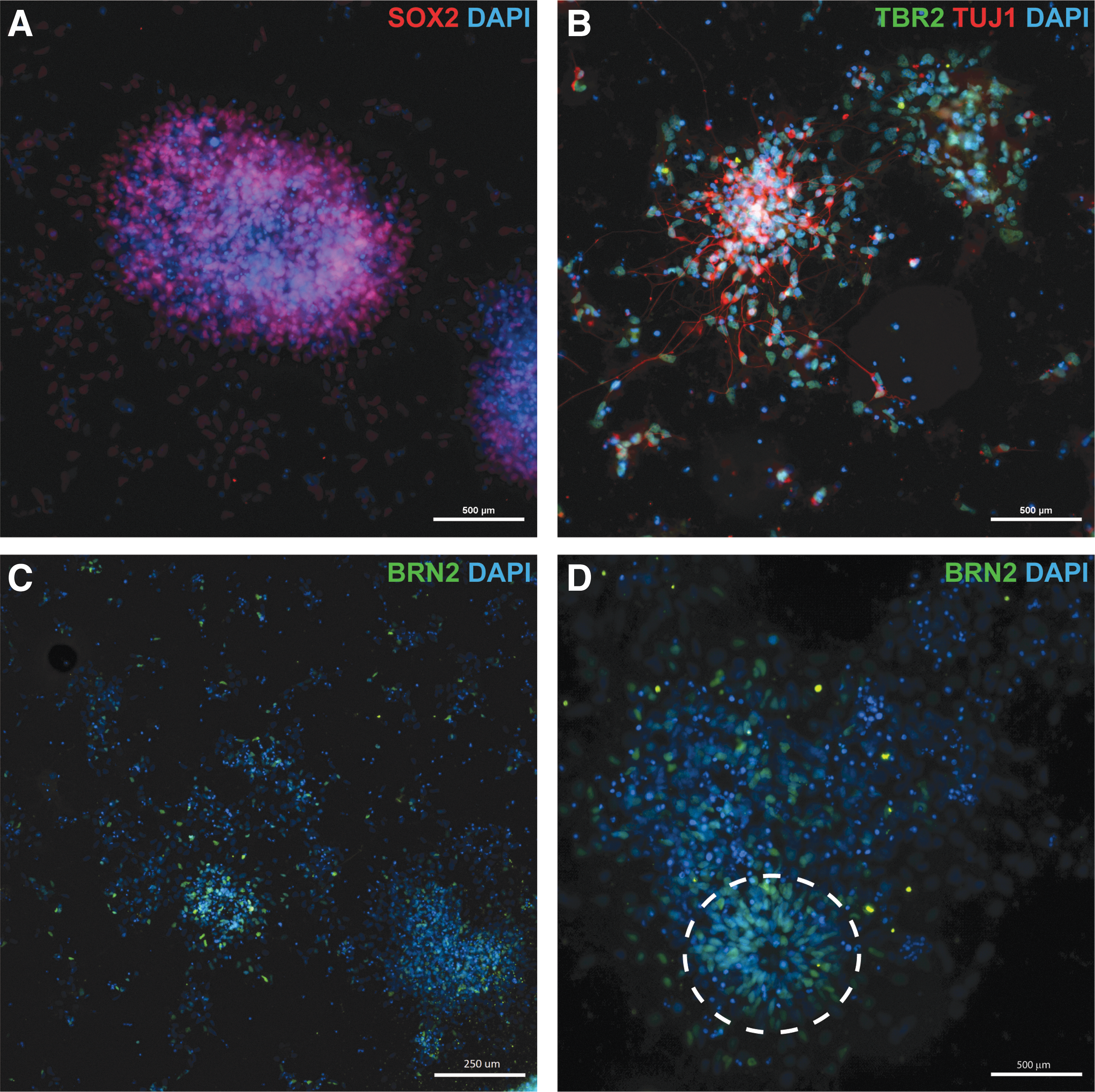
Neuronal differentiation at 14 days postinduction.
Positive immunostaining for BRN2 confirmed that the mature neurons were excitatory cortical neurons (Fig. 3C). Neural rosettes, the in vitro equivalent of neural tube formations that are commonly observed in human and mouse neuronal differentiation cultures [1,2,23,26], were observed (Fig. 3D). Neural rosettes from mouse and human pluripotent stem cells consist of NPCs that give rise to cortical neurons [1,2,23]. Similarly, neural rosettes derived from equiPSCs consisted of cells positive for BRN2 (Fig. 3D). Thus, while the majority of the cells were still NPCs, expression of the mature neuronal marker BRN2 reflects that a proportion of the cells were differentiating toward a terminal cortical fate, which is in agreement with the qPCR data.
Neuronal cultures for both clones A and K were maintained for an additional 3 weeks (total of 5 weeks of neuronal differentiation) and subsequently loaded with Fluo-4 AM calcium dye to assess neuronal functionality. Spontaneous calcium transients were observed in neurons derived from both clones A and K (Fig. 4A, B). In instances where connections between adjacent neurons were observed, calcium transients were propagated unidirectionally from one neuron to an adjacent neuron, presumably via synapses (Fig. 4A).
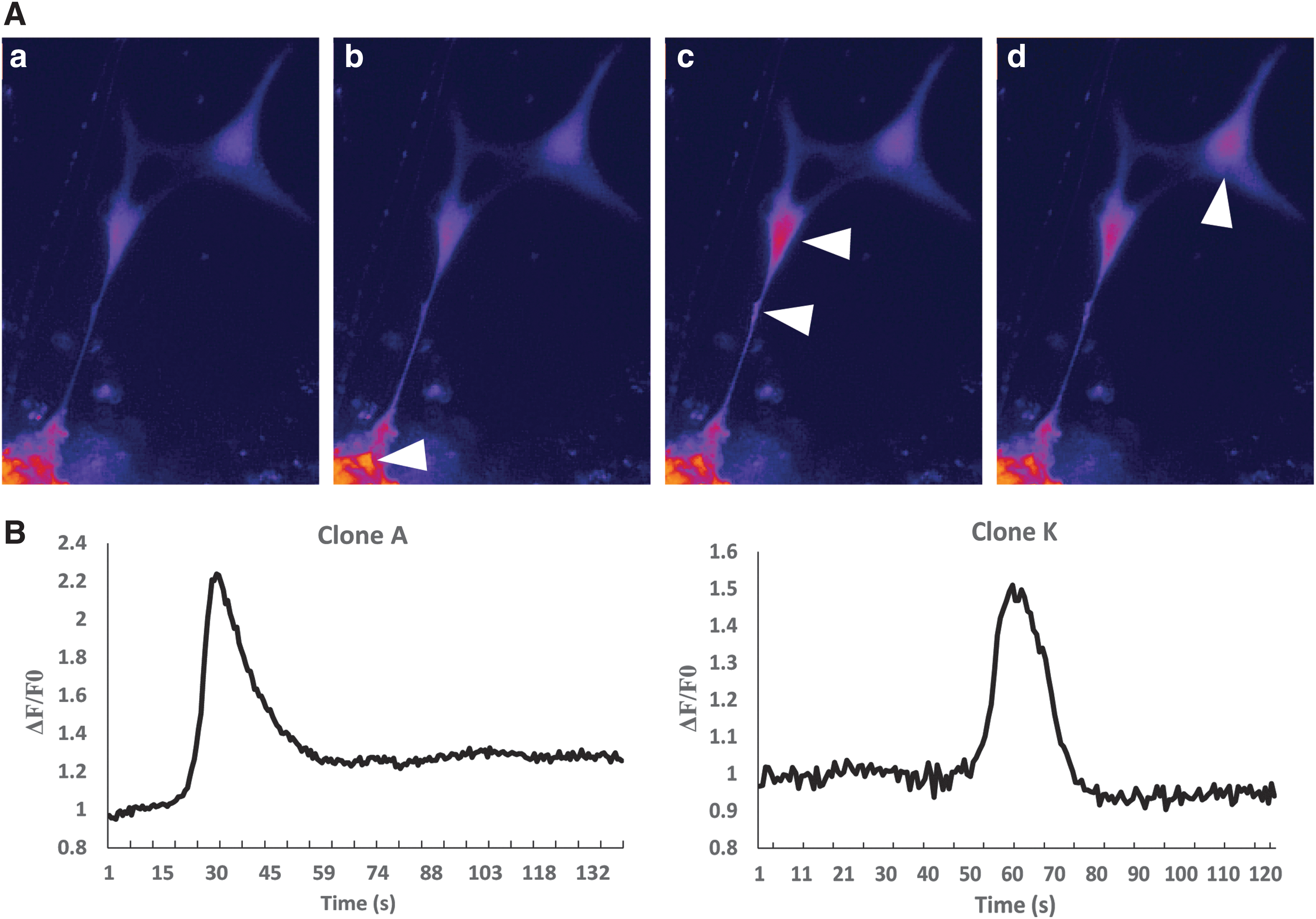
Spontaneous neuronal activity in equiPSC-derived neurons.
Viral infection
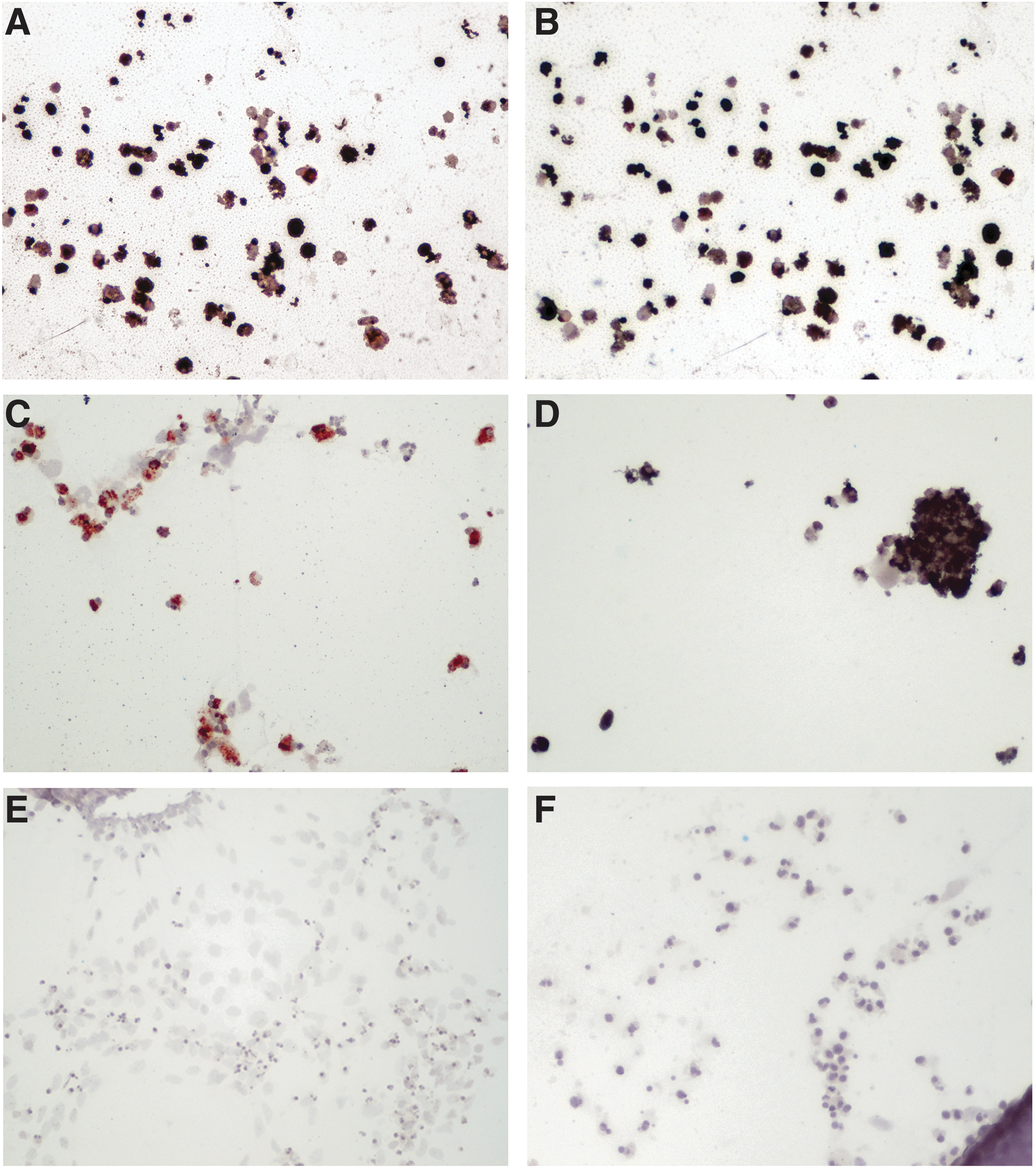
All three strains of WNV and MVEV grew to high titres in neuronal cells derived from both equiPSC clones A and K (Table 2). In the case of the WNV strains, this was also reflected in increasing numbers of NS1-positive cells over the 120 h of incubation (Fig. 5), all of which coexpressed the neuronal marker TUJ1, confirming that WNV infection was specific to neurons (Fig. 6). There was less concordance between virus titres and ICC results for MVEV, but this may be due to cell loss either through apoptosis/necrosis or cell detachment. In contrast, FRV showed restricted replication in the neuronal cells; that is, while virus NS1 could be detected by ICC in a small number of cells, indicating replication, the virus titre did not increase beyond input virus, but rather decreased in accordance with thermal inactivation (Table 2). This is in line with the finding that although FRV infects horses, as judged by seroepidemiological studies, the virus is not known to cause disease in this, or any other, vertebrate species [13]. Finally, there was no detectable replication of BgV in the neuronal cells, which was expected considering its very restricted growth in vertebrate cells in vitro, and its suspected mosquito–marsupial transmission cycle [14].

Examples of WNV replication in equiPSC-derived neurons demonstrated by immunocytochemistry for the flavivirus NS1.
WNV exclusively infects neurons. Neuronal cultures were immunolabeled for neuronal differentiation (anti-TUJ1; black signal) and infection with WNV (flavivirus-NS1-specific mAb 4G4; red signal). The cells were lightly counterstained with dilute Meyer's hematoxylin.
On a scale of: 0 = no NS1/E signal; + = a few NS1/E-positive cells; ++ = moderate numbers of NS1/E positive cells; +++ = large numbers of NS1/E positive cells.
No colonies left, only scattered cells present, but all NS1-positive.
FRV, Fitzroy River virus; MVEV, Murray Valley encephalitis virus; ND, not done; NS1, nonstructural protein-1; TCID, tissue culture infective dose; WNV, West Nile virus.
In contrast to the high replication rates of WNV and MVEV in the equiPSC-derived neurons (Table 2) and in undifferentiated equiPSCs (data not shown), WNV and MVEV barely grew to detectable titres above input virus and no cpe was detected in the primary equine fibroblasts. There was no detectable replication of FRV and BgV in these cells (data not shown).
Discussion
Although horses are susceptible to neurotropic viruses that can infect humans, such as WNV and MVEV, there are currently no in vitro models enabling the study of virus replication or transmission. To address this deficit, we took advantage of equiPSCs previously generated in our laboratory [6] and subjected these cells to a dual SMAD-inhibition neuronal differentiation protocol widely used for human pluripotent stem cells [1]. The equiPSCs engaged in much more rapid neuronal differentiation than that observed in human pluripotent stem cells, but was similar to that observed by Sharma et al., who generated motor neurons from equine iPSCs [5]. Six days after transitioning into neuronal differentiation medium, cells expressed a range of markers associated in humans with excitatory neurons in the upper cortical layer, including EMX1, CUX1, BRN2, SATB2, and TBR1. After 2 weeks, the equiPSCs had formed extensive TUJ1-positive, fasciculated neuronal networks. Expression of BRN2 within neural rosettes and by individual neurons confirmed the differentiation of excitatory cortical neurons. After 5 weeks of neuronal differentiation, neurons displayed spontaneous calcium transients and, where connections between adjacent neurons were observed, calcium transients were propagated unidirectionally, presumably from pre- to postsynaptic neurons.
To explore the suitability of the equiPSC-derived neurons as a platform for the study of viral neuropathic diseases in the horse, we examined their susceptibility to infection with flaviviruses that are known to be neurotropic in horses, including WNV and MVEV. We observed that the three strains of WNV and MVEV grew to high titres and caused neuronal cell death, suggesting that equiPSC-derived neurons permit ready replication of these viruses. Significantly, FRV and BgV, that do not cause pathology in horses, did not replicate in the equiPSC-derived neurons. In contrast, WNV and MVEV grew poorly in primary equine fibroblasts. These results are in concordance with in vivo findings of target cell tropism: the flaviviruses target neurons, leukocytes, pericytes, and epithelial cells, but flavivirus replication has never been described in fibroblasts, regardless of virus strain or host species studied [12,18,20,27,28].
While the route(s) of neuroinvasion of the neurotropic flaviviruses remains elusive [27] and may depend on factors such as virus strain, age of host and host-species, as well as the route of virus challenge in the case of experimental infections, it appears that many different types of neurons can become infected with WNV and MVEV, including cortical neurons, and it may to some extent be a stochastic process [27]. At the time that horses show clinical signs of flavivirus encephalitis, the virus has all but ceased to replicate in the brain, as judged by both immunohistochemistry and reverse transcription (RT)-PCR [28]; however, as judged by the distribution of inflammatory leukocyte infiltrates, all parts of the brain can be involved [12,28]. Thus, the sensitivity of our equiPSC-derived neurons, which display a cortical identity, to both WNV and MVEV, appears to confirm that they are eminently suitable as an in vitro model to study neuron–virus interactions.
Considering the economic impact of the equine industry in countries such as the United States and Australia, totaling $112 billion and $8 billion, respectively, the availability of cell-based therapeutics and assays is surprisingly limited. In this study, we show that equiPSCs rapidly generate cortical neurons that permit replication of WNV and MVEV and that they also express Ephrin B3, the receptor via which henipaviruses enter mammalian neurons [29]. Given the significant impact that both flavivirus and henipavirus infections can have on equine and, in some instances, human populations, our study represents a significant advance in the establishment of in vitro horse neuronal models that can be used to study, and treat or prevent, neurotropic viral infections in horses.
Footnotes
Acknowledgments
The authors thank Cheryl Johansen for providing FRV, Caitlin O'Brien for growing stock of FRV, Agathe Colmant for BgV stock, Jessica Harrison for stock of MVEV51, and Roy Hall for providing mAbs 4G2 and 4G4. This study was supported by an Alister Rogers Hendra Foundation grant (D.W.) and the Australian Research Council (ARC-LP120100686; H.B.O.).
Author Disclosure Statement
No competing financial interests exist.