
Abstract
Coculture of hematopoietic stem cells (HSC) with primary stromal cells from HSC niches supports the maintenance and expansion of HSC and progenitors ex vivo. However, a major drawback is the availability of primary human samples for research and clinical applications. We investigated the use of in vitro derived osteoblasts as a new source of feeder cells and characterized the molecular pathways that mediate their growth-promoting activities. First, we compared the growth and differentiation modulating activities of mesenchymal stromal cells (MSC)-derived osteoblasts (M-OST) with those of their undifferentiated precursor on umbilical cord blood (UCB) progenitors. Feeder-free cultures were also included as baseline control. Cell growth and expansion of hematopoietic progenitors were significantly enhanced by both feeder cell types. However, progenitor cell growth was considerably greater with M-OST. Coculture also promoted the maintenance of immature CD34+ progenitor subsets and modulated in a positive fashion the expression of several homing-related cell surface receptors, in a feeder-specific fashion. Serial transplantation experiments revealed that M-OST coculture supported the maintenance of long-term lympho-myeloid reconstituting HSC that provided engraftment levels that were generally superior to those from MSC cocultures. Mechanistically, we found that coculture with M-OST was associated with enhanced beta-catenin (β-Cat) activity in UCB cells and that abrogation of β-Cat/T-cell factor activity blunted the growth-promoting activity of the M-OST coculture. Conversely, Notch inhibition reduced UCB cell expansion, but to a much lesser extent. In conclusion, this study demonstrates that M-OST are excellent feeder cells for HSC and progenitors, and it identifies key molecular pathways that are responsible for the growth-enhancing activities of osteoblasts on UCB progenitors.
Introduction
C
The potential of MSC coculture for the expansion of HSC/progenitors is well established, whereas that provided by osteoblasts remains less well elucidated. Osteoblasts are derived from MSC and are an integral part of the endosteal HSC niche [4]. Previous studies showed that co-culture of LinnegSca-1+c-kit+ (LSK) murine BM cells with primary osteoblasts improved not only cell expansion but also engraftment activity compared with coculture with Dexter stromal cells [5,6]. Moreover, immature osteoblasts appear to be the most potent in this regard [6,7].
Osteoblasts derived in vitro from MSC (M-OST) with osteogenic differentiation medium provide an excellent source of osteoblasts at distinct stages of maturation and share similar patterns of gene expression and morphology as primary human osteoblasts [8,9]. We previously showed that serum-free medium (SFM) conditioned by immature M-OST promoted stronger production of CD34+ cells and multipotent progenitors (long-term culture initiating cells) and committed progenitors [colony-forming cells (CFCs)] to that achieved in either SFM or SFM conditioned with MSC [7,10,11]. Moreover, progenitors produced in osteoblast conditioned medium cultures provided superior platelet engraftment in transplant experiments [11]. Altogether, these studies raise the intriguing possibility that immature M-OST could be excellent feeder cells for the expansion of progenitors with strong short- and long-term engraftment properties. An important aim of this work was to test this hypothesis.
Several independent cell signaling pathways have been implicated in the regulation of HSC in stem cell niches, including the Wnt and Notch signaling pathways [1,12]. These highly conserved developmental signaling pathways may also mediate some of the regulatory effects of feeder cells on stem and progenitors in coculture [13 –15]. Wnt proteins form a large family of 19 secreted glycoproteins that bind to Wnt receptors composed of the Frizzled receptor family (10 members) and the low-density lipoproteins-related proteins (Lrp). Wnt proteins have been implicated in many developmental biological processes, including hematopoiesis [12], osteogenic differentiation, as well as skeletal development and enhancement of bone mass [16,17].
In the absence of canonical Wnt signaling, beta-catenin (β-Cat) is phosphorylated by a destruction complex composed of GSK-3b/Axin/APC, which flags β-Cat for ubiquitin proteasome destruction [18]. Canonical Wnt signaling inhibits the destruction of β-Cat, allowing it to translocate into the nucleus where it interacts with members of the T cell factor/lymphoid enhancer factor (TCF/LEF) family of transcription factors, which then act as a transcriptional activator complex [19]. Regulation of hematopoiesis by Wnt remains highly controversial, having been implicated in improving the proliferation and repopulation capacity of HSC, blocking multilineage differentiation, exhausting the HSC pool, or even being altogether dispensable (reviewed in Refs. [12,18]). For instance, conditional deletion of β-Cat showed that although HSC can form without it, β-Cat-deficient HSC had significant defect in self-renewal and engraftment activity [20].
Conversely, adult hematopoiesis and HSC transplantation activity were reported to be largely intact when Wnt secretion was blocked in Porcn mutants or via pharmacological inhibition [21]. One study showed that many of the aforementioned effects could be explained by the gradient of Wnt signaling levels [22]. Differences in methodology and cross-talk between molecular pathways are certainly responsible for many of these discrepancies. Osteoblasts secrete many Wnt proteins [23,24], but whether Wnt proteins can support hematopoiesis in coculture settings with osteoblasts remains uninvestigated.
Similarly, Notch signaling has well-established roles in embryogenesis and hematopoiesis [25 –27]. It is now clear that Notch signaling is dispensable for the maintenance of HSC under steady state [27]. However, Notch can also act as a strong promoter of HSC expansion ex vivo [28 –30], and Notch-mediated expansion of UCB progenitors has been shown to accelerate hematopoietic recovery in clinical trials [31,32]. Indeed, inhibition of Notch signaling with γ-secretase inhibitors abrogates many of the supportive effects of coculture with osteoblasts and endothelial cells, primary elements of endosteal and vascular stem cell niches, respectively [28,29].
The principal aims of this study were first to compare the growth modulatory activities of M-OST with their parental undifferentiated MSC on UCB progenitors, and then to investigate the molecular pathway mediating the growth-promoting activity of M-OST.
Materials and Methods
Isolation and culture of human MSC and production of M-OST
The research described herein was approved beforehand by the Canadian Blood Services research ethical board. Human BM MSC were established from purchased fresh BM mononuclear cells (Lonza, Walkersville, MD) as previously described [11]. A total of 4 plastic-adherent MSC lines were generated from independent samples and used between passages 2 and 8. Immature M-OST were produced by culturing MSC for 6 days in osteogenic differentiation medium with media changes every 2 days as previously described [7].
Isolation of CD34+-enriched cells
Human UCB samples were obtained from the Canadian Blood Services' Cord Blood for Research Program after obtaining institutional review board approval and written informed consent. Mononuclear cells were isolated with Ficoll-Paque PLUS (GE Healthcare, Inc., Baie D'Urfe, Quebec, Canada) gradient, and UCB CD34+ cells were isolated by positive selection from pooled CB units (2–4) with EasySep™ Human Cord Blood CD34 Positive Selection Kit (StemCell Technologies, Vancouver, British-Columbia, Canada) by following the manufacturer's instructions (final CD34+ purity 90% ± 4%).
Coculture of CD34+ cells with MSC or M-OST
Semi-confluent MSC or M-OST differentiated for 6 days were rinsed twice with phosphate-buffered saline (PBS), and CD34+ cells were plated at a density of 10,000–15,000 cells/mL. All cocultures were done in CellBind multi-well plates (Corning, Inc., Corning, NY). The SFM consisted of Iscove's modified Dulbecco's medium with 20% serum substitute solution (BIT; StemCell Technologies), 20 μg/mL low-density lipoprotein (StemCell Technologies), and 100 μM β-mercaptoethanol (pH 6.0; Sigma, St.-Louis, MO). All UCB cultures were supplemented with the cytokine cocktail “optimized megakaryocyte progenitor cocktail” (OMPC), developed for the expansion of megakaryocyte and myeloid progenitors and composed of 10 ng/mL stem cell factor (SCF), 11 ng/mL Flt-3 ligand (FL), and 35 ng/mL thrombopoietin (TPO) [33]. All cytokines were purchased at Peprotech (Rocky Hill, NJ). One volume of fresh media was added at day 4 of culture. In selected cultures, inhibitor of β-Cat responsive transcription 3 (iCRT3; Sigma) and RO4929097 (Selleck Chemicals, Houston, TX) were added at day 0 and refreshed every 2nd day. The concentrations of RO4929097 tested were based on the fact that 2 μM of RO4929097 was sufficient to reduce transcript levels of Notch downstream targets Hes-1 and Deltex by 80%–90%, and that a higher concentration was found to be toxic [7]. The insulin-like growth factor 1 receptor (IGF-1R) inhibitor AG-1024 (Selleck Chemicals) was added to cultures at 10 μM at day 0 and 3. DMSO vehicle controls were prepared by using the corresponding dilution identical to the highest drug concentration. Cultures were done in duplicate and incubated in a humidified atmosphere (5% CO2) at 37°C. Net productions in the different cultures are presented as number produced per starting seeded cells. Unless specified otherwise, results were repeated in at least 3 independent biological experiments. CFC were measured by using MethoCult H4434 (StemCell Technologies), and colonies were counted after 14 days by using an Olympus CKX41 inverted microscope (Olympus Corporation, Shinjuku, Tokyo, Japan). CFC assay was performed for expanded UCB cells after 6 days of culture or for fresh murine BM samples as indicated.
Flow cytometry analysis
UCB cells were phenotyped by flow cytometry by using an FACS-Attune (ThermoFisher, Waltham, MA). At least 10,000 events were acquired for each sample, with dead cell and debris gated out by forward and side-scatter gating and/or Sytox stain (Invitrogen, Mississauga, ON, Canada). The antibodies used included CD235a-phycoerythrine (PE), CD41a (GPIIb)-fluoresceine isothiocyanate (FITC), CD14-allophycocyanine (APC), CD34-PE, anti-CD45-APC, CD45RA-APC, CD38-FITC, CD90-Percp-Cy7, CD162-Alexa647 (PSGL-1), CLA-FITC, CD11a-FITC, and CD184-APC (CXCR4). All antibodies were purchased at Becton Dickinson Pharmingen (Mississauga, Ontario, Canada). AlexaFluor-488 Annexin V and Sytox were used for viability analysis of M-OST cells incubated in SFM with OMPC and/or DMSO, iCRT3, or RO4929097 for 3–6 days by following the manufacturer's instructions (Invitrogen). Intracellular staining for flow cytometry was performed as follows: CD34+-stained samples were fixed with PBS with 1% paraformaldehyde for 20 min at room temperature. Rinsed samples were then treated with PBS + 0.2% Triton X-100 + 2% fetal bovine serum (FBS) for 5 min at room temperature. Samples were then centrifuged, resuspended in 100 μL of PBS + 0.2% Triton X-100 + 2% FBS, and stained with intracellular β-Cat-APC (R&D Systems, Minneapolis, MN) antibodies for 20 min. Afterward, the samples were washed with PBS + 0.2% Triton X-100 + 2% FBS and resuspended in 500 μL of PBS + 2% FBS. Fluorescent minus one controls were used to set gates.
RNA isolation and cDNA synthesis
RNA was isolated from whole culture of CD34+ cells pre-expanded for 5 days in SFM cultures, and it was then incubated for an extra day in control SFM or in coculture with M-OST. RNA was isolated from RNeasy Mini kit (Qiagen, Hilden, Germany) by following the manufacturer's protocol. Isolated RNA was reverse transcribed by using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). Up to 500 ng of RNA was used per synthesis of a 20 μL reaction.
Quantitative real-time polymerase chain reaction
All quantitative real-time polymerase chain reaction (qPCR) experiments were done in triplicate and performed on the CFX96 Real-Time System by using 10 μL reaction mixtures containing SsoAdvanced Universal SYBR Green Supermix (Bio-Rad). The thermal cycling program used for all genes except GAPDH was: 95°C (3 min), then 40 cycles of 95°C (10 s), 60°C (10 s), and 72°C (30 s), ending with a melt curve analysis program of 95°C (10 s), 65°C (5 s), and 95°C (5 s). The GAPDH primers required an annealing temperature of 63°C instead. Genes tested were WNT2, WNT3a, WNT4, WNT5a, WNT16, DKK1, CJUN, CFOS, CL100, GAPDH, and HPRT1. Primer efficiency was assessed with a dilution series using pooled cDNA from all samples, whereas melt curve analysis confirmed primer specificity. Gene expression was determined by using the ΔΔCt method, with normalization to GAPDH and HPRT1. For statistical analysis, each biological replicate was normalized by using the squared difference of the mean method and outliers were tested for by using the Thompson tau technique [34].
Mice transplantation assays and engraftment analysis
Sub-lethally irradiated (300 cGy 137Cs, Gammacell Exactor 40; Best Thetratonics, Canada) 8-week-old NOD.Cg-Prkdcscid Il2rgtm1Wjl /SzJ (NSG) mice (Jackson Laboratory, Bar Harbor) were transplanted intravenously with the total progeny of 1.0 × 104 UCB CD34+ cells expanded for 7 days. Each group contained a total of five mice (two independent lines tested). Mice injected with saline solution served as negative controls for human engraftment analyses. Platelet analyses were done in two steps: The concentration of murine platelets in diluted blood was measured with rat anti-mCD41-FITC stain, and the proportions of human and murine platelets were determined in platelet-rich plasma stained with species-specific anti-CD41a antibodies [11]. BM harvested from flushed tibias and femurs was analyzed by cytometry and CFC assay as previously described [7]. Frozen BM samples representing 85% of the collected BM from primary recipients were thawed [35], rinsed, and transplanted in secondary mice (two primaries per MSC line tested). Monoclonal antibodies (Becton Dickinson Pharmingen) used were anti-human CD41a (GPIIb)-APC (clone HIP8), CD33-APC (WM53), CD19-PE (HIb19), CD45-APC (HI30), and mouse rat anti-mCD41-FITC (MWReg30).
Statistical analysis
Statistical analyses were performed by using GraphPad InStat version 3.00 (La Jolla, CA). Multigroup comparisons were done by two-tail paired ANOVA, whereas two group comparisons were done with two-tail paired Student's t-tests. P values smaller than 0.05 were considered significant.
Results
M-OST increase the expansion of immature UCB cell subsets and committed progenitors
In our original study, coculture of UCB CD34+ cells with MSC or M-OST was investigated only in a non-contact setting [11]. Herein, we sought to characterize the properties of M-OST as a feeder layer through a parallel comparison to undifferentiated MSC. All hematopoietic cultures were supplemented with the cytokine cocktail OMPC, composed of SCF, FL, and TPO [33], and feeder-free SFM culture was used as an additional control.
Expansion of total nucleated cell (TNC) was increased when UCB CD34+ cells were cocultured with undifferentiated MSC when compared with feeder-free SFM cultures. However, cell growth was even greater when UCB MSC had first undergone osteogenic differentiation (Fig. 1A). Interestingly, the retention of CD34+, expressed on hematopoietic stem and progenitors, was significantly improved in both cocultures (Fig. 1B, C), such that the output of CD34+ cells was increased three to fourfold in cocultures (Fig. 1D). Comparable observations were made for the immature subset CD34+/CD45RA− and to a lesser extent for CD34+/CD90+/CD45RA− (Fig. 1C, D). The impact of coculture on commitment and differentiation of CD34+cells was also assessed. Coculture with both MSC and M-OST significantly enhanced the production of CD14+ monocytic cells while reducing that of CD41+ megakaryocytes, but it had no impact on erythroblasts (Fig. 1E). The net production of myeloid CFC progenitors after expansion was also investigated. Myeloid progenitor yield was the lowest in feeder-free SFM cultures, and it was the highest in M-OST cocultures (Fig. 1F) due largely to increases in burst-forming-unit erythroid. CFC outputs were also slightly increased in MSC cocultures; however, the differences versus SFM cultures failed to be significant.
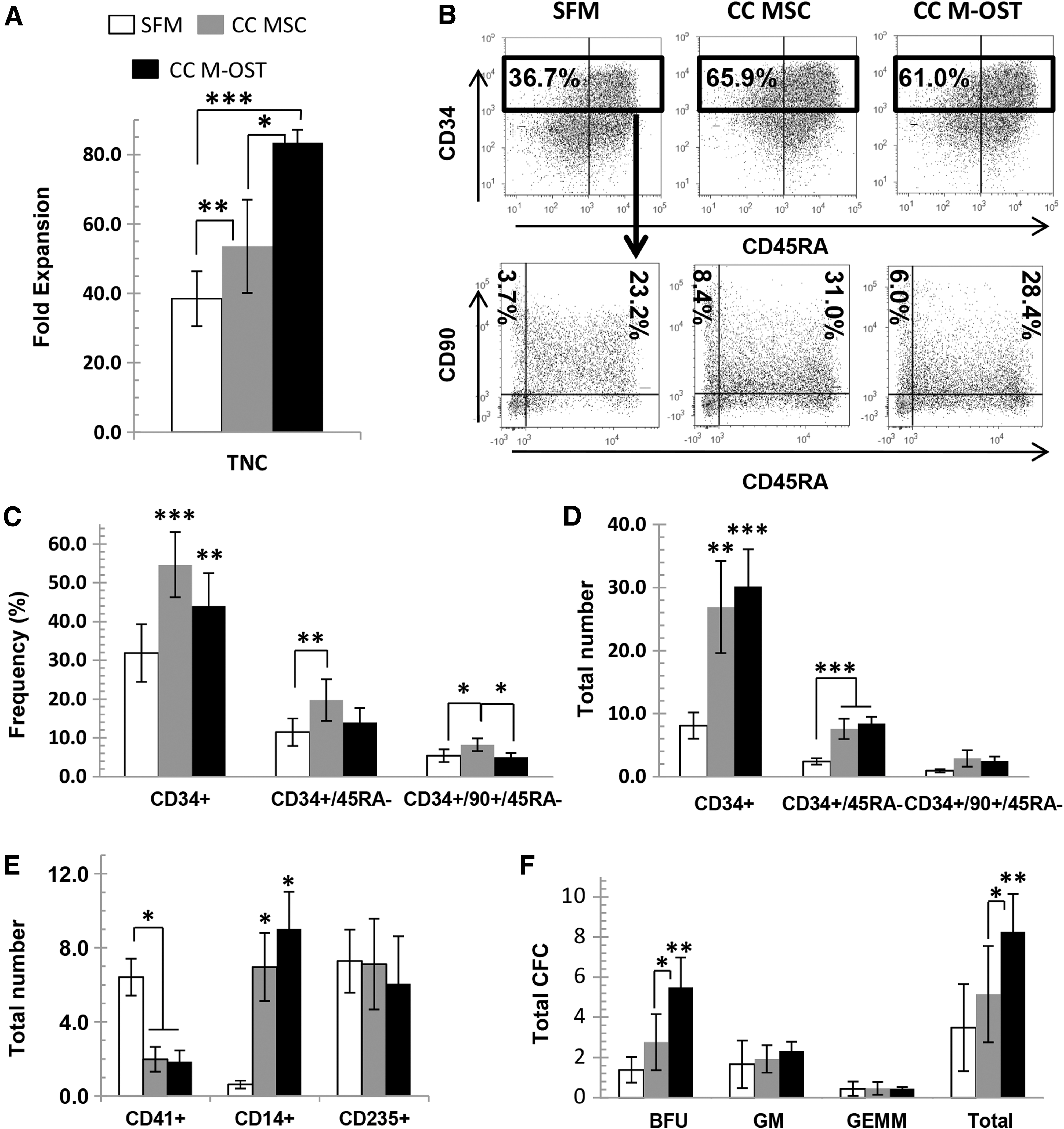
Production of UCB CD34+ primitive subsets is increased in coculture with M-OST.
Coculture of CD34+ cells with stromal cells has a major impact on the expression of cell adhesion molecules and CXCR4
Engraftment is dictated, in part, by the capacity of the transplanted stem and progenitors to home to the BM, an intricate process mediated by cell adhesion molecules, chemokines, and their receptors. In previous work, we found that UCB CD34+ cells cultured in osteoblast conditioned medium had increased expression of C-X-C chemokine receptor type 4 (CXCR4) and lymphocyte function-associated antigen 1 (LFA-1/CD11A) [11]. Hence, we assessed whether similar changes occurred in M-OST cocultures.
No significant differences in the frequency of CXCR4+ and CD11A+ TNC were seen between SFM and M-OST cocultures (data not shown). CD162/PSGL-1 was ubiquitously expressed in all cultures, but the frequency of fucosylated PSGL-1 (CLA+) was increased twofold in M-OST cocultures (data not shown). Interestingly, the frequency of CD34+ cells expressing CXCR4, fucosylated PSGL-1 (CLA+), and CD11A was greater in M-OST cocultures (Fig. 2A). This increase was partially due to the increased proportion of CD34+ with CLA and the increased retention of CD34+ cells in cocultures (Fig. 2A, B). Consistent with results of Fig. 1, M-OST cocultures produced significantly higher amounts of most CD34+ subsets examined (Fig. 2C).

Fucosylation of PSGL-1 on UCB CD34+ cells is increased in M-OST and MSC cocultures.
Next, we investigated whether coculture with MSC would lead to similar modulation on UCB CD34+ cells. Interestingly, significant differences were observed for CXCR4 and fucosylated PSGL-1. Cocultures with MSC had a reduced frequency of CD34+CXCR4+ cells and proportion of CXCR4+ CD34+ cells versus M-OST cocultures (Fig. 2D, E), whereas the opposite was observed for CD34+CLA+ (Fig. 2D, E). Conversely, the net numbers of several CD34+ subsets produced were superior in M-OST cocultures (Fig. 2F).
M-OST support the maintenance of hematopoietic progenitors with long-term engraftment activity
Assessment of hematopoietic progenitors with in vitro assays provides some information on the nature and properties of the progenitor cells in culture. However, these fail to assess HSC function. The latter was investigated through serial reconstitution transplant experiments into immunodeficient mice (Fig. 3A). Groups of NSG mice were transplanted with the total progeny of 104 UCB CD34+ cells expanded for 1 week in SFM, MSC cocultures, or M-OST cocultures. Two independent MSC donors were tested to prevent donor-derived effects.

Impact of cocultures on engraftment activities of expanded UCB progenitors.
The graft contribution to leukocyte engraftment was investigated at 4, 12, and 20 weeks post-transplant. No significant differences were observed between the three groups. However, human leukocyte frequencies tended to be superior in M-OST recipients versus MSC recipients at 4 and 12 weeks (Fig. 3B). At 20 weeks, leukocyte frequencies were superior in coculture cohorts when compared with SFM recipients (Fig. 3B). Similarly, no significant differences were observed for human platelet levels in the three groups; however, human platelet levels were typically superior in M-OST recipients (Fig. 3C). The latter was reminiscent of what we had previously observed with osteoblast conditioned medium-expanded progenitors [11].
Human BM engraftment was investigated at 23 weeks post-transplantation to focus on the contribution of long-term engrafting cells (Table 1). Lympho-myeloid engraftment was confirmed in all three cohorts; however, MSC-cocultured UCB recipients showed much greater variation in engraftment. Indeed, one recipient had an engraftment level fourfold greater than the mean of its littermates (range of 8%–75% hCD45+ in MSC vs. 11%–47% in M-OST recipients). Despite this bias, a slight advantage in human engraftment was evident in M-OST-cocultured UCB recipients for most cell subpopulations investigated (Table 1). A similar rise in the net number of human myeloid progenitors present in the M-OST recipients was also observed (Table 1).
Mean ± SEM. No significant differences detected between groups based on ANOVA analysis.
CFCs, colony-forming cells; MSCs, mesenchymal stromal cells.
Secondary transplantation was carried out to further test the self-renewal and reconstitution activity of expanded grafts from both cocultures. Frozen BM samples from primary recipients with the highest level of human engraftment were thawed and transplanted in secondary mice (two primaries per MSC line tested). Human BM engraftment was assessed at week 11 (Fig. 3D). Only one out of four MSC recipients showed significant levels of human chimerism in serial transplants. In contrast, three out of four M-OST recipients were positive (Fig. 3E).
Taken together, these results demonstrate that UCB CD34+ expanded in M-OST coculture possess long-term engraftment activities that are similar, if not superior, to those expanded in MSC cocultures. The remainder of the study focused on M-OST cocultures and the characterization of the molecular pathways modulating the effects of M-OST on UCB CD34+ progenitors.
Wnt canonical signaling in UCB CD34+ cells is stimulated by M-OST
The previous results show that coculture of UCB CD34+ cells with M-OST increases the production of UCB progenitors with good engraftment activity. Next, we wanted to investigate signaling pathways that support the growth-promoting activities of M-OST on UCB CD34+ cells. Since Wnt signaling has been implicated in the regulation of stem and progenitor cells as well as osteogenesis, we investigated the potential implication of the Wnt pathway in M-OST-UCB cocultures.
First, we assessed the expression of selected Wnt genes by qPCR in M-OST. Transcripts for Wnt3, Wnt4, Wnt5a, Wnt9b, and Wnt10b were found to be expressed at low levels in M-OST [36]. In addition, consistent with active Wnt signaling in M-OST, Axin2 transcripts were upregulated by almost threefold in M-OST versus MSC (P < 0.01, data not shown). Next, we measured β-Cat levels, a central signal mediator of the canonical Wnt pathway. We detected a 25% increase in the relative level of β-Cat in CD34+ cells in M-OST coculture and a 15% increase in TNC (Fig. 4B, C, P < 0.001). Expression of several Wnt/β-Cat downstream target genes was also increased in UCB cells cocultured with M-OST. These included c-Jun (2.5-fold) [37], c-Fos (3-fold) [38], and CL100 (2-fold) [38], whereas expression of the Wnt target and inhibitor Dkk1 was decreased (2-fold, Fig. 4C) [39]. Altogether, these results demonstrate increased levels of β-Cat in UCB cells cocultured with M-OST, increased expression of several Wnt/β-Cat downstream target genes, and decreased expression of Wnt antagonist Dkk1 in cocultured UCB cells.
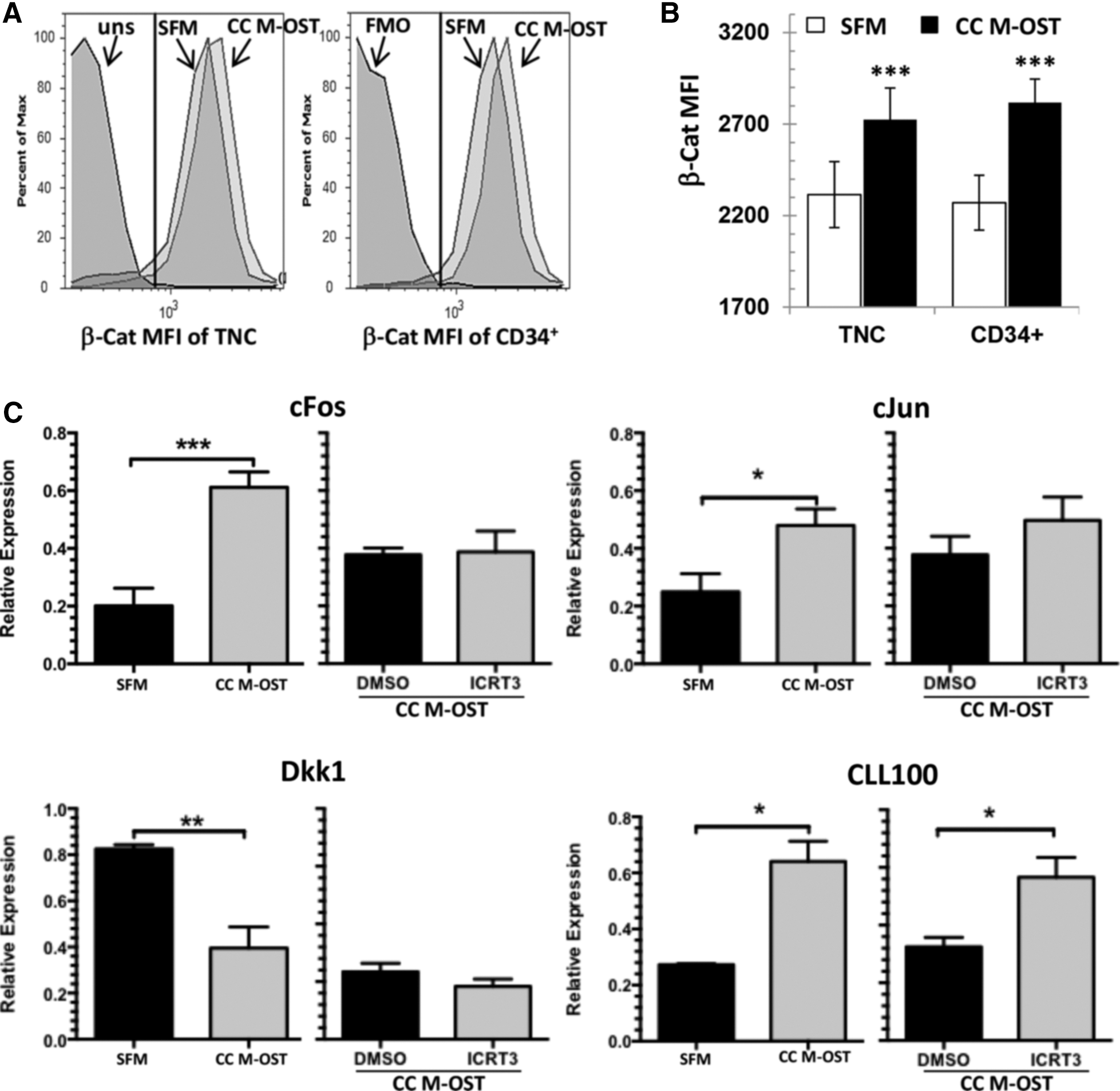
Coculture of UCB CD34+ cells with M-OST is associated with increased Wnt canonical signaling.
The growth-promoting activity of M-OST cocultures on UCB CD34+ is largely dependent on β-Cat/TCF activity
The previous results pointed to elevated canonical Wnt/β-Cat signaling in M-OST cocultures. Hence, we evaluated whether the growth and differentiation activities were modulated by β-Cat-dependent pathways. This was achieved by treating cultures with the specific and potent Wnt inhibitor iCRT3, which blocks the interaction of β-Cat with its transcriptional coactivator TCFs [40]. Consistent with β-Cat/TCF inactivation, the expression of three downstream targets was downregulated in cocultured UCB cells after iCRT3 treatment (Fig. 4C, DMSO vs. iCRT3).
Viability of M-OST determined by Annexin V/Sytox stains was not significantly impacted by iCRT3 (data not shown). However, iCRT3 induced a dose-dependent decrease in the expansion of UCB TNC, CD34+, and CD34+/CD38−/CD45RA− cells in M-OST cocultures (Fig. 5A–C). In contrast, reductions observed in SFM cultures were far more subtle and were not significant when compared with the DMSO control (ie, 0 μM iCRT3) and high concentration of iCRT3 (50 μM) for all subpopulations with the sole exception of CD34+/CD38−/CD45RA− cells (Fig. 5C). Of note, cell growth with l2.5 μM of iCRT3 was slightly enhanced versus the vehicle control in both cultures. This could be due to the fact that the amount of DMSO in the vehicle control (0 μM of iCRT3) corresponded to the DMSO amount used for the highest iCRT3 concentration, which was four times greater than in the 12.5 μM condition. Alternatively, it is possible that light canonical Wnt inhibition modulates β-Cat to levels that favor cell growth [22].
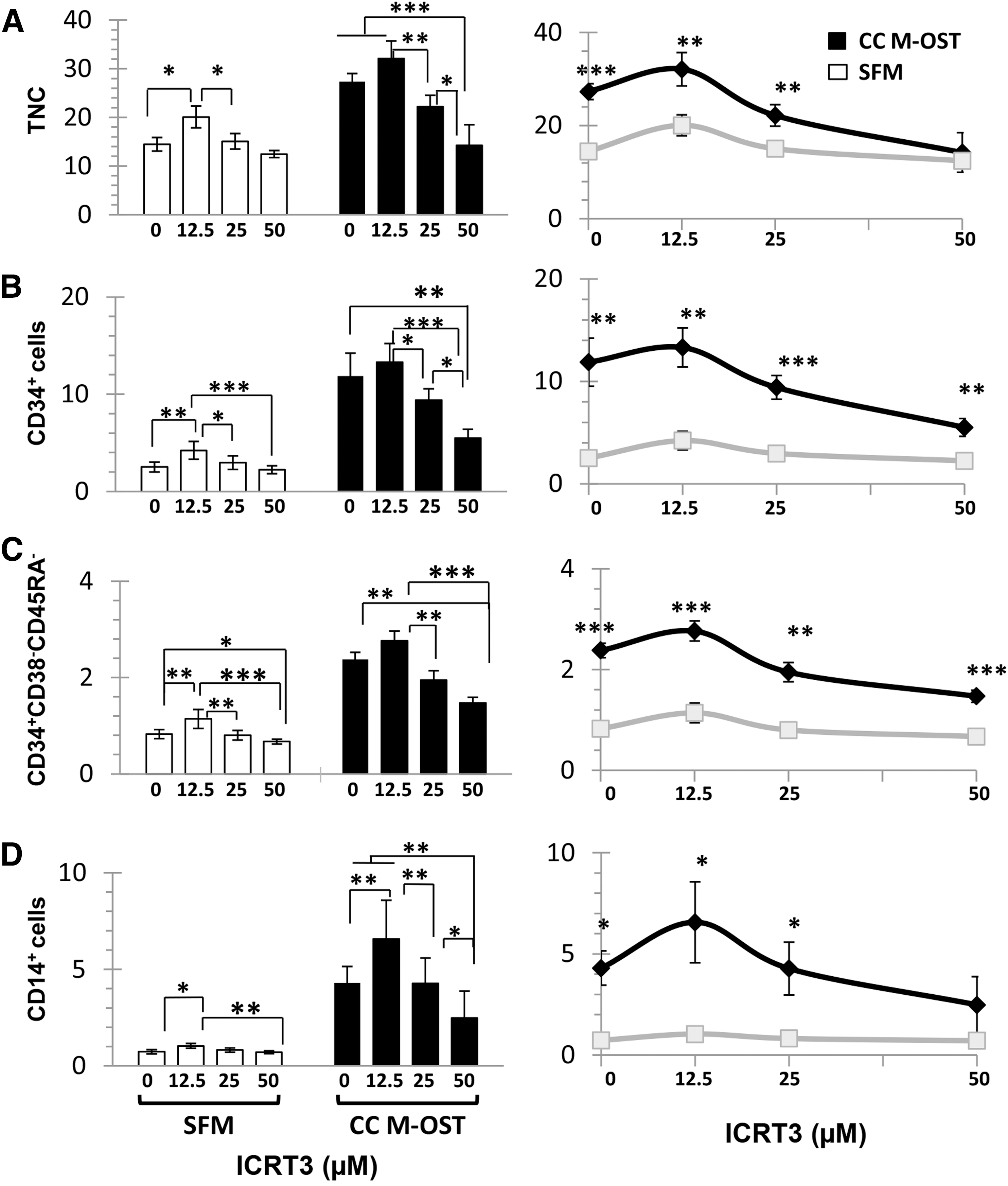
Impact of β-Cat inhibition on the growth-promoting activities of M-OST cocultures on UCB CD34+ cells. Production of TNC
Under high iCRT3 concentrations, the net production of TNC and CD14+ cells in cocultures approached that seen in the control SFM condition with or without iCRT3 (Fig. 5A, D, right graphs). Conversely, the production of CD34+ and CD34+/CD38−/CD45RA− cells, though clearly reduced by iCRT3 treatment, remained superior in M-OST cocultures (Fig. 5B–D, right graphs). These results suggest that the growth-promoting activities of M-OST on UCB CD34+ cells are mediated, in large part, by β-Cat downstream pathways, and that other pathway(s) must also be implicated.
The growth-promoting activity of M-OST cocultures on UCB CD34+ is not dependent on IGF-1R signaling
β-Cat activation is most often associated with Wnt canonical signaling. However, the cell signaling cascade downstream of IGF-II/IGF-1R has also been shown to be a potent activator of β-Cat in oligodendroglial bladder carcinoma cells [41,42]. Since IGF-II is highly secreted by M-OST (K. Law & O. Hovey, unpublished results), we tested whether the β-Cat level in UCB cells cocultured with M-OST was modulated by IGF-II/GF-1R signaling. Inhibition of IGF-1R with the selective kinase inhibitor AG-1024 had a minor impact on the expansion of UCB TNC, CD34+ subsets, and CD14+ cells in M-OST cocultures (Fig. 6). AG-1024 also had no major impact on intracellular β-Cat levels (data not shown). These results rule out the IGF-II/IGF-1R signaling pathway as an essential upstream activator of β-Cat in M-OST cocultures.

Impact of IGF-1R inhibition on the growth-promoting activities of M-OST cocultures on UCB CD34+ cells. Production of TNC, CD34+ cells, CD34+/CD38−/CD45RA− cells, and CD14+ cells per starting cell in M-OST cocultures supplemented with DMSO or 10 μM of AG-1024. Mean ± SEM, n = 3; no significant differences. IGF-1R, insulin-like growth factor 1 receptor.
Notch signaling is a minor contributor to the growth-promoting activity of M-OST
We previously showed that transcripts for several Notch ligands, including Jagged 1 and 2, Delta like 1, and Delta like 4, are expressed in M-OST [7]. Given the opportunity for cell contact in coculture, we assessed the implication of Notch signaling on the growth-promoting activity of M-OST by using the γ-secretase inhibitor RO4929097. As with iCRT3, viability of M-OST was not significantly impacted by the γ-secretase inhibitor (data not shown). Notch inhibition moderately reduced the growth of UCB cells in both low (2 μM) and high (10 μM) concentrations of RO4929097 in cocultures, but only at high concentrations in SFM cultures (Fig. 7A). RO4929097 also significantly reduced CD34+ and CD34+/CD38−/CD45RA− cell outputs in both culture conditions (Fig. 7A–C), but it had no effect on CD14+ cells (Fig. 7D). Finally, the net production of TNC and CD34+ subsets remained superior in M-OST cocultures compared with SFM (Fig. 7, right graphs). Hence, Notch signaling is a minor contributor that is required to maximize the expansion of UCB CD34+ cells and CD34+/CD38−/CD45RA− cells in M-OST coculture.
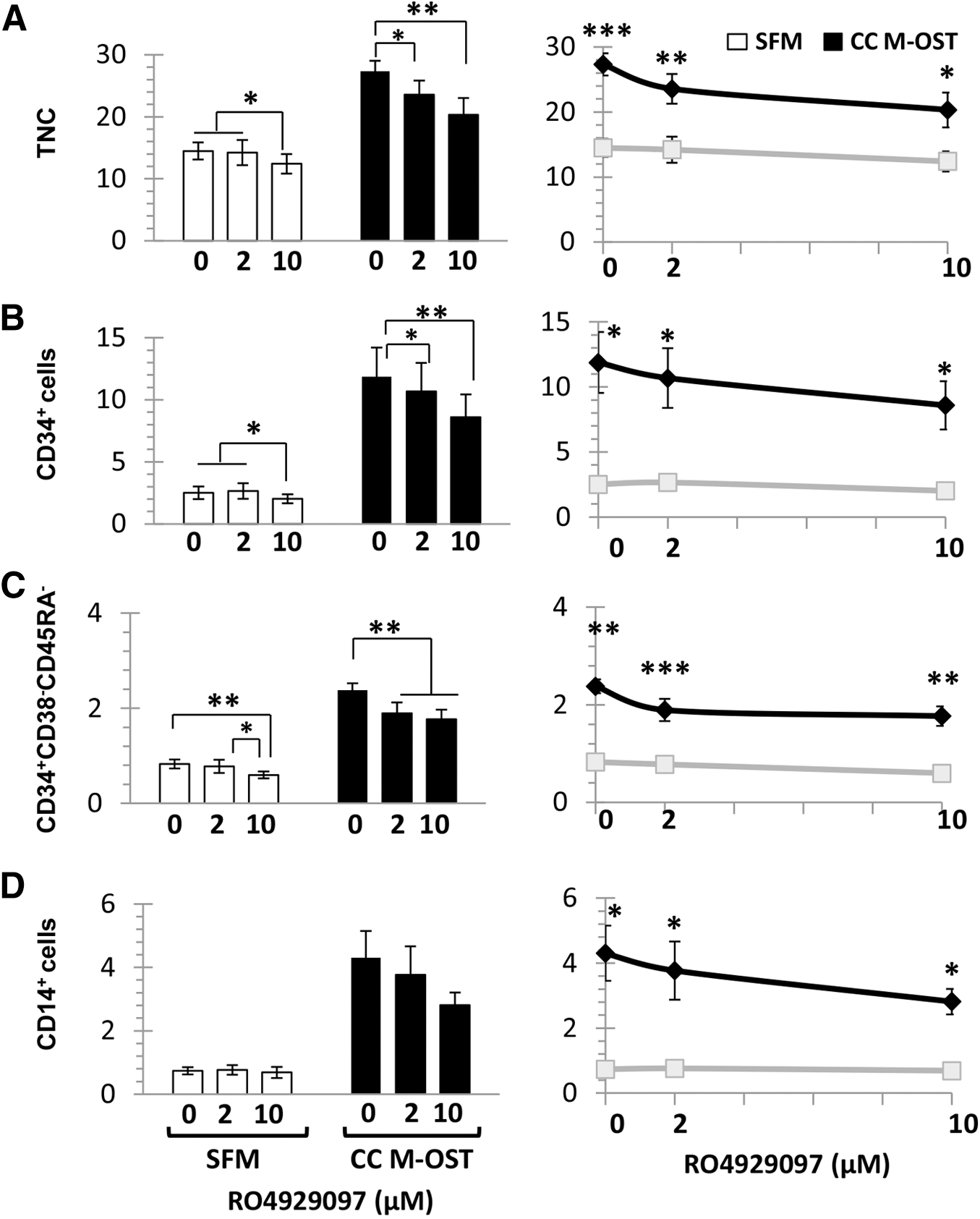
Impact of γ-secretase inhibition on the growth-promoting activities of M-OST cocultures on UCB CD34+ cells. Production of TNC
Discussion
Expansion of HSCs and progenitors is a challenge due to the strong differentiation pressure induced by the ex vivo environment on HSC, and the natural tendency of HSC to lose their stemness state when entering the cell cycle. Under this paradigm, we investigated whether M-OST could serve as feeder cells, and we characterized the molecular mechanisms responsible for their potent growth-promoting activities. Notably, no study had compared the feeder cell capacity of MSC-derived osteoblasts with their MSC precursor, and the molecular mechanisms mediating the growth-promoting activities of osteoblasts on hematopoietic progenitors remained poorly understood.
Coculture of UCB progenitors with MSC and osteoblasts, two cell types located in HSC niches, improved the maintenance and production of CD34+ cells. Moreover, both feeder cells supported greater output of more primitive CD34+ subsets enriched in stem and progenitor cells (ie, CD34+/CD45RA− and CD34+/CD90+/CD45RA− cells) [43]. However, overall UCB cell growth was the highest in M-OST cocultures followed by MSC cocultures and, finally, SFM. These findings are supported by data recently reported with primary osteoblasts [5,24], and they confirm those previously reported with MSC [44 –50]. These also reveal an important distinction between M-OST cocultures and cultures done with osteoblast conditioned medium. Indeed, although both systems favor strong proliferation and greater progenitor expansion, the frequencies of immature UCB subsets in M-OST cocultures are significantly superior [7]. Another important difference was that inhibition of IGF-1R had no significant impact on the growth of UCB CD34+ cells in coculture (K. Law & O. Hovey, unpublished results). Altogether, this reinforces the concept that cell contact in addition to paracrine factors between niche elements and CD34+ cells promotes the maintenance of phenotypically defined stem and progenitor subpopulations [51 –53].
Interestingly, our study also revealed that MSC and M-OST differentially modulated the expression of CXCR4 and fucosylated PSGL-1 (CLA) on CD34+ cells. Indeed, although the proportion of CLA+ UCB TNC and CD34+ was higher in both cocultures, it was the highest in MSC cocultures. Conversely, the opposite was observed for CXCR4 since it was the highest in M-OST. Both increased fucosylation of UCB CD34+ cells and higher CXCR4 expression improve homing to the marrow and have been associated with increased engraftment in xenotransplantation and in a human trial [54 –56]. Whether coculture of CD34+ cells with M-OST or MSC promotes the homing of UCB progenitors as previously reported for fucosylated UCB progenitors is an interesting hypothesis that remains untested at this time [55].
An initial comparison of the serial engraftment activities of UCB progenitors expanded in coculture revealed that UCB HSC and progenitors expanded in M-OST cocultures had engraftment activities that may be superior to those expanded with their MSC precursors. This prospect is supported by the greater levels of engraftment in the periphery (platelet and leukocytes), and the trend of higher human BM chimerism in primary and secondary transplants. The improved platelet engraftment is reminiscent of that reported with progenitors expanded in osteoblast conditioned medium, which was superior to the engraftment seen with SFM- or MSC-conditioned medium [11]. However, we recognize that these results represent a first step and will need to be confirmed in future studies.
In an effort to elucidate the molecular pathways mediating the growth-promoting activity of M-OST on UCB CD34+ cells, we evaluated the contribution of the Wnt/β-Cat and Notch pathways. Wnt signaling is an important, yet contested, pathway that is shown to influence CD34+ cell growth ex vivo [13]. It is presently accepted that tight regulation of Wnt is required for growth promotion of HSC in cultures [22]. We confirmed expression of Wnt genes in both MSC and M-OST, and we also showed that UCB CD34+ cells cocultured with M-OST showed increased levels of β-Cat as well as downstream gene targets, including c-Fos and c-Jun. Increased levels of such AP-1 related transcripts in UCB cells are consistent with the increased cell expansion achieved in M-OST cocultures.
Increases in β-Cat levels are often regarded as a hallmark of Wnt canonical pathway activation, although cross-talk between the non-canonical and canonical pathways has been demonstrated [57]. Results in this study provide an important missing functional insight into the role of Wnt/β-Cat in osteoblast-mediated growth regulation of hematopoietic progenitors recently brought forward by gene profiling. Indeed, a recent study reported increased expression of genes associated with “regulation of Wnt receptor signaling pathway” and “cell proliferation” in UCB CD34+ cells cocultured with primary osteoblasts [24]. Herein, we demonstrated that the growth-promoting activity of osteoblasts on UCB CD34+ cells is largely mediated by β-Cat/TCF, since its inhibition profoundly reduced UCB progenitor growth and CD14+ cells while having a minimal effect in SFM cultures.
As previously indicated, a complex interplay of regulatory activities exists between osteoblasts, Wnt proteins, and hematopoiesis [1,12,58]. Our results are partially consistent with the reduced number of CFC recovered in transgenic mice overexpressing the Wnt-inhibitor Dkk1 in the endosteal niche [59]. Repression of Wnt signaling was also associated with a loss of HSC activity in serial transplant. On the other hand, Wnt canonical signaling has previously been reported to down-modulate expression of CXCL12/SDF-1 in MSC [60], and SDF-1 was previously shown to modulate HSC quiescence in stem cell niches and in cultures [61,62]. Consistent with this model, we found a 5.4-fold reduction in SDF-1 transcript levels in M-OST versus undifferentiated MSC (P = 0.05). Therefore, it is tempting to speculate that reduced SDF-1/CXCR4 signaling in UCB CD34+ M-OST cocultures may also contribute to the increased CD34+ cells growth.
Conversely, other studies have implicated Notch signaling in osteoblast-mediated regulation of HSC and progenitors [5,25,26], and γ-secretase inhibition was previously shown to down-modulate Notch signaling and to reduce osteoblast-mediated expansion of LSK cells [5]. In our study, γ-secretase inhibition led to a reduction of growth in both culture settings, but expansion remained superior in M-OST cocultures. Nonetheless, the net outputs of UCB CD34+ and CD34+/CD38−/CD45RA− cells in M-OST cocultures were reduced even with a low concentration of the inhibitor, suggesting that Notch signaling may still contribute to the growth-promoting activities of M-OST coculture.
MSC have been isolated from many tissues aside from the BM, but the capacity of those MSC to support hematopoiesis ex vivo remains poorly investigated [63]. Whether M-OST derived from tissues other than BM share the same properties as BM M-OST will need to be investigated to further define the functional overlap between MSC from distinct sources. In conclusion, this study demonstrated that the capacity of osteoblasts to raise the growth and expansion of UCB CD34+ progenitors is mediated in large part by Wnt/β-Cat and to a lesser extent by the Notch-dependent pathways. Moreover, our results showed that M-OST promoted the expansion of HSC-enriched subpopulations and CFC progenitors in a similar fashion to what was originally reported with primary osteoblasts. Future work will be required to further quantify the modulating activity of M-OST on the engraftment activities of human HSC.
Footnotes
Acknowledgments
The authors wish to thank the staff at Canadian Blood Services (CBS) Cord Blood for Research Program for their precious help. This work was funded through a CBS and the Canadian Institutes of Health and Research partnership grant (RN210152), which is funded in part by Health Canada, a division of the federal government of Canada. As a condition of this support, this article must contain the statement “The views expressed herein do not necessarily represent the views of the federal government.”
Author Disclosure Statement
No competing financial interests exist.