
Abstract
Stromal cell-derived factor-1α (SDF-1α) drives endothelial colony-forming cell (ECFC) homing and incorporation within neovessels, thereby restoring tissue perfusion in ischemic tissues and favoring tumor vascularization and metastasis. SDF-1α stimulates ECFC migration by activating the Gi-protein-coupled receptor, CXCR4, and then engaging the phosphoinositide 3-kinase (PI3K)/AKT signaling pathway. Sporadic evidence showed that SDF-1α may also act through an increase in intracellular Ca2+ concentration ([Ca2+]i) in bone marrow-derived hematopoietic progenitor cells and fully differentiated endothelial cells. Of note, recent evidence demonstrated that intracellular Ca2+ signals play a key role in controlling the proangiogenic activity of ECFCs. The present investigation was, therefore, undertaken to assess whether and how SDF-1α induces ECFC motility by triggering intracellular Ca2+ signals. We found that SDF-1α caused a dose-dependent increase in [Ca2+]i that was inhibited by ADM3100, a selective CXCR4 antagonist. Pharmacological manipulation revealed that the Ca2+ response to [Ca2+]i was shaped by an initial intracellular Ca2+ release through inositol-1,4,5-trisphosphate receptors (InsP3Rs), followed by a sustained phase of extracellular Ca2+ entry through store-operated Ca2+ channels. InsP3-dependent Ca2+ release and store-operated Ca2+ entry (SOCE) were both necessary for SDF-1α-induced extracellular signal-regulated kinases 1/2 (ERK 1/2) and AKT phosphorylation. Finally, SDF-1α employed intracellular Ca2+ signals, ERK 1/2, and PI3K/AKT to promote ECFC migration in vitro and neovessel formation in vivo. These data, therefore, provide the first evidence that SDF-1α induces ECFC migration through the Ca2+-dependent activation of the ERK 1/2 and PI3K/AKT pathways.
Introduction
E
An increase in intracellular Ca2+ concentration ([Ca2+]i) is emerging as a key determinant of the proangiogenic activity of ECFC [11,12]. ECFC proliferation and tube formation are tightly controlled by the interaction between the inositol-1,4,5-trisphosphate (InsP3)-induced Ca2+ depletion of the endoplasmic reticulum (ER), the largest endogenous Ca2+ reservoir in these cells [13], and the subsequent store-operated Ca2+ entry (SOCE) across the plasma membrane [14]. Accordingly, vascular endothelial growth factor (VEGF) stimulates phospholipase Cγ (PLCγ) to cleave off InsP3 from phosphatidylinositol-4,5-bisphosphate (PIP2), thereby inducing Ca2+ release from the ER. The following drop in ER Ca2+ levels prompts is detected by Stim1, a sensor of ER Ca2+ concentration, which undergoes a conformation rearrangement, oligomerizing, and repositioning from the bulk ER to ER-plasma membrane contact sites. Herein, Stim1 interacts with and gates two Ca2+ permeable channels, namely Orai1 and Transient Receptor Potential (TRP) Canonical 1 (TRPC1), which altogether mediate SOCE [11,14 –17]. The dynamic interplay between InsP3-induced ER Ca2+ release and SOCE results in the generation of intracellular Ca2+ oscillations, which promote ECFC proliferation and tube formation by inducing the nuclear translocation of the Ca2+ sensitive transcription factor, NF-κB [16]. Of note, SDF-1α has been shown to stimulate migration through an increase in [Ca2+]i in BM-derived hematopoietic progenitor and stem cells [18 –20], but whether this mechanism also occurs in ECFCs is still unknown. Therefore, a better understanding of the signal transduction pathways that guide ECFCs toward the site of neovessel formation is mandatory to improve the therapeutic outcome of ECFC-based regenerative medicine [2,5].
The present investigation sought to assess the role played by intracellular Ca2+ signaling in SDF-1α-induced ECFC migration. We demonstrated that SDF-1α activates CXCR4 to cause a dose-dependent biphasic increase in [Ca2+]i. The Ca2+ response to SDF-1α required an initial rapid phase of Ca2+ release through InsP3 receptors (InsP3Rs), which was followed by a slower phase of Ca2+ entry through store-operated channels (SOCs). SDF-1α-induced Ca2+ signals led to the recruitment of the ERK 1/2 and PI3K/AKT signaling pathways, which were both involved in ECFC migration and neovascularization in vivo. These data shed further light on the molecular mechanisms, whereby SDF-1α controls ECFC migration and could be useful to either boost or inhibit CXCR4 signaling for therapeutic purposes in ischemic and cancer patients, respectively.
Materials and Methods
Isolation and cultivation of ECFCs
Blood samples (40 mL) collected in EDTA (ethylenediaminetetraacetic acid)-containing tubes were obtained from healthy human volunteers from 22 to 28 years of age. The Institutional Review Board at “Istituto di Ricovero e Cura a Carattere Scientifico Policlinico San Matteo Foundation” in Pavia approved all protocols and specifically approved this study. Informed written consent was obtained according to the Declaration of Helsinki of 1975 as revised in 2008. We focused on the so-called ECFCs, a subgroup of EPCs, which are found in the CD34+ CD45− fraction of circulating mononuclear cells (MNCs), exhibit robust proliferative potential, and form capillary-like structures in vitro [21]. To isolate ECFCs, MNCs were separated from peripheral blood by density gradient centrifugation on lymphocyte separation medium for 30 min at 400 g and washed twice in EBM-2 with 2% FCS. A median of 36 × 106 MNCs (range 18–66) was plated on collagen-coated culture dishes (BD Biosciences) in the presence of the Endothelial Cell Growth Medium EGM-2 MV Bullet Kit (Lonza) containing endothelial basal medium (EBM-2), 5% fetal bovine serum (FBS), recombinant human (rh) EGF, rhVEGF, rhFGF-B, rhIGF-1, ascorbic acid, and heparin, and maintained at 37°C in 5% CO2 and humidified atmosphere. Nonadherent cells were discarded after 2 days and thereafter, medium was changed three times a week. The outgrowth of ECs from adherent MNCs was characterized by the formation of a cluster of cobblestone-shaped cells. That ECFC-derived colonies belonged to endothelial lineage was confirmed as described in [14,22]. In more detail, ECFC-derived colonies were characterized by staining them with anti-CD31, anti-CD105, anti-CD144, anti-CD146, anti-von Willebrand Factor, anti-CD45, and anti-CD14 monoclonal antibodies, and by assessment of capillary-like network formation in the in vitro Matrigel assay.
For our experiments, we have mainly used endothelial cells obtained from early passage ECFCs (P1-3, which roughly encompasses a 15–18 day period) with the purpose to avoid (or maximally reduce) any potential bias due to cell differentiation. However, to make sure that the phenotype of the cells did not change throughout the experiments, in preliminary experiments we tested the immunophenotype of ECFCs at different passages and we found no differences [17]. We also tested whether functional differences occurred when early (P2) and late (P6) passage-ECFCs were used by testing the in vitro capacity of capillary network formation in a Matrigel assay and found no differences between early and late passage ECFCs-derived cells.
Solutions
Physiological salt solution (PSS) had the following composition (in mM): 150 NaCl, 6 KCl, 1.5 CaCl2, 1 MgCl2, 10 Glucose, 10 Hepes. In Ca2+-free solution (0Ca2+), Ca2+ was substituted with 2 mM NaCl, and 0.5 mM EGTA was added. Solutions were titrated to pH 7.4 with NaOH. In Mn2+-quenching experiments, 200 μM MnCl2 was added to the 0Ca2+ external solution. The osmolality of PSS as measured with an osmometer (Wescor 5500, Logan, UT) was 338 mmol/kg.
[Ca2+]i measurements
ECFCs were loaded with 4 μM fura-2 acetoxymethyl ester (Fura-2/AM; 1 mM stock in dimethyl sulfoxide) in PSS for 1 h at room temperature. After washing in PSS, the coverslip was fixed to the bottom of a Petri dish and the cells observed by an upright epifluorescence AxioLab microscope (Carl Zeiss, Oberkochen, Germany), usually equipped with a Zeiss × 40 Achroplan objective (water-immersion, 2.0 mm working distance, 0.9 numerical aperture). ECFCs were excited alternately at 340 and 380 nm, and the emitted light was detected at 510 nm. A first neutral density filter (1 or 0.3 optical density) reduced the overall intensity of the excitation light and a second neutral density filter (optical density = 0.3) was coupled to the 380 nm filter to approach the intensity of the 340 nm light. A round diaphragm was used to increase the contrast. The excitation filters were mounted on a filter wheel (Lambda 10; Sutter Instrument, Novato, CA). Custom software, working in the LINUX environment, was used to drive the camera (Extended-ISIS Camera, Photonic Science, Millham, United Kingdom) and the filter wheel, and to measure and plot online the fluorescence from 10 up to100 rectangular “regions of interest” (ROI). Each ROI was identified by a number. Since cell borders were not clearly identifiable, a ROI may not include the whole cell or may include part of an adjacent cell. Adjacent ROIs never superimposed. [Ca2+]i was monitored by measuring, for each ROI, the ratio of the mean fluorescence emitted at 510 nm when exciting alternatively at 340 and 380 nm (shortly termed “ratio”). An increase in [Ca2+]i causes an increase in the ratio [17]. Ratio measurements were performed and plotted online every 3 s. The experiments were performed at room temperature (22°C).
Western blotting
Around 5 × 105 ECFCs/condition were harvested and suspended in PSS and pretreated or not with BAPTA-AM (30 μM), BTP2 (10 μM), or 2-APB (50 μM), for 30 min at 37°C and 5% CO2. Subsequently, ECFCs were stimulated or not with 10 ng/mL SDF-1α, at 37°C and 5% CO2. After different time points (5-20-40 min) samples were lysed with Hepes-glycerol lysis buffer (Hepes 50 mM, NaCl 150 mM, 10% glycerol, 1% Triton X-100, MgCl2 1.5 mM, EGTA 1 mM, NaF 10 mM, Na3VO4 1 mM, 1 μg/mL leupeptin, 1 μg/mL aprotinin) on ice for 30 min and lysates clarified by centrifugation at 15,700 g, for 15 min at 4°C [23]. Samples containing equal amounts of proteins were subjected to 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF Membrane (Bio-Rad, Milan, Italy). Membranes were incubated using the following affinity-purified antibodies: rabbit polyclonal anti-phospho-AKT (1:1,000; Cell Signaling Technology), rabbit polyclonal anti-AKT (1:1,000; Cell Signaling Technology), rabbit monoclonal anti-phospho-ERK 1/2 (clone AW39) (1:1,000; Merck Millipore), mouse monoclonal anti-ERK 1/2 (clone 137F5) (1:2,000; Cell Signaling Technology), rabbit monoclonal anti-phospho-Src Family (clone D49G4) (1:1,000; Cell Signaling Technology), mouse monoclonal anti-Src (clone L4A1) (1:2,000; Cell Signaling Technology), rabbit monoclonal anti-phospho-p38 (clone E229) (1:2,000; Abcam), mouse monoclonal anti-p38 (1:2000, Abcam), mouse monoclonal anti-β-actin (clone DM1) (1:5,000; Sigma), following the conditions recommended by the manufacturers. Immunoreactive bands were detected by horseradish peroxidase-labeled secondary antibodies (Bio-Rad), using enhanced chemiluminescence reagent (Merck-Millipore). Prestained protein ladders (Bio-Rad) were used to estimate molecular weights.
Cell migration assay
Cell migration assay was performed, with the transwell migration chamber system (Merck Millipore, Milan, Italy), as previously published [23 –26]. Briefly, 96-well plates with polycarbonate inserts having 0.3 cm2/well membrane area with 8 μm pore size were seeded with 5 × 103 ECFC/well in the upper chambers. The lower chamber was filled with culture medium supplemented or not with 10 ng/mL SDF-1α (PeproTech, London, United Kingdom). The cells were left to migrate for 16 h at 37°C and 5% CO2. At the end of the incubation cells remaining on the upper face of the filters were removed by a cotton wool swab. Inserts were then washed three times with PBS and cells on the underside of the membrane were fixed with 4% PFA and stained with Hoechst 33258 (Sigma). Finally, the membranes were washed again, cut out with a scalpel, and mounted onto glass slides. The assay was performed in five independent experiments for each condition. Images were acquired using an Olympus BX51 microscope (Olympus, Deutschland GmbH, Hamburg, Germany). The number of cells that had migrated was counted by analyzing the entire area of the membranes. Data are expressed as mean ± SD number of cells migrated per analyzed field.
In vivo Matrigel plug assay
Experiments involving animals were ratified by the Italian Ministry of Health and have been done in accordance with the applicable Italian laws (D.L.vo 26/14 and following amendments) and the institutional guidelines. In vivo studies were carried out on nonobese diabetic/severe combined immunodeficient (NOD/SCID) interleukin-2 receptor g (IL-2Rg)–null (NSG) 6- to 8-week-old mice. Mice were bred and housed under pathogen-free conditions in the animal facility at the European Institute of Oncology–Italian Foundation for Cancer Research (FIRC) Institute of Molecular Oncology (IEO-IFOM, Milan, Italy). Mice were injected subcutaneously into the laterodorsal abdominal region with 0.3 mL of Matrigel basement membrane matrix (BD Biosciences) mixed with 250,000 human ECFCs containing either PBS or SDF-1α (10 ng/mL) combined with BAPTA (30 μM) or BTP2 (10 μM). Twelve days later, Matrigel plugs were removed, fixed in 4% phosphate-buffered formalin, embedded in paraffin, and sectioned for immunohistochemical staining. Briefly, slides were deparaffinized by xylene and rehydrated in graded alcohol. Sections were incubated with blocking solution (2% BSA in TBS) and stained with primary antibody (anti-human CD31, clone JC70A; DAKO). Slides were incubated with biotinylated secondary antibody for 30 min at room temperature. After washing, sections were incubated in peroxidase substrate solution (DAB; DAKO), rinsed in water, and counterstained with Hematoxylin. Images were acquired with a ScanScope XT scanner (Leica) and analyzed with Aperio Digital Pathology software.
Statistics
As to Ca2+ imaging, all data have been collected from ECFCs deriving from at least three different donors. The amplitude of SDF-1α-evoked Ca2+ peak was measured as the difference between the ratio at the peak of intracellular Ca2+ mobilization and the mean ratio of 1 min baseline before the peak. Pooled data are given as mean ± SE and statistical significance (P < 0.05) was evaluated by the Student's t-test for unpaired observations [14,17]. ANOVA, followed by the Bonferroni post hoc t-test, was performed for statistical analysis of cell migration. A value of P < 0.05 was considered statistically significant.
Chemicals
EBM and EGM-2 were purchased from Lonza (Walkersville, MD, USA). Fura-2/AM was obtained from Molecular Probes (Molecular Probes Europe BV, Leiden, The Netherlands). N-(4-[3,5-bis(trifluoromethyl)-1H-pyrazol-1-yl]phenyl)-4-methyl-1,2,3-thiadiazole-5-carboxamide (BTP2) was purchased from Calbiochem (La Jolla, CA). SDF-1α was provided by PeproTech (United Kingdom). All other chemicals were obtained from Sigma Chemical Co. (St. Louis, MO).
Results
SDF-1α elicits an increase in [Ca2+]i in human ECFCs
Previous work showed that SDF-1α could cause an increase in [Ca2+]i in both BM-derived hematopoietic cells [18 –20] and fully differentiated endothelial cells [27,28]. Consistently, 10 ng/mL SDF-1α induced a biphasic increase in [Ca2+]i in human ECFCs (Fig. 1A). This signal consisted of a rapid Ca2+ transient, followed by a plateau phase of intermediate amplitude between the baseline and the initial peak. The [Ca2+]i returned to resting level upon agonist removal of the bath. The Ca2+ response to SDF-1α was abrogated by pretreating the cells with the bicyclam ADM3100 (Fig. 1A, B; 5 μM, 30 min) (n = 75, from three separate experiments), a selective CXCR4 inhibitor, which has recently been shown to interfere with SDF-1α in ECFCs [8]. The dose–response relationship showed that SDF-1α-evoked Ca2+ signal occurred at 1 ng/mL, the percentage of responding cells and the magnitude of the initial peak achieving a maximum at 10 ng/mL. ECFC sensitivity to SDF-1α was reduced by further increasing its concentration to 50 ng/mL, as both the percentage of activated cells and the magnitude of the Ca2+ peak were significantly lower at this dose (Fig. 2). Therefore, in the following experiments, ECFCs were always stimulated with 10 ng/mL SDF-1α. Collectively, these observations indicate that SDF-1α induces a biphasic Ca2+ signal in human ECFCs that closely resembles that induced by this chemokine in other cell types [18,28].
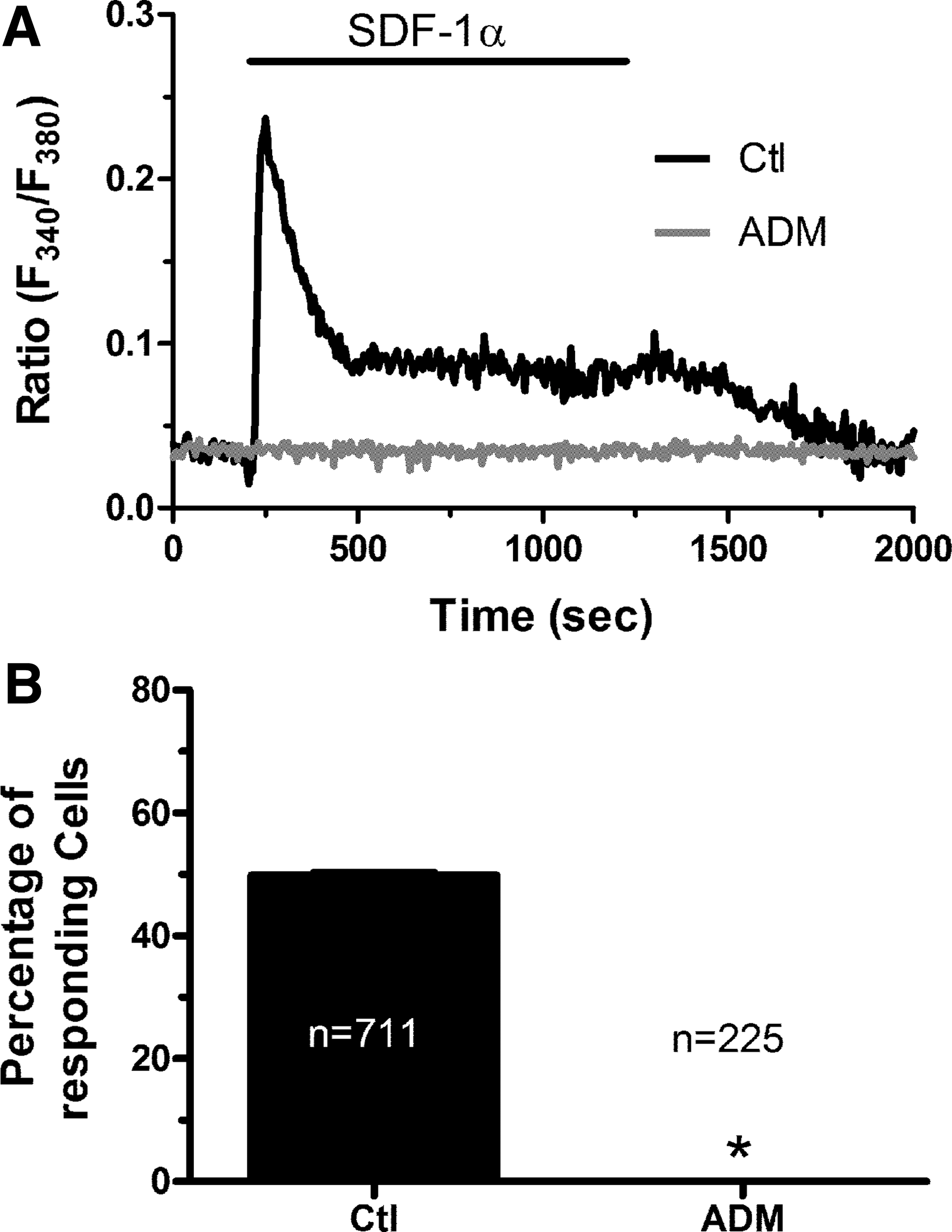
SDF-1α causes an increase in intracellular [Ca2+]i in endothelial colony-forming cells by activating CXCR4.

Dose–response relationship of the Ca2+ response to SDF-1α in endothelial colony-forming cells.
SDF-1α-evoked Ca2+ signals require both intracellular Ca2+ release and extracellular Ca2+ entry
The signaling machinery that underlies SDF-1α-evoked Ca2+ signals is unclear as no study has deeply addressed this issue [20]. Several studies showed that agonists-induced Ca2+ signals in ECFCs are shaped by the interaction between InsP3-induced Ca2+ release from the ER and SOCE [14,16,29]. To assess the sources of the Ca2+ response to SDF-1α, ECFCs were challenged in the absence of Ca2+ in the extracellular solution (0Ca2+). Under these conditions, SDF-1α elicited a transient increase in [Ca2+]i (Fig. 3B), whose amplitude was however no different as compared with that recorded under control conditions (Fig. 3A, C). The subsequent restoration of extracellular Ca2+ caused a second increase in [Ca2+]i which was indicative of SOCE activation (Fig. 3B), as extensively demonstrated elsewhere [14,30]. Accordingly, SDF-1α was removed from the perfusate 100 s before readdition of extracellular Ca2+ to inhibit the synthesis of intracellular second messengers, such as diacylglycerol and arachidonic acid, which might occur upon CXCR4 activation. Control experiments, carried out by removing and restoring extracellular Ca2+ in the absence of SDF-1α in the bath, did not reveal any detectable change in [Ca2+]i (not shown) [14]. These experiments show that the biphasic Ca2+ response to SDF-1α requires endogenous Ca2+ release to drive the initial Ca2+ peak and suggest that SOCE maintains the following plateau. This hypothesis was further corroborated by the observation that removal of extracellular Ca2+ during the plateau phase of SDF-1α-evoked Ca2+ signal caused a rapid and reversible decline in [Ca2+]i to prestimulatory levels (Fig. 3D). A pharmacological approach was then undertaken to further corroborate this model.
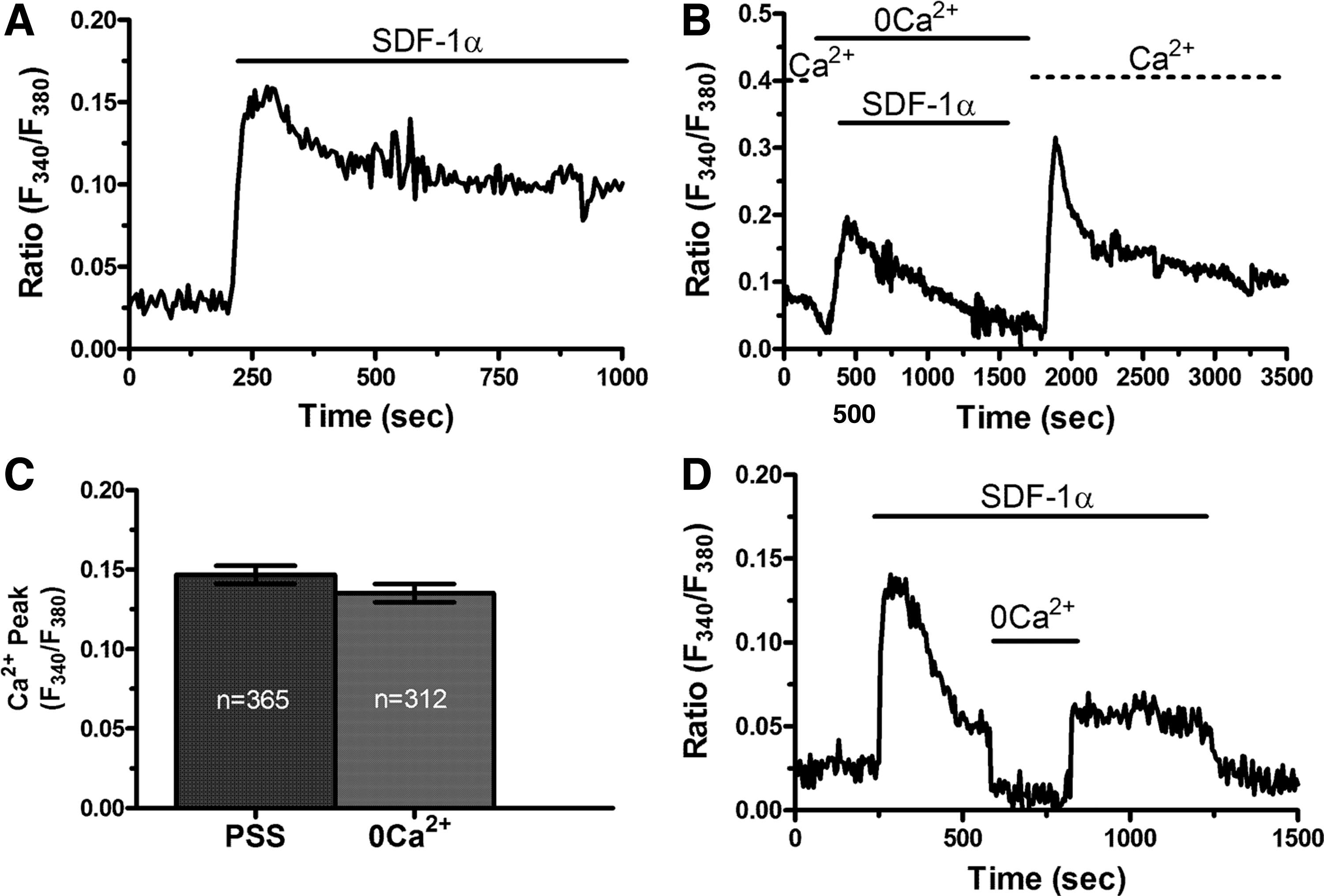
The Ca2+ response requires both intracellular Ca2+ release and extracellular Ca2+ entry in endothelial colony-forming cells.
SDF-1α-evoked Ca2+ signals require InsP3-depedent Ca2+ release and SOCE activation
The ER represents the main endogenous Ca2+ store in ECFCs and is known to release intraluminal Ca2+ through ER-embedded InsP3Rs [11,12]. CXCR4 signals by recruiting a variety of intracellular effectors [31], including PLCβ2 [32]. Of note, PLCβ2 is the main PLC isoform engaged by pertussis toxin-sensitive Gi-protein-coupled receptors in human ECFCs [33]. Consistent with this notion, U73122 (10 μM, 30 min), an aminosteroid, which selectively inhibits PLC in ECFCs [14,16], abrogated the Ca2+ response to SDF-1α (Fig. 4A), whereas its inactive structural analog, U73343 (10 μM, 30 min), was without effect (Fig. 4A, D). Moreover, 2-aminoethoxydiphenyl borate (2-APB; 50 μM, 30 min), a widely employed InsP3R inhibitor, also suppressed SDF-1α-evoked intracellular Ca2+ release (Fig. 4B, D). The 2-APB has been reported to affect Orai and TRP Vanilloid channels [34,35]. However, the effect of 2-APB was probed under 0Ca2+ conditions, which ruled out any effect on extracellular Ca2+ influx [16]. Finally, depleting the ER Ca2+ stores with cyclopiazonic acid (CPA; 10 μM), which inhibits Sarco–Endoplasmic Reticulum Ca2+-ATPase (SERCA), thereby preventing Ca2+ sequestration and leading to intraluminal Ca2+ leakage into the cytosol, abrogated the onset of the Ca2+ response to SDF-1α in the absence of external Ca2+ (Fig. 4C, D). InsP3-induced emptying of the ER Ca2+ pool is known to lead to SOCE activation in ECFCs [14,16]. Moreover, the experiments depicted in Fig. 3B strongly hinted at SOCE as the main pathway for Ca2+ entry in SDF-1α-stimulated cells. In agreement with this hypothesis, BTP2 (10 μM, 30 min), a selective SOCE inhibitor in ECFCs [14,17,36], mimicked the effect of extracellular Ca2+ removal by curtailing the Ca2+ response to SDF-1α without affecting the amplitude of the initial Ca2+ peak (Fig. 5A, B). To further confirm this mechanism, ECFCs were challenged with CPA (10 μM) for 30 min in the continuous presence of external Ca2+. As discussed elsewhere [13,29,37], this maneuver depletes the InsP3-dependent Ca2+ store and fully activates SOCE. Therefore, any Ca2+ signal induced by SDF-1α under these conditions would reflect the recruitment of signaling pathways other than InsP3Rs and SOCs, such as store-independent channels. However, SDF-1α failed to cause any detectable increase in [Ca2+]i when added at 30 min after CPA (Fig. 5C). Altogether, these experiments demonstrate that the Ca2+ response to SDF-1α is shaped by the concerted interplay between InsP3-dependent Ca2+ release and SOCE.

Role of the PLCβ/InsP3R signaling pathway in the onset of the Ca2+ response to SDF-1α in endothelial colony-forming cells.
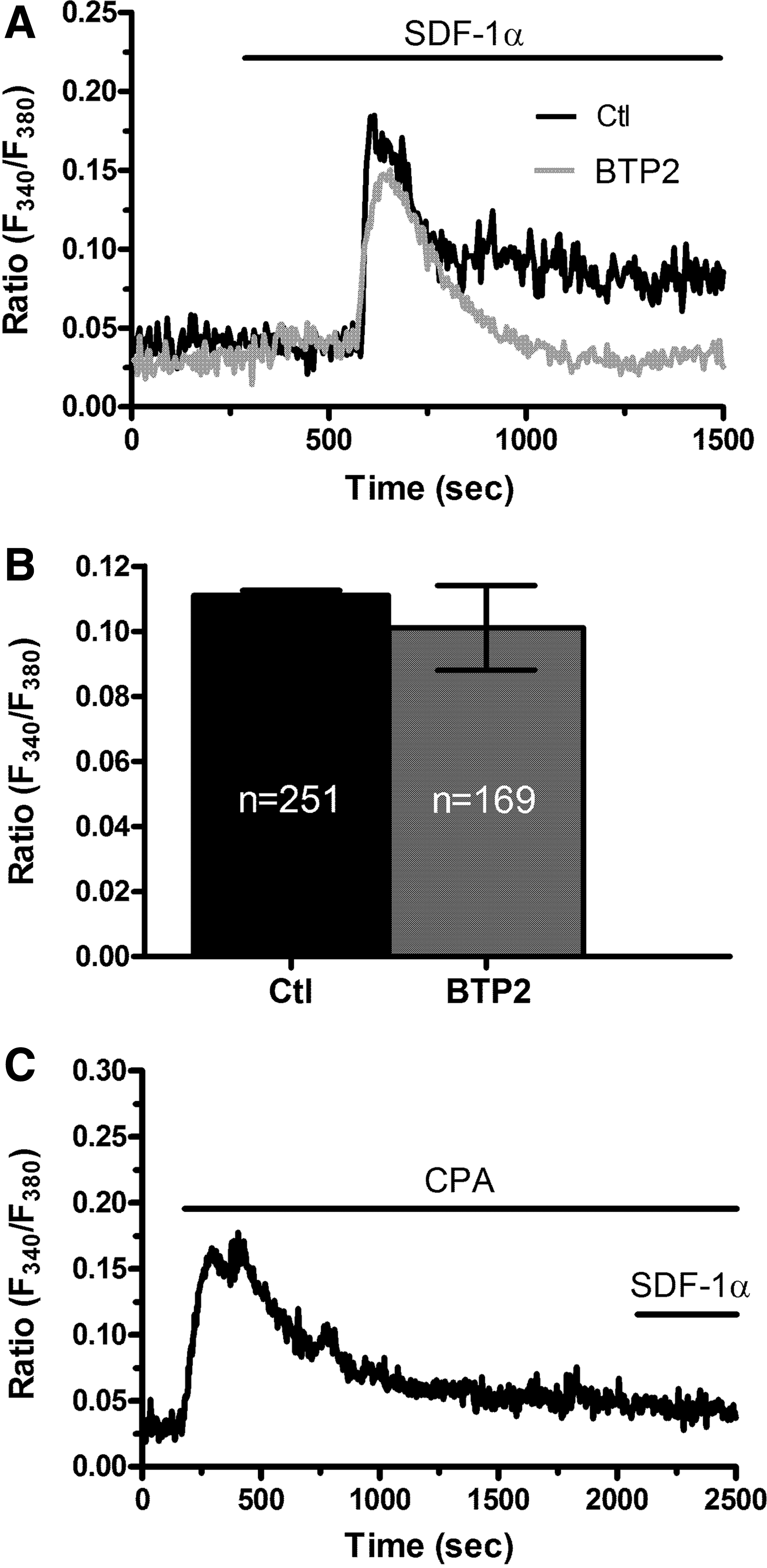
Store-operated Ca2+ entry sustains the plateau phase during the Ca2+ response to SDF-1α in endothelial colony-forming cells.
SDF-1α requires the increase in [Ca2+]i to activate the PI3K/AKT and ERK signaling pathways
The SDF-1α receptor CXCR4 has previously been shown to engage multiple signal transduction pathways, including ERK 1/2, PI3K/AKT, Src-related kinases, and p38 [31,38,39]. We found that SDF-1α stimulated ERK 1/2 and AKT phosphorylation in a time-dependent manner (Fig. 5A), whereas it did not activate p38 and Src (Fig. 6A). Phospho-ERK 1/2 and AKT levels achieved a peak at 20 min and 5 min after SDF-1α stimulation, respectively. These results are consistent with those previously obtained on ECFCs [10]. In principle, both pathways may be recruited through an increase in [Ca2+]i [23,40]. To address this issue in ECFCs, we examined SDF-1α-induced AKT and ERK phosphorylation in the absence and presence of blockers of the concomitant Ca2+ signal. SDF-1α-induced phosphorylation of ERK 1/2 and AKT was inhibited by BAPTA (30 μM, 30 min) (Fig. 6B), a membrane-permeable buffer of intracellular Ca2+ levels [23], and by 2-APB (50 μM, 30 min) (Fig. 6C) at both 5 and 20 min poststimulation. Conversely, BTP2 (10 μM, 30 min) inhibited SDF-1α-induced AKT phosphorylation at each time tested, whereas it reduced ERK 1/2 phosphorylation only at 20 min (Fig. 6B). Collectively, these data demonstrate that SDF-1α-evoked Ca2+ signal is necessary to recruit both the PI3K/AKT and ERK 1/2 signaling pathways.
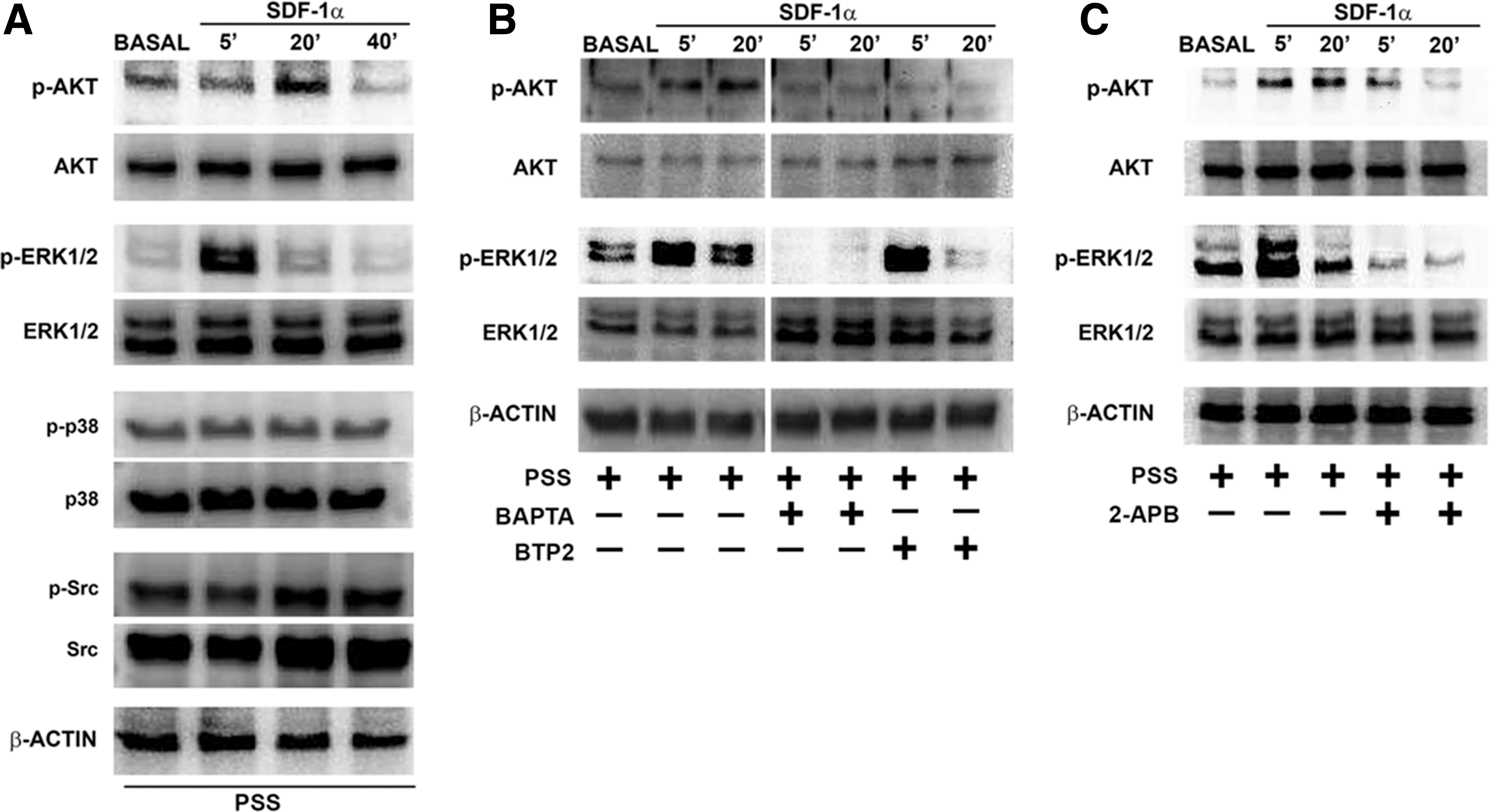
SDF-1α-evoked Ca2+ signals mediate ERK 1/2 and AKT phosphorylation in endothelial colony-forming cells.
SDF-1α-induced ECFC migration requires the Ca2+-dependent activation of PI3K/AKT and ERK 1/2
To assess whether intracellular Ca2+ signaling underpins the chemotactic effect of SDF-1α on ECFCs, we pretreated the cells with either 2-APB (50 μM, 30 min) or BTP2 (10 μM, 30 min) to, respectively, block InsP3Rs and SOCE. As shown in Fig. 7, both treatments significantly (P < 0.05) reduced SDF-1α-evoked ECFC migration. The same result was obtained when ECFCs were preincubated with PD98059 (10 μM, 30 min) and LY294002 (10 μM, 30 min), which, respectively, inhibit the ERK 1/2 and PI3K/AKT signaling pathways [10,41] (Fig. 7A). Altogether, these data demonstrate that SDF-1α stimulates ECFC migration by promoting the Ca2+-dependent activation of PI3K/AKT and ERK 1/2 phosphorylation cascades. Interestingly, in all the different treatments, the number of cells that had passed on the underside of the membrane was similar to that observed in the absence of SDF-1α, thus suggesting that SDF-1α-induced Ca2+ signals support active endothelial cell migration, rather than baseline motility, which is probably due to passive cell diffusion through the membrane pores. Conversely, as recently demonstrated in [10], SDF-1α did not promote ECFC proliferation (Supplementary Fig. S1; Supplementary Data are available online at

SDF-1α requires intracellular Ca2+ signals, ERK 1/2 and Akt phosphorylation, to induce endothelial colony-forming cell migration in vitro and neovessel formation in vivo.
SDF-1α-induced Ca2+ signals drive ECFC homing in vivo
To assess whether SDF-1α-induced Ca2+ signals drive ECFC migration in vivo, we finally exploited the Matrigel plug assay, which is an established quick strategy to assess newly formed neovessels in vivo. As summarized in Fig. 7B and more widely illustrated in Supplementary Fig. S2, SDF-1α (10 ng/mL) caused a significant (P < 0.05) increase in ECFC-dependent neovessel formation. In agreement with the in vitro cell migration assay, pretreating the cells with either BAPTA (30 μM, 30 min) or BTP2 (10 μM, 30 min), dramatically prevented SDF-1α-induced in vivo vascularization. These data, therefore, strongly suggest that SDF-1α uses intracellular Ca2+ signaling to drive ECFC homing in vivo.
Discussion
The chemokine SDF-1α promotes vascular regeneration and tumor neovascularization by inducing ECFC mobilization from their vascular niches, by controlling their correct homing to the target organ and by favoring their incorporation within developing neovessels [8 –10,42]. ECFCs are gathering growing attention, among the different EPC subsets described in literature [6,43], due to their unique endothelial phenotype, which renders them the most suitable cellular tool for regenerative medicine [2,4,5] and the most attractive target for antiangiogenic therapies [3,44]. SDF-1α stimulates ECFCs by binding to its cognate receptor, CXCR4, which may in turn recruit multiple signal transduction pathways [31,38,39], including the PI3K/AKT and ERK 1/2 cascades [9,10]. Herein, we demonstrated for the first time that SDF-1α increases the [Ca2+]i in ECFCs by first inducing intracellular Ca2+ release from ER-embedded InsP3Rs and then activating SOCE. SDF-1α-evoked Ca2+ signals are in turn necessary to engage the ERK 1/2 and PI3K/AKT pathways and promote ECFC migration in vitro and neovessel formation in vivo. This information extends our knowledge of SDF-1α signaling and could be exploited to enhance the therapeutic outcome of ECFC-based therapies and of antiangiogenic treatments.
It has long been known that hypoxic conditions, such as those occurring in ischemic tissues or growing tumors, cause a massive release in circulation of several chemokines, pivotal among which is SDF-1α [3,45]. There is wide evidence supporting the notion that ECFCs reside throughout the vascular endothelium and in the BM vascular niches maintaining vascular integrity and stability [1,7,43,46]. SDF-1α stimulates ECFC mobilization from their vascular niches, so that, once in circulation, ECFCs migrate along SDF-1α gradient to either restore local tissue perfusion or contribute to the angiogenic switch, which supports tumor growth and metastasis [8,9,47]. CXCR4 plays a key role in transducing the proangiogenic effects of SDF-1α. For instance, a recent study demonstrated that the migratory activity was enhanced in CXCR4-overexpressing ECFCs [10], whereas either the pharmacological [8] or genetic [48] inhibition of CXCR4 prevented SDF-1α-induced ECFC migration to hypoxic sites. Finally, ADM3100 was recently found to inhibit ECFCs-dependent vascularization in a murine model of prostate cancer [47]. Sporadic reports had previously shown that CXCR4 was able to increase the [Ca2+]i in several cell types [20,31]. SDF-1α-evoked elevations in [Ca2+]i was mainly used as a functional assay to monitor CXCR4 activation [49,50], but some investigations suggested that this Ca2+ signal was required to induce cell motility in both BM-derived hematopoietic progenitor and stem cells [18,51]. Moreover, SDF-1α-induced endothelial cell migration was associated to an intracellular Ca2+ signal [27,28]. Herein, we showed that SDF-1α caused an increase in [Ca2+]i in ECFCs that was fully suppressed by ADM3100, a selective CXCR4 antagonist. The Ca2+ signal consisted of a rapid Ca2+ peak followed by a plateau phase, as also observed in BM-derived hematopoietic progenitor and stem cells [18,51] and in mature endothelial cells [27,28]. Consistently, CXCR4 mediated the SDF-1α-evoked intracellular Ca2+ waves recorded in other cell types, such as human immune cells [50], glioblastoma cells [52], and platelets [53]. It should, however, be pointed out that the signaling machinery responsible for this biphasic Ca2+ increase has never been thoroughly investigated before [20]. The dose–response relationship recorded in ECFCs displayed a prominent desensitization of the Ca2+ signal at high concentrations, which is consistent with the ligand-dependent internalization of CXCR4 that readily occurs when SDF-1α dose increases beyond 10 ng/mL [54 –56]. A similar finding was also reported in human retinal endothelial cells [28]. Therefore, ECFCs represent a suitable model to investigate the signaling machinery that underlies SDF-1α-induced Ca2+ signals and their physiological role.
As mentioned earlier, the components of the Ca2+ toolkit involved in the Ca2+ response to SDF-1α have remained quite elusive for a long time [20]. The following pieces of evidence demonstrate that the Ca2+ response to SDF-1α is shaped by InsP3-dependent Ca2+ release followed by SOCE activation. First, the increase in [Ca2+]i adopts a biphasic pattern, which is typical of the Ca2+ signals elicited upon G-protein-coupled receptor stimulation in vascular endothelial cells [57] and ECFCs [14]. It has long been known that these intracellular Ca2+ dynamics reflect an initial phase of InsP3-dependent Ca2+ release from the ER, followed by sustained Ca2+ entry across plasmalemmal SOCs, which maintain the increase in [Ca2+] as long as the agonist is present in the bath [57]. As expected, SDF-1α caused a transient, rather than prolonged, increase in [Ca2+]i under 0Ca2+ conditions in ECFCs. Second, the Ca2+ response to SDF-1α was abrogated by the pharmacological inhibition of PLC. CXCR4 has recently been shown to recruit PLCβ2 through the Gβγ dimer [32,38], thereby driving the homing and engraftment of hematopoietic stem progenitor cells [32]. Of note, PLCβ2 is the main effector of Gi-protein-dependent Ca2+ release in ECFCs [33]. Third, the depletion of the ER Ca2+ content with CPA and the selective InsP3R inhibition with 2-APB prevented SDF-1α-evoked Ca2+ signals. We have recently demonstrated that ECFCs lack RyRs [13], whereas they express all the three known InsP3R isoforms [58]. Likewise, SDF-1α-induced migration in hematopoietic progenitor and stem cells was prevented by thapsigargin, another selective SERCA inhibitor [51]. Fourth, the pharmacological blockade of SOCs with BTP2 mimicked the effect of Ca2+ removal, by reducing the amplitude and curtailing the duration of the Ca2+ response to SDF-1α. Previous studies, both from ours [30,35,36] and other research groups [59,60], have clearly shown that BTP2 specifically targets SOCE in both endothelial cells and their more immature precursors. For instance, ECFCs lack TRPC3 and TRPC5, which may be blocked by BTP2 in heterologous expression systems [61], and its effect is independent on TRPM4 activation [36]. Moreover, SDF-1α did not evoke any increase in [Ca2+]i after full SOCE activation with CPA. Under these conditions, SDF-1α can only rely on store-independent channels to trigger Ca2+ inflow. For instance, we have recently shown that arachidonic acid was still able to elicit Ca2+ entry by activating store-insensitive TRPV4 channels in CPA-treated ECFCs [13]. The fact that SDF-1α could not promote Ca2+ influx after full SOCE activation further confirms that this pathway was the only one to mediated Ca2+ entry upon CXCR4 stimulation. Similar to ECFCs, SDF-1α-evoked Ca2+ entry and migration were suppressed by BTP2 in BM-derived CD133+ hematopoietic stem and progenitor cells [18], whereas SKF96365, a less specific SOC inhibitor, dampened SDF-1α-evoked Ca2+ entry and migration in human retinal endothelial cells [28]. Therefore, our insightful characterization of the Ca2+ response to SDF-1α in ECFCs fills a gap in our understanding of CXCR4 signaling by putting a number of isolated observations together and could pave the way for future investigations of the same pathway in other cell types.
Next, we assessed whether the intracellular Ca2+ signal was necessary to recruit the multiple signal transduction pathways that are known to be coupled to CXCR4. In particular, SDF-1α engaged both the ERK 1/2 and PI3K/AKT pathways, whereas it did not stimulate either p38 or Src. These observations are entirely consistent with those previously reported by Oh et al. in human ECFCs [10]. However, we further demonstrated that phospho-AKT and phospho-ERK 1/2 levels were significantly reduced upon inhibition of the concomitant increase in [Ca2+]i with the Ca2+-chelating agent, BAPTA. Moreover, SDF-1α-induced AKT and ERK 1/2 phosphorylations were significantly reduced in the presence of either 2-APB, which blocks InsP3Rs, or BTP2, which inhibits SOCE. Of note, BTP2 reduced ERK 1/2 phosphorylation only at 20 min poststimulation, whereas 2-APB was effective already at 5 min after SDF-1α treatment. These findings strongly suggest that, while InsP3Rs and SOCs are necessary to recruit the PI3K/AKT pathway from the very beginning of the process, SOCs are not important to engage the ERK 1/2 cascade, but to maintain its activation over time. This study provided, therefore, the first evidence that SDF-1α requires InsP3-dependent Ca2+ release and SOCE to engage the ERK 1/2 and the PI3K/AKT pathways. Accordingly, previous studies investigated these signaling mechanisms, that is, InsP3Rs and SOCE, separately in SDF-1α-stimulated cells, despite the fact that both the ERK 1/2 and AKT may be phosphorylated in a Ca2+-dependent manner [23,40,62]. Intracellular Ca2+ may indeed activate the small GTPase Ras, which in turn recruits both PI3K and ERK 1/2, through the Ca2+-dependent Ras guanine nucleotide-releasing proteins (RasGRPs) or Ras guanine nucleotide release-inducing factors (RasGRFs) [63,64]. Moreover, the PI3K class II α-isoform (PI3K-C2α) may be directly activated by an increase in [Ca2+]i [65]. Therefore, intracellular Ca2+ signaling represents a suitable mechanistic link between CXCR4 and the molecular determinants of SDF-1α-induced cell migration. Accordingly, the chemotactic effect of SDF-1α on ECFCs was dampened by the pharmacological inhibition of the concomitant intracellular Ca2+ wave (with either 2-APB or BTP2) and of the ERK 1/2 and PI3K/AKT phosphorylation cascades. These data remarkably extend our knowledge on the molecular mechanisms whereby SDF-1α controls ECFC migration [8–10] and definitively hint at Ca2+ signaling as an essential requirement for SDF-1α-induced cell motility. These observations were further confirmed in vivo by exploiting the Matrigel plug assay, which clearly showed that SDF-1α-induced in vivo vascularization was remarkably reduced by inhibiting the accompanying intracellular Ca2+ wave with either BAPTA or BTP2. These data represent the first demonstration that SOCE drives ECFC homing in vivo and further support the role of Ca2+ signaling in sustaining ECFC recruitment to the target organ originally proposed for PLCβ2 in [33]. In addition, the Ca2+ response to SDF-1α could also mediate ECFC retention within the BM in the presence of high concentrations of this chemokine [66]. Accordingly, it has long been known that, in the absence of any ischemic insult, EPCs are maintained within the BM stem cell niche by the interaction between CXCR4 and SDF-1α, which is massively secreted by the same BM stem cells. Thus, depending on the SDF-1α source, CXCR4-dependent Ca2+ signals might control either ECFC permanence within the BM or ECFC egression into peripheral circulation.
In conclusion, our data demonstrate that SDF-1α induces ECFC migration by stimulating an increase in [Ca2+]i. The Ca2+ response is initiated by InsP3-mediated ER-dependent intracellular Ca2+ mobilization and maintained by SOCE activation. The Ca2+ signal, in turn, leads to the recruitment of the Ca2+-dependent downstream effectors, PI3K and ERK 1/2, which ultimately drive cell motility. These results are likely to provide the framework to decipher the components of the Ca2+ toolkit engaged by SDF-1α in other types of proangiogenic cell types, such as BM-derived hematopoietic progenitor cells and endothelial cells. Moreover, the Ca2+ signalosome could represent a novel target to improve the therapeutic outcome of ECFC-based therapies. For instance, it has been shown that priming ECFCs with SDF-1α before transplantation improved their regenerative ability in a mouse model of hindlimb ischemia [67]. Moreover, ex vivo transduced ECFCs with AKT displayed enhanced proangiogenic activity and significantly improved cardiac performance and reduced negative cardiac remodeling in a mouse model of acute myocardial infarction [68]. Future studies might therefore challenge the hypothesis that the genetic manipulation of the components of the Ca2+ toolkit activated downstream CXCR4 could improve the efficacy of ECFCs-based therapy of ischemic diseases [4,30]. Finally, SDF-1α plays a key role in driving ECFCs to sites of neovessel formation in growing tumors [48,69] and is, therefore, a very promising target for antiangiogenic treatments of VEGF-resistant tumors. As the Ca2+ toolkit is dramatically affected in tumor-derived ECFCs [12,22], which renders them quite insensitive to VEGF [22,70,71], future studies will have to ascertain whether intracellular Ca2+ signaling drives SDF-1α-induced migration also in these cells.
Footnotes
Acknowledgments
The authors do thank Drs. Silvia Dragoni and Elisa Bonetti for their valuable contribution to the early phases of this investigation. CADB fellowship was funded by Collegio Ghislieri, Pavia Progetto “Progressi in Biologia e Medicina.” The work was supported by Cariplo Foundation (2013-0717) to A.B., Collegio Ghislieri. They also thank Prof. Gianni Guidetti, University of Pavia, for the kind gift of LY294002.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.