
Abstract
Retinal disorders represent the main cause of decreased quality of vision and even blindness worldwide. The loss of retinal cells causes irreversible damage of the retina, and there are currently no effective treatment protocols for most retinal degenerative diseases. A promising approach for the treatment of retinal disorders is represented by stem cell-based therapy. The perspective candidates are mesenchymal stem cells (MSCs), which can differentiate into multiple cell types and produce a number of trophic and growth factors. In this study, we show the potential of murine bone marrow-derived MSCs to differentiate into cells expressing retinal markers and we identify the key supportive role of interferon-γ (IFN-γ) in the differentiation process. MSCs were cultured for 7 days with retinal extract and supernatant from T-cell mitogen concanavalin A-stimulated splenocytes, simulating the inflammatory site of retinal damage. MSCs cultured in such conditions differentiated to the cells expressing retinal cell markers such as rhodopsin, S antigen, retinaldehyde-binding protein, calbindin 2, recoverin, and retinal pigment epithelium 65. To identify a supportive molecule in the supernatants from activated spleen cells, MSCs were cultured with retinal extract in the presence of various T-cell cytokines. The expression of retinal markers was enhanced only in the presence of IFN-γ, and the supportive role of spleen cell supernatants was abrogated with the neutralization antibody anti-IFN-γ. In addition, differentiated MSCs were able to express a number of neurotrophic factors, which are important for retinal regeneration. Taken together, the results show that MSCs can differentiate into cells expressing retinal markers and that this differentiation process is supported by IFN-γ.
Introduction
T
MSCs are adult stem cells that can be isolated from most tissues of the organism and used as autologous cells. The main sources of MSCs are bone marrow and adipose tissue [4]. MSCs are able to migrate to the site of injury and differentiate into multiple cell types, including adipose, cartilage, and bone cells [5], or even transdifferentiate into neuronal [6,7], corneal [8], retinal cells [9], and other cell types. Moreover MSCs can suppress an inflammatory response by production of soluble immunomodulatory molecules or cell to cell contact [10]. Most of these regulatory molecules are produced at a higher level after the activation of MSCs by inflammation stimuli [11,12]. MSCs also produce a number of growth or trophic factors, which play an important role in the regeneration at the site of tissue injury [13]. Considering all these properties, MSCs are a promising candidate for stem cell-based therapy of retinal degenerative diseases.
In our previous studies, we have shown that bone marrow-derived MSCs are able to differentiate into corneal epithelial cells [14] and adipose tissue-derived MSCs were differentiated into neuron-like cells [15]. We also observed that mouse MSCs inhibit the acute phase of inflammation in an alkali-injured eye [16] and support the regeneration and healing of the ocular surface after alkali burn [17]. In the present study, we have characterized the potential of bone marrow-derived MSCs to differentiate into cells expressing retinal markers in cultures simulating the environment of diseased retina, and we have identified the key supportive role of interferon-γ (IFN-γ) in this differentiation process.
Materials and Methods
Mice
Female BALB/c mice (aged 8–14 weeks) were used in the experiments. The mice were obtained from the breeding unit of the Institute of Molecular Genetics of the Czech Academy of Sciences, Prague. The use of animals was approved by the local Animal Ethics Committee of the Institute of Experimental Medicine, Prague.
Isolation, culture, and purification of MSCs
MSCs were isolated from the bone marrow of female BALB/c mice. The bone marrow was flushed out from the femurs and tibias, and a single-cell suspension was prepared using tissue homogenizer. The cells were seeded at a concentration of 4 × 106 cells/mL in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, St. Louis, MO) containing 10% fetal bovine serum (FBS; Gibco BRL, Grand Island, NY), antibiotics (100 U/mL of penicillin, 100 μg/mL of streptomycin), and 10 mM HEPES buffer (referred as a complete DMEM) in 75-cm2 tissue culture flasks (Techno Plastic Products, Trasadingen, Switzerland). After a 48-h incubation at 37°C in an atmosphere of 5% CO2, the nonadherent cells were washed out and the remaining adherent cells were cultured for an additional 2 weeks at the same conditions. The adherent cells were harvested by incubation with 1 mL of 0.5% trypsin for 5 min and then gently scraped. The resulting cell suspension was cultured for 15 min with CD11b MicroBeads and CD45 MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's instructions. The cell suspension was then immunodepleted of CD11b+ and CD45+ contaminating cells using a magnetic activated cell sorter (MACS; Miltenyi Biotec). The purity and differentiation potential of MSCs were then characterized.
Phenotypic characterization of MSCs by flow cytometry
The cells were washed in phosphate-buffered saline (PBS) containing 0.5% bovine serum albumin and were incubated for 30 min with the following anti-mouse monoclonal antibodies (mAb): allophycocyanine (APC)-labeled anti-CD44 (clone IM7; BD PharMingen, San Jose, CA), phycoerythrin (PE)-labeled anti-CD73 (cloneTY/11.8; eBioscience, San Diego, CA), PE-labeled anti-CD105 (clone TY/11.8; eBioscience), APC-labeled anti-CD11b (clone M1/70; BioLegend, San Diego, CA), fluorescein isothiocyanate (FITC)-labeled anti-CD45 (clone 30-F11; BioLegend), or PE-labeled anti-CD31 (clone MEC 13.3; BD PharMingen). Cells stained with PE-labeled rat IgG2a (clone RTK2758; BioLegend), APC-labeled rat IgG2b (clone RTK4530; BioLegend), or FITC-labeled rat IgG2b (clone RTK4530; BioLegend) were used as negative controls. Dead cells were stained with Hoechst 33258 fluorescent dye (Invitrogen, Carlsbad, CA) added to the samples 10 min before flow cytometry analysis. Data were collected using an LSRII cytometer (BD Biosciences, Franklin Lakes, NJ) and analyzed using FlowJo software (Tree Star, Ashland, OR). The morphological characteristics and differentiation potential of purified MSCs have been described in detail elsewhere [16,18].
Preparation of tissue extracts
The posterior segments of the mouse eyes (containing retina) were harvested in serum-free DMEM on ice (1 segment/100 μL of DMEM). The posterior segments were used because of a small size of the mouse eye and difficulty to prepare pure retinal tissue in a sufficient quantity. We always tried to minimize the presence of nonretinal tissue. Similarly, small pieces (corresponding in size to the samples of the posterior eye tissue) were collected from the muscle, heart, or lung tissue. The samples were thoroughly homogenized and frozen at −80°C. The homogenate was thawed and frozen three times and centrifuged at 425 g for 10 min. The supernatant was filtered through a 0.22 μm filter (Millipore, Billerica, MA) and stored in aliquots at −80°C.
Preparation of supernatants from stimulated splenocytes
Mouse spleen was homogenized to a single-cell suspension and the cells were adjusted to a concentration 1.3 × 106 cells/mL. The cells were stimulated with 1 μg/mL of Concanavalin A (Con A; Sigma-Aldrich) in RPMI-1640 medium (Sigma-Aldrich), containing 10% of FBS, antibiotics, 5 × 10−5 M 2-mercaptoethanol, and 10 mM HEPES buffer (referred as a complete RPMI-1640 medium) at 37°C. The supernatants were harvested after a 48-h incubation, centrifugated, filtered through a 0.22 μm filter, and stored in aliquots at −80°C.
Preparation of supernatants from T cells, B cells, and macrophages
The single-cell suspensions of spleen cells were prepared in a complete RPMI-1640 medium. The B cells were isolated by positive selection using a CD19 MicroBead Isolation Kit (Miltenyi Biotec), and T cells were isolated by negative selection using a Pan T cell Isolation Kit (Miltenyi Biotec). The macrophages were obtained by flushing the peritoneal cavity and washing out the nonadherent cells. Purified T cells (cultured in the presence of macrophages as a source of antigen-presenting cells at a ratio of 20:1) were stimulated with Con A (1 μg/mL). Purified B cells or macrophages were stimulated with 5 μg/mL of lipopolysaccharide (LPS; Sigma-Aldrich). The supernatants were harvested after a 48-h incubation, centrifugated, filtered through 0.22 μm filter, and stored at −80°C.
Differentiation of MSCs
MSCs were seeded at a concentration of 7 × 104 cells/mL in a 12-well tissue culture plate (Nunc, Roskilde, Denmark) and were cultured for 2, 4, or 7 days in 1 mL of complete DMEM together with retinal extract (30% of the volume), supernatants from Con A-stimulated splenocytes (30% of the volume) or with a combination of the extract and supernatant. Half of the culture medium was exchanged after 3 days of differentiation with a fresh DMEM containing 30% of extract and 30% of supernatant, thus the final composition of the medium remained the same as at the beginning of differentiation process.
To identify the supportive molecule in the supernatants, MSCs were cultured with retinal extract, and the supernatant from Con A-stimulated spleen cells was replaced by DMEM containing IL-2, IL-6, IL-10, IL-17, IFN-γ, or TGF-β (all cytokines were purchased from PeproTech, Rocky Hill, NJ). The final concentration of cytokines in cultures was 20 ng/mL.
Detection of gene expression by real-time polymerase chain reaction
The expression of genes for retinal markers and growth factors was detected using real-time polymerase chain reaction (PCR). MSCs were cultured for 2, 4, or 7 days, untreated or in the presence of retinal extract, supernatant from stimulated splenocytes or both together. The total RNA was extracted using TRI Reagent (Molecular Research Center, Cincinnati, OH) according to the manufacturer's instructions. One microgram of RNA was treated with deoxyribonuclease I (Promega, Madison, WI) and used for subsequent reverse transcription. The first-strand cDNA was synthesized using random hexamers (Promega) in a total reaction volume of 25 μL using M-MLV Reverse Transcriptase (Promega).
Quantitative real-time PCR was performed in a StepOnePlus real-time PCR system (Applied Biosystems, Foster City, CA) using SYBR Green (Applied Biosystems) as previously described [14]. The sequences of the primers for glyceraldehyde-3-phosphate dehydrogenase (Gapdh), rhodopsin (Rho), S antigen (Sag), recoverin (Rcvr), retinaldehyde binding protein (Rlbp), calbindin 2 (Calb2), retinal pigment epithelium 65 (Rpe65), pigment epithelium-derived factor (Pedf), nestin (Nes), transforming growth factor beta (Tgf-β), interleukin 6 (Il-6), nerve growth factor (Ngf), and glial cell-derived neurotrophic factor (Gdnf) genes used for amplification are presented in Table 1.
Calb2, calbindin 2; Gapdh, glyceraldehyde-3-phosphate dehydrogenase; Gdnf, glial cell-derived neurotrophic factor; Il-6, interleukin 6; Nes, nestin; Ngf, nerve growth factor; Pedf, pigment epithelium-derived factor; Rcvr, recoverin; Rho, rhodopsin; Rlbp, retinaldehyde-binding protein; Rpe65, retinal pigment epithelium 65; Sag, S antigen; Tgf-β, transforming growth factor beta.
The relative gene expression was normalized by the endogenous control Gapdh. The PCR parameters included denaturation at 95°C for 3 min followed by 40 cycles at 95°C for 20 s, annealing at 60°C for 30 s, and elongation at 72°C for 30 s. Fluorescence data were collected at each cycle after an elongation step at 80°C for 5 s and were analyzed using StepOne Software version 2.2.2 (Applied Biosystems). The possibility of the presence of RNA in retinal extract was excluded by a negative gel electrophoresis (data not shown), and the extracts were also tested as control for PCR.
Neutralization of IFN-γ in supernatants from stimulated splenocytes
MSCs were differentiated in the presence of retinal extract and supernatant from stimulated splenocytes. The supernatant was either untreated or preincubated for 10 min with neutralization antibody anti-IFN-γ (PeproTech) at a concentration of 5 μg/mL before being added to the culture. The expression of gene for Rho was determined after a 7-day incubation by real-time PCR.
Immunostaining with antirhodopsin antibody
The untreated or differentiated MSCs were fixed with 4% paraformaldehyde for 1 h and then permeabilized using 0.1% Triton X-100 for 20 min. The samples were incubated with anti-mouse FITC-labeled mAb anti-Rho (clone 4D2; Abcam, Cambridge, United Kingdom) for 2 h. After rinsing with PBS, the cells were fixed on glass slides with Mowiol 4–88 (Calbiochem, San Diego, CA), and nuclei were stained with 4′,6′-diamidino-2-phenylindole (DAPI) dye. The samples were visualized by fluorescent microscope (Leica, Wetzlar, Germany).
Statistical analysis
The results are expressed as the mean ± SD. Comparisons between the two groups were analyzed by Student's t-test, and multiple comparisons were performed by ANOVA. A value of P < 0.05 was considered statistically significant.
Results
Phenotypic characterization of MSCs
The phenotype of MACS-separated MSCs was characterized by flow cytometry. The cells were positive for CD44, CD73, and CD105, which are markers attributed to murine MSCs, and were negative for leucocyte markers CD11b, CD45, and CD31 (Fig. 1). In addition, the growing MSCs had a typical fibrocyte-like morphology, adhered to plastic and were able to undergo adipogenic and osteogenic differentiation, as we have described previously [18].

Phenotypic characterization of MSCs. Representative histograms show the flow cytometry analysis of CD11b, CD45, CD31
Detection of retinal markers in differentiated MSCs
MSCs were cultured with retinal extract and/or supernatant from stimulated splenocytes to simulate the environment of the damaged retina. The expression of genes for retinal markers was detected by real-time PCR. First, we selected six retinal markers Rho, Sag, Rcvr, Rlbp, Rpe65, and Calb2, which were strongly expressed in the retina, but were not or only weakly expressed by MSCs (Fig. 2). Two other tested markers, Pedf and Nes, were expressed in a higher level in MSCs than in the retina and therefore were not used in the next studies.
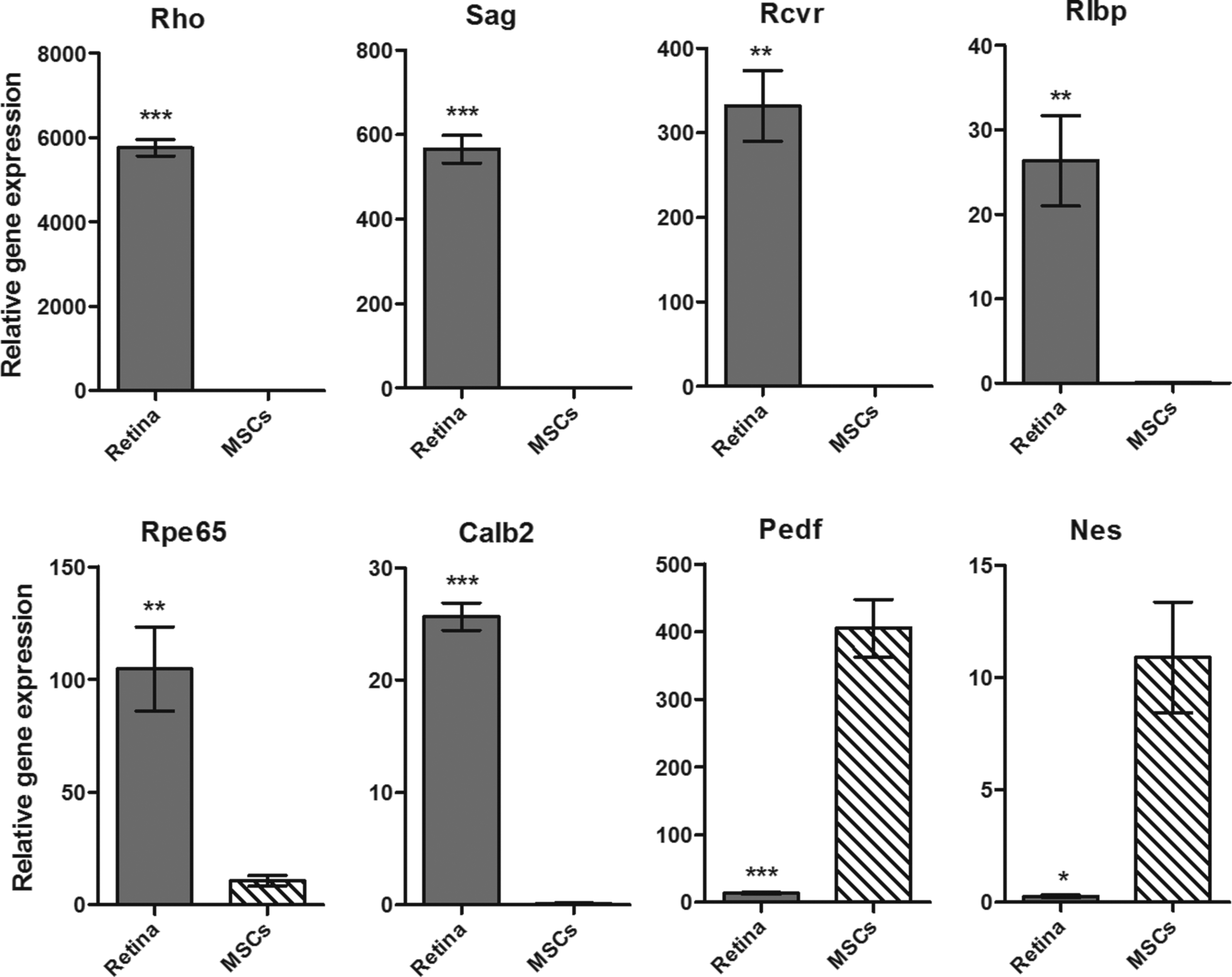
The expression of genes for retinal markers in MSCs and retina. The expression of rhodopsin (Rho), S antigen (Sag), recoverin (Rcvr), retinaldehyde-binding protein (Rlbp), retinal pigment epithelium-specific protein 65 (Rpe65), calbindin 2 (Calb2), pigment epithelium-derived factor (Pedf), and nestin (Nes) genes was determined by real-time PCR in untreated MSCs and the retinal tissue. Each bar represents the mean ± SD from three independent determinations. Values with asterisks are significantly different (*P < 0.05, **P < 0.01, ***P < 0.001) from untreated MSCs. PCR, polymerase chain reaction.
Untreated MSCs and MSCs cultured with supernatant from Con A-stimulated spleen cells expressed undetectable or only very low levels of retinal markers. The level of expression of retinal markers slightly increased in the presence of retinal extract, but was significantly enhanced after culturing of MSCs with retinal extract and supernatant together. As demonstrated in Fig. 3, MSCs expressed significant levels of genes for markers typical for photoreceptors (Rho, Sag, Rcvr), horizontal and bipolar cells (Calb2), Muller cells (Rlbp), and RPE cells (Rlbp, Rpe65). These suggested that our differentiation protocol enabled MSCs to differentiate into cells of multiple retinal layers. For further detailed analysis of the differentiation process, we selected photoreceptor marker Rho, which had the highest level of expression in differentiated MSCs. The number of Rho-positive cells increased with the time of differentiation, and using flow cytometry we detected that 7%–15% of cells expressed rhodopsin protein on day 7 of differentiation (data not shown).

The expression of retinal markers in differentiated MSCs. MSCs were cultured untreated (-), with supernatant from stimulated splenocytes (Sup), with retinal extract (Ext), or with supernatant and retinal extract together (Ext+Sup). The expression of rhodopsin (Rho), S antigen (Sag), recoverin (Rcvr), retinaldehyde-binding protein (Rlbp), retinal pigment epithelium-specific protein 65 (Rpe65), and calbindin 2 (Calb2) genes was detected after 7 days of differentiation by real-time PCR. Each bar represents the mean ± SD from three independent determinations. Values with asterisks are significantly different (**P < 0.01, ***P < 0.001) from untreated MSCs.
The kinetic of expression of the retinal genes during the differentiation process
The expression of gene for Rho in untreated MSCs is undetectable and was already upregulated after 2-day cultivation of cells with retinal extract or with extract in combination with supernatant. After 4 and 7 days of differentiation, the expression of the Rho gene was gradually increasing, especially in the cultures containing both retinal extract and spleen cell supernatant (Fig. 4A). The expression of other retinal markers displayed a similar trend as the rhodopsin. The fold changes of gene expression of differentiated MSCs (cultivated with retinal extract and supernatant) relative to untreated MSCs are summarized in Fig. 4C.
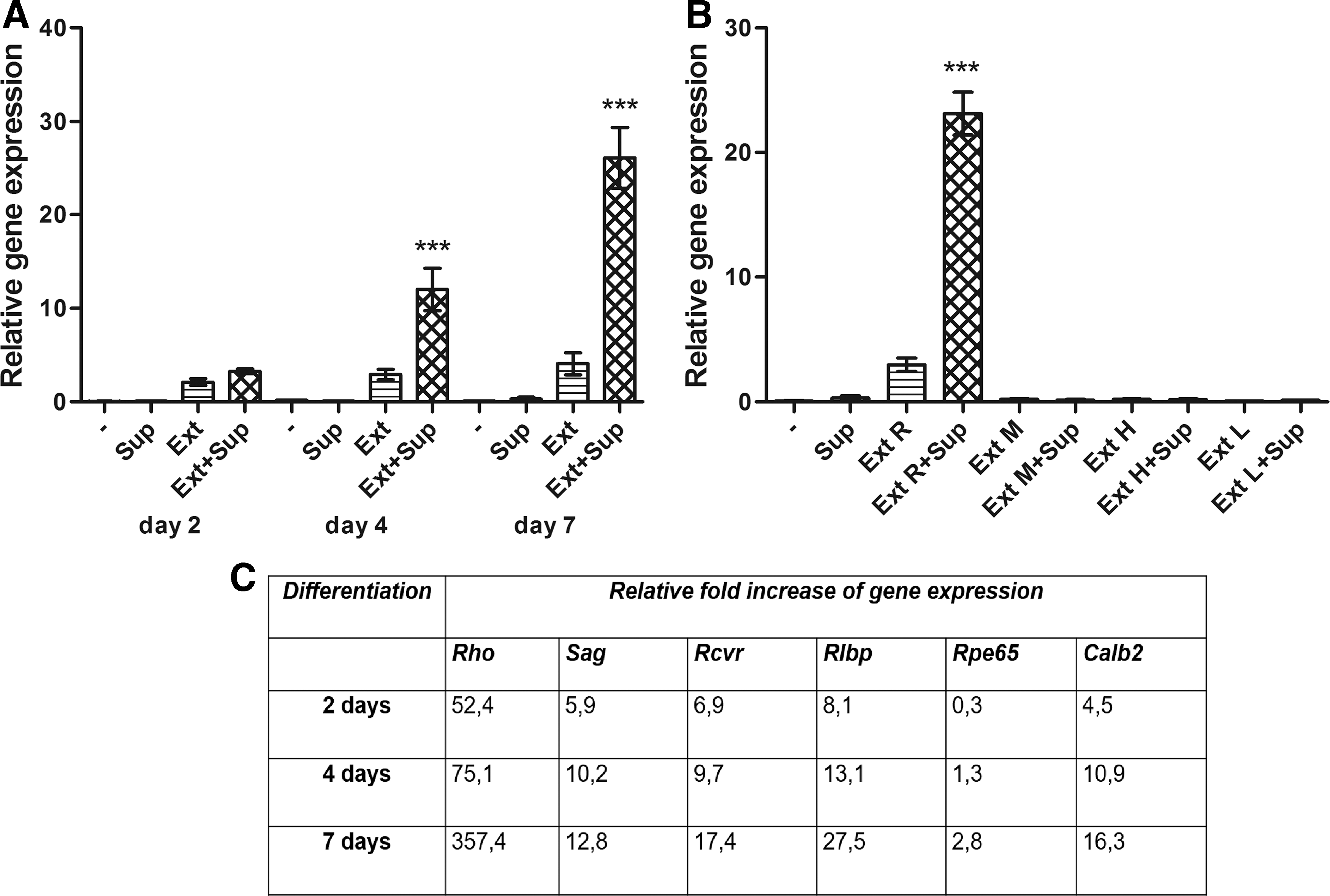
The kinetics of expression of the retinal genes and the specific role of retinal extract during the differentiation process. MSCs were cultured untreated (-), with supernatant from stimulated splenocytes (Sup), with retinal extract (Ext) or with supernatant and retinal extract together (Ext+Sup). The expression of the Rho gene was determined by real-time PCR after 2, 4, and 7 days of incubation
The specific role of retinal extract in the differentiation process
To demonstrate the tissue specificity of retinal extract in the differentiation process, the extracts from retina, muscle, heart, and lung were compared in differentiation protocol. The significant increase in the Rho gene expression was detected only in cultures containing retinal extract and not in MSCs differentiated in the presence of muscle, heart, or lung extract with or without the spleen cell supernatant (Fig. 4B).
The role of IFN-γ in the differentiation process
We observed that the combination of retinal extract and supernatant from Con A-stimulated splenocytes represented the optimal conditions for retinal differentiation of MSCs. To identify a molecule in the supernatant, which is responsible for the increased expression of retinal markers, the supernatants were prepared from Con A-stimulated spleen cells, from unstimulated or Con A-stimulated T cells, unstimulated or LPS-stimulated B cells, and from unstimulated or LPS-stimulated macrophages. MSCs were cultured for 7 days with these supernatants or with retinal extract and these supernatants. As demonstrated in Fig. 5A, the significant increase in the Rho gene expression occurred only in cultures containing retinal extract and supernatant from Con A-stimulated spleen cells or Con A-stimulated purified T cells. The supernatants from unstimulated cells or from mitogen-stimulated B cells or macrophages did not have a supportive effect on MSC differentiation.
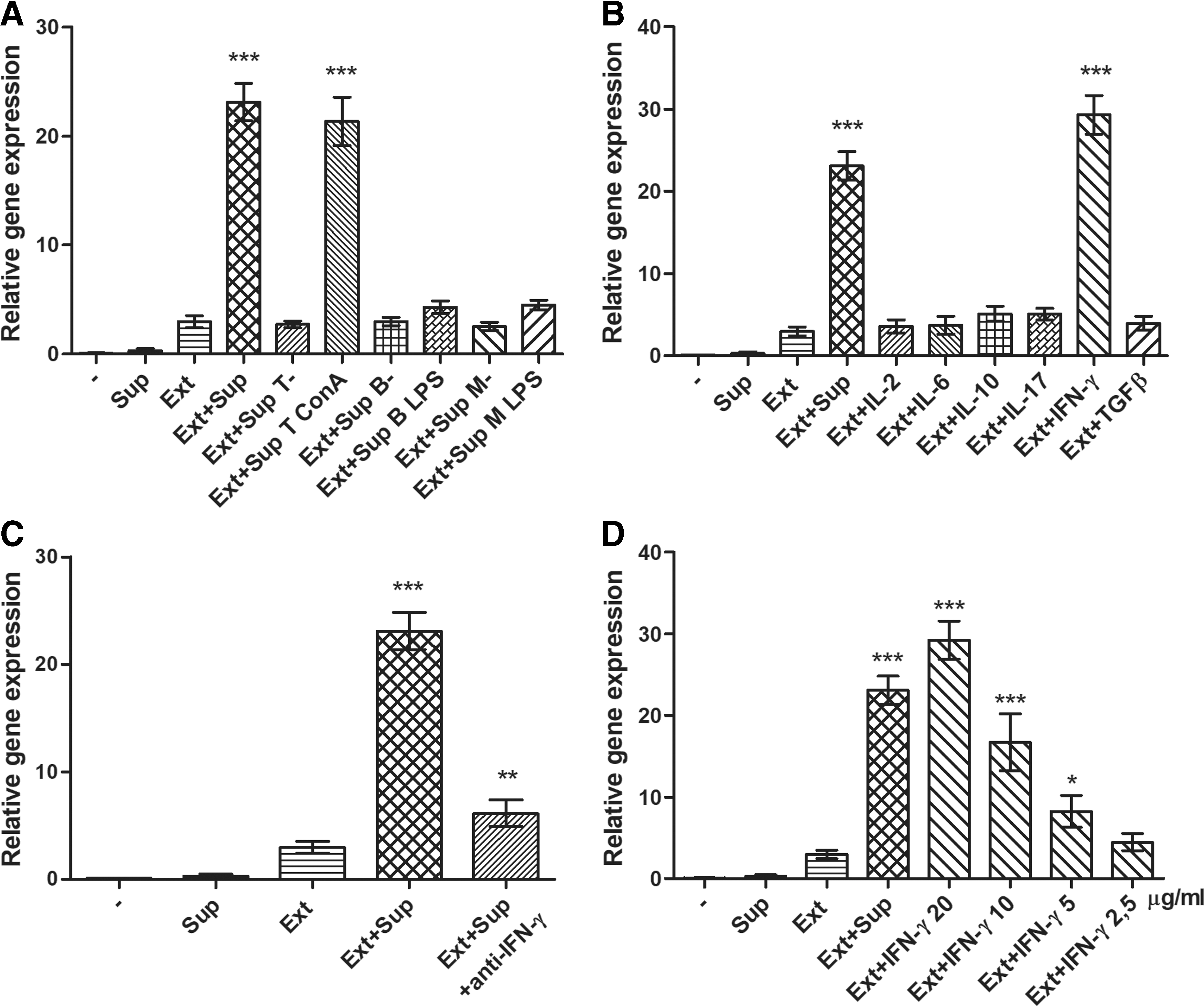
The effect of IFN-γ in the differentiation process. The effect of supernatant from stimulated splenocytes (Sup) was compared with supernatants from unstimulated T cells (Sup T-), B cells (Sup B-), or macrophages (Sup M-) and Con A-stimulated T cells (Sup T ConA), LPS-stimulated B cells (Sup B LPS), and LPS-stimulated macrophages (Sup M LPS)
These observations indicated that the molecule supporting retinal differentiation is a product of activated T cells. Therefore, we cultured MSCs with retinal extract in the presence of various T-cell cytokines such as IL-2, IL-6, IL-10, IL-17, IFN-γ, and TGF-β. As shown in the Fig. 5B, the enhanced expression of the Rho gene occurred only in the presence of IFN-γ, whereas other cytokines were without any supportive effect. Similarly, none of the cytokines from a wider panel of tested cytokines and growth factors (IL-1, 2, 4, 6, 7, 10, 12, 13, 15, 17, TGF-β, TNF-α, IGF-I, EGF, HGF, KGF, LIF, NGF, or FGF) supported the differentiation of MSCs into cells expressing retinal markers (data not shown).
To confirm the supportive role of IFN-γ in a differentiation process, MSCs were cultured with retinal extract and supernatant from Con A-stimulated splenocytes with added neutralization antibody anti-IFN-γ. The anti-IFN-γ antibody completely abrogated the supportive role of the supernatant on the Rho gene expression (Fig. 5C). To demonstrate a dose-dependent effect of IFN-γ on MSC differentiation, IFN-γ at the concentrations 2.5–20 ng/mL was added to the cultures of MSCs with retinal extract. As demonstrated in Fig. 5D, the expression of Rho gene was enhanced by IFN-γ in a dose-dependent manner (Fig. 5D).
The expression of genes for growth factors and cytokines in differentiated MSCs
The expression of genes for NGF, GDNF, IL-6, and TGF-β was tested in untreated MSCs, and MSCs differentiated for 7 days with retinal extract and supernatant from activated splenocytes. The level of expression of genes for NGF, GDNF, and IL-6 was significantly increased in differentiated MSCs in comparison with untreated MSCs (Fig. 6). On the contrary, the expression of the Tgf-β gene remained on the same level in both untreated and differentiated cells (Fig. 6).
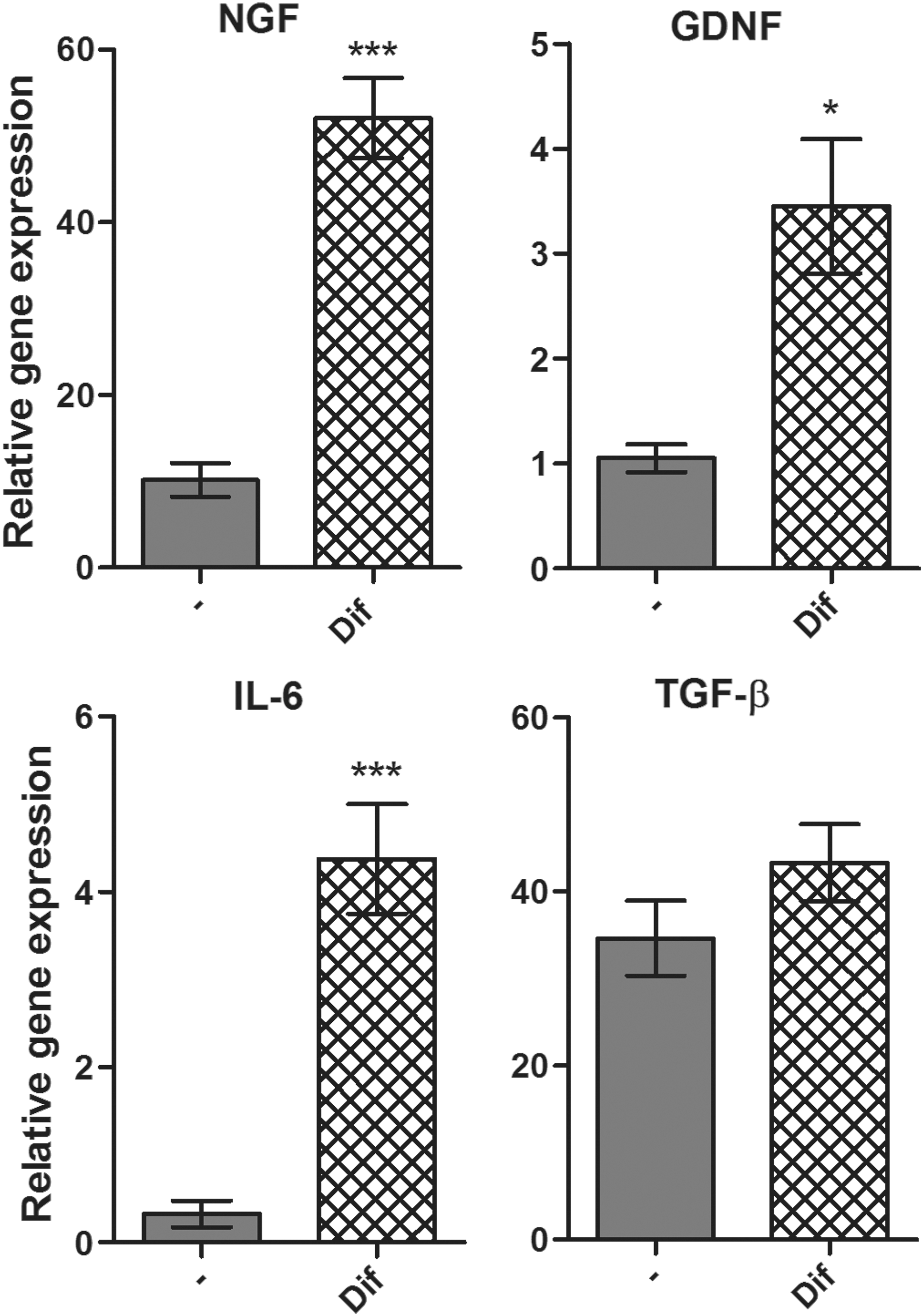
The expression of genes for growth factors in untreated and differentiated MSCs. The expression of Ngf, Gdnf, Il-6, and Tgf-β genes was detected in untreated MSCs (-) and MSCs differentiated with retinal extract and supernatant from splenocytes (dif) by real-time PCR after 7 days. Each bar represents the mean ± SD from three independent determinations. Values with asterisks are significantly different (*P < 0.05, ***P < 0.001) from untreated MSCs.
Detection of Rho protein in differentiated cells by immunocytochemistry
The differentiation potential of MSCs was confirmed by immunostaining for the Rho protein. While untreated MSCs were negative for Rho (Fig. 7A), MSCs cultured for 7 days with retinal extract and supernatant from activated spleen cells were clearly positive for the Rho protein (Fig. 7B).
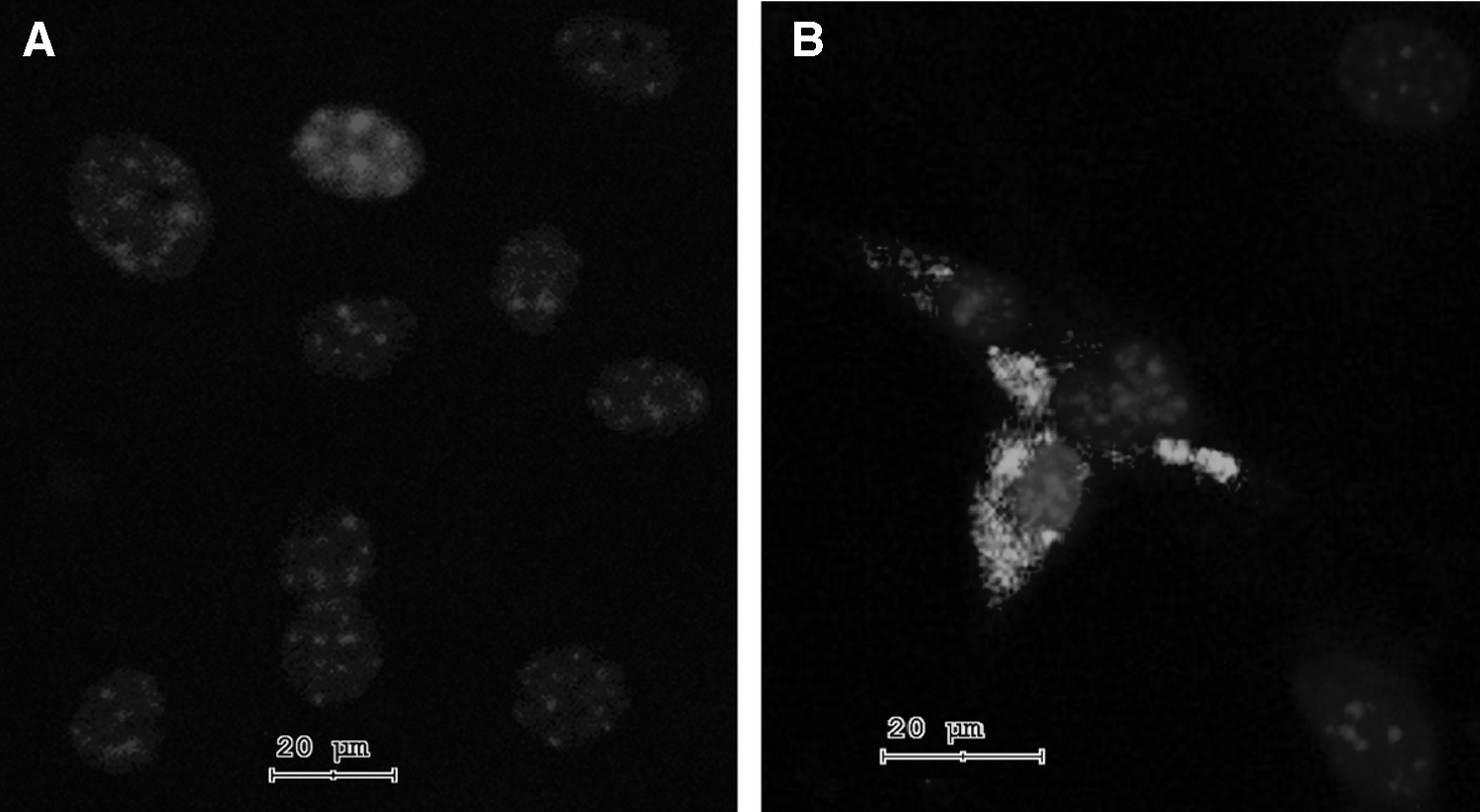
Demonstration of Rho protein by immunocytochemistry. The expression of Rho protein in untreated MSCs
Discussion
There is still an absence of effective treatment protocols for sight-threatening degenerative retinal diseases. For this reason, the potential application of stem cell therapy represents a great promise. MSCs, with their ability to differentiate into multiple cell types are a perspective source of replacement and regeneration of damaged retinal cells.
It has been shown that MSCs are able to differentiate into various retinal cell types [3]. MSCs isolated from rat conjunctiva after culturing in the presence of taurine expressed markers characteristic for photoreceptors and bipolar cells [19]. Taurine, together with activin A and epidermal growth factor, was used in another study to differentiate MSCs to photoreceptors. The cells differentiated for 8–10 days expressed the Rho and Rlbp genes [9]. The same authors also showed that MSCs injected into the subretinal space are able to integrate into the retina and express markers specific for photoreceptors. Other studies have demonstrated that transplantation of MSCs into the damaged retina induced expression of markers typical for photoreceptors, bipolar cells, and amacrine cells [20 –22] in grafted MSCs. There are also several studies showing the differentiation of MSCs into RPE cells [21,23], which are important for the nourishment of photoreceptors, and disorders of RPE cells result in photoreceptor dysfunction. Human MSCs were also differentiated into RPE-like cell types after culturing with RPE cells in vitro [24].
In our study, to differentiate MSCs, we simulated the environment of diseased or injured retina. The retinal extract mimicked the environment of the damaged retinal tissue, and the supernatant from stimulated lymphocytes simulated the inflammation at the site of injury. MSCs cultured in the presence of retinal extract and supernatant from Con A-stimulated spleen cells expressed markers characteristic not only for one type of retinal cells but also for several types of retinal cells (photoreceptors, bipolar, and horizontal cells, Müller cells and RPE cells). This could be an advantage since retinal degenerative diseases often affect multiple retinal layers and various cell types. In our protocol, the retinal extract ensures the specificity of differentiation. The induction (or increase) of retinal gene expression in MSCs occurred only in the presence of retinal extract. As specificity control, similarly prepared extracts from muscle tissue, heart, or lung did not induce the expression of genes for retinal markers, even in the presence of supernatants.
We observed that MSCs cultured in the presence of retinal extract and supernatant expressed a significantly higher level of photoreceptor marker Rho than MSCs differentiated only with the extract. In this respect, it has been shown that MSCs need, for activation and higher production of inducible molecules, stimulation with proinflammatory factors [12,16]. To search for a molecule responsible for the increased expression of photoreceptor marker Rho, we cultured MSCs with retinal extract and supernatant from unstimulated or mitogen-stimulated T cells, B cells, or macrophages. The level of the Rho gene expression was enhanced only in the culture containing retinal extract and supernatant from Con A-stimulated spleen cells or T cells. This finding indicated that the molecule supporting MSC differentiation is a T-cell product. For further characterization of putative molecule supporting MSC differentiation, we cultured MSCs with retinal extract and a panel of T-cell cytokines. The enhanced levels of the Rho gene expression were detected only in cultures with retinal extract and IFN-γ.
The key role of IFN-γ in the differentiation of MSCs into cells expressing the Rho gene was verified by neutralization antibody anti-IFN-γ. The differentiation of MSCs in cultures containing retinal extract and supernatant from Con A-stimulated spleen cells was completely abrogated by the adding of the neutralization anti-IFN-γ antibody. These results identify IFN-γ as a molecule playing a key supportive role in the differentiation of MSCs into cells expressing retinal markers. The role of IFN-γ in differentiation process has been indicated in some other models. For example, Croitoru-Lamoury et al. [25] demonstrated that IFN-γ inhibited adipogenic and osteogenic differentiation of MSCs, but increased the expression of neural markers in differentiated cells [25]. The ability of IFN-γ to support neuronal differentiation of neural stem cells was observed by Wong et al. [26].
In accordance with the published data [27 –29], we have shown that MSCs expressed genes for neurotrophic factors NGF and GDNF and for cytokines IL-6 and TGF-β. The level of expression of genes Ngf, Gdnf, and Il-6 was significantly enhanced in differentiated MSCs, which suggests their higher potential for regeneration of retinal cells. It was demonstrated that the supernatants from light-injured retina significantly promote secretion of neurotrophic factors by MSCs and slow down the process of apoptosis in damaged retinal cells [30]. Another study showed that secretion of neurotrophic factors by MSCs promoted viability of photoreceptors in vitro, and also supported their survival after subretinal transplantation of MSCs in a retinal degeneration model [31]. Thus, MSCs differentiated according to our protocol have a higher secretory activity than untreated MSCs and may have a better regenerative potential than primary MSCs.
In conclusion, we have demonstrated the key supportive role of IFN-γ in the differentiation of MSCs into the cells expressing retinal markers. Moreover, it was shown that differentiated MSCs are a potent source of neurotrophic factors, which are important for the regeneration of damaged retinal cells. All these properties make MSCs a promising candidate for stem cell-based therapy of retinal degenerative diseases.
Footnotes
Acknowledgments
This work was supported by the project 80815 from the Grant Agency of Charles University, grant 17-04800S from the Grant Agency of the Czech Republic and by the projects SVV 244-260435, CZ.1.05/1.1.00/02.0109, CZ.2.16/3.1.00/21528, and NPUI: LO1309.
Author Disclosure Statement
No competing financial interests exist.