
Abstract
Calreticulin (CALR) is a chaperone protein that localizes primarily to the endoplasmic reticulum (ER) lumen where it is responsible for the control of proper folding of neo-synthesized glycoproteins and the retention of calcium. Recently, mutations affecting exon 9 of the CALR gene have been described in approximately 40% of patients with myeloproliferative neoplasms (MPNs). Although the role of mutated CALR in the development of MPNs has begun to be clarified, there are still no data available on the function of wild-type (WT) CALR during physiological hematopoiesis. To shed light on the role of WT CALR during normal hematopoiesis, we performed gene silencing and overexpression experiments in hematopoietic stem progenitor cells (HSPCs). Our results showed that CALR overexpression is able to affect physiological hematopoiesis by enhancing both erythroid and megakaryocytic (MK) differentiation. In agreement with overexpression data, CALR silencing caused a significant decrease in both erythroid and MK differentiation of human HSPCs. Gene expression profiling (GEP) analysis showed that CALR is able to affect the expression of several genes involved in HSPC differentiation toward both the erythroid and MK lineages. Moreover, GEP data also highlighted the modulation of several genes involved in ER stress response, unfolded protein response (UPR), and DNA repair, and of several genes already described to play a role in MPN development, such as proinflammatory cytokines and hematological neoplasm-related markers. Altogether, our data unraveled a new and unexpected role for CALR in the regulation of normal hematopoietic differentiation. Moreover, by showing the impact of CALR on the expression of genes involved in several biological processes already described in cellular transformation, our data strongly suggest a more complex role for CALR in MPN development that goes beyond the activation of the THPO receptor and involves ER stress response, UPR, and DNA repair.
Introduction
C
In the cytoplasm, CALR has been reported to be associated with the C-terminal tail of alpha integrins [3] and has been demonstrated to affect cell adhesion through the induction of adhesion molecule expression and its involvement in beta-catenin-associated pathways [4]. Interestingly, at the cell surface, CALR has been described to function as “eat me signal” regulating recognition and removal of apoptotic cells by professional phagocytes [5].
Recently, two independent groups reported the discovery of mutations in the CALR gene in myeloproliferative neoplasms (MPNs), specifically in 60–80% of JAK2- and MPL-unmutated essential thrombocythemia (ET) and primary myelofibrosis (PMF) patients [6,7]. So far, 36 different types of CALR mutants have been reported in MPNs [6]. All these mutations, consisting of deletions and/or insertions, lead to a 1-bp frameshift generating an alternative reading frame, resulting in the loss of most of the acidic C-terminal domain and the KDEL signal, which might lead to protein mislocalization and aberrant protein function and stability.
Recently, several reports shed light on the function of CALR mutants in ET and PMF: Marty et al. [8] established a retroviral mouse model for CALRins5 and CALRdel52, showing that CALR mutations are able to induce the development of an ET-like phenotype, and in case of CALRdel52, the progression to myelofibrosis. Moreover, Chachoua et al. demonstrated the necessity of the thrombopoietin (THPO) receptor MPL for CALR-mediated cellular transformation [9]. Finally, two research groups have shown physical interaction of CALR mutants and MPL; CALR mutations generate a novel C-terminal domain that enables the N-terminal domain of CALR to interact with the extracellular domain of MPL, causing its activation [10,11].
So far, the role of CALR mutations in the development of MPN has been only partially elucidated, and no data are available on the physiological function played by CALR during normal hematopoiesis. The only report available is focused on the study of CALR during stress erythropoiesis [12]: the authors demonstrated that in normal proerythroblasts, the CALR C-terminal domain undergoes conformational changes in response to Ca2+ that activate the nuclear export of the glucocorticoid receptor (GR), resetting the stress response and allowing the cells to undergo terminal differentiation.
In this study, to unravel the physiological role played by CALR in normal hematopoiesis, overexpression and gene silencing experiments have been performed. Our results demonstrated that CALR expression is able to induce the differentiation of hematopoietic stem progenitor cells (HSPCs) toward the erythroid and megakaryocytic (MK) lineages. GEP analysis pointed out the activation mediated by CALR of several signaling pathways involved in erythroid and MK development and in the regulation of HSC self-renewal. Moreover, our GEP data showed that CALR also affects the expression of genes involved in DNA repair, and in ER stress response and unfolded protein response (UPR). These data suggest a more complex role for CALR in the development of the MPN disease that goes beyond the interaction and activation of the THPO receptor.
Materials and Methods
Ethics statement
Human CD34+ cells were purified upon donor's informed written consent from umbilical cord blood (CB) samples, collected after normal deliveries, according to the institutional guidelines for discarded material (Clearance of Ethical Commitee for Human experimentation of Florence: Comitato Etico Area Vasta dell'Azienda Ospedaliero-Universitaria Careggi, approval date: April 22, 2011, approval file number # 2011/0014777).
Human CD34+ HSPCs purification
CB CD34+ cells were purified as previously described [13]. After immunomagnetic separation, CD34+ cells were seeded in 24-well plates at 5 × 105/mL in Iscove's modified Dulbecco's medium (IMDM) (GIBCO, Grand Island, NY) containing 20% human serum (HS) (Bio-Whittaker, Walkersville, MD), SCF (50 ng/mL), Flt3-ligand (Flt3 L) (50 ng/mL), THPO (20 ng/mL), IL-6 (10 ng/mL), and IL-3 (10 ng/mL) (all from Miltenyi Biotec) and transduced with the retroviral vector LXIDN or electroporated 24 h later.
Retroviral vectors packaging
The human CALR cDNA (NM_004343) was synthesized and cloned into retroviral vector LXIDN [14]. Packaging line for LCALRIDN was generated by transinfection in the ecotropic Phoenix and amphotropic GP+envAm12 cells, as previously described [14]. Viral titers were assessed by flow cytometry analysis of a truncated version of low-affinity nerve growth factor receptor (DeltaNGFR, DNGFR) expression percentage upon infection of CB CD34+ cells.
Hematopoietic cell transduction and purification
Transduction of CB CD34+ cells was performed 24 h after isolation. Retroviral transduction was performed by four cycles of infection (one every 12 h) with viral supernatant with the addition of polybrene (8 μg/mL), 20% HS, and human cytokines (SCF, Flt3-l, THPO, IL-6, and IL-3 as described above) in retronectin-coated plates. Untreated 24-well plates were coated with retronectin (10 μg/cm2) (Takara Bio, Inc., Japan) following the manufacturer's protocol. To achieve optimal expansion and infection of primary CD34+ cells, the retronectin-coated plates were preincubated with retroviral supernatant for 4 h, and then CD34+ cells were seeded in 24-well plate at 3 × 105 cells/mL (1 mL/well) in fresh viral supernatant.
After transduction, CB CD34+ cells were maintained in the above described liquid culture conditions for additional 36 h. Transduced CD34+ cells were subsequently purified by means of immunomagnetic selection (EasySep “Do-It-Yourself” Selection Kit; StemCell Technologies) using the anti-human p75-NGFR mouse monoclonal antibody (BD Biosciences). Purity of the NGFR+ cell fraction was assessed by flow cytometry after labeling with PE-conjugated anti-NGFR monoclonal antibody (Miltenyi) 48 h postpurification and was always >90%.
Nucleofection of CD34+ cells
Human CD34+ cells were transfected by using the 4D-Nucleofector™ System (Lonza) as previously reported [15]. Briefly, starting from the day after CD34+ cell purification, each sample was electroporated thrice, once every 24 h, with a small interfering RNA (siRNA) targeting human CALR mRNAs (Life Technologies, siRNA ID s115). For each electroporation, 4 × 105 CD34+ cells were resuspended in 100 μL of P3 Primary Cell Solution (Lonza), containing 3 μg of siRNA, and pulsed with the program DS112. To exclude nonspecific effects caused by interfering RNA (RNAi) nucleofection, a sample transfected with a nontargeting siRNA (NTsiRNA; Life Technologies) was included. Cells were analyzed 48 h after the last nucleofection for both cell viability and CALR mRNA and protein expression.
CD34+ cell culture conditions
For liquid culture differentiation assays, 24 h after the last nucleofection or immediately after NGFR-positive cell isolation (hereafter reported as postnucleofection and posttransduction, respectively), CD34+ cells were plated (5 × 105/mL) in the following: (a) multilineage differentiation medium: IMDM added with 20% BIT serum substitute (bovine serum albumin, insulin, and transferrin; StemCell Technologies), supplemented with SCF (50 ng/mL), Flt3 L (50 ng/mL), THPO (20 ng/mL), IL-6 (10 ng/mL), and IL-3 (10 ng/mL) [15]; (b) MK differentiation medium: serum-free medium SYN-H (ABCell-Bio, Paris, France) supplemented with SCF (5 ng/mL), THPO (50 ng/mL), IL11 (40 ng/mL), IL3 (2 ng/mL), and IL6 (1 ng/mL) [16]; (c) erythroid differentiation medium: IMDM added with 20% BIT serum substitute (StemCell Technologies) supplemented with erythropoietin (EPO; 0.4 U/mL; R&D Systems) and SCF (50 ng/mL; Miltenyi) [17]; (all cytokines from Miltenyi Biotec). The medium was replaced every 3 days.
Methylcellulose and collagen clonogenic assays
The methylcellulose assay was carried out by plating CD34+ cells in MethoCult™ GF H4434 (StemCell Technologies, Inc., Vancouver), as previously described [18]. MK colony-forming units (CFU-MKs) were assayed in a collagen-based medium, using a commercial MK assay detection kit (MegaCult-C; StemCell Technologies, Inc.) as previously reported [18]. CFU-MKs were scored according to the manufacturer's protocol as small (3–21 cells, deriving from more mature megakaryocyte progenitors), medium (21–49 cells), and large (>50 cells, arising from more primitive MK progenitors) colonies based on their size, which reflects the maturation stage of the progenitor cells giving rise to each colony.
Morphological and immunophenotypic analysis
Differentiation of CD34+ cells was monitored by morphological analysis of May-Grunwald-Giemsa-stained cytospins and by flow cytometric analysis of differentiation markers' expression (Glycophorin A [GPA], CD41, and CD42b) on day 9, 12, and 14 after the last nucleofection or retroviral infection. Images were captured by using an Ax10scopeA1 microscope equipped with AxioCam ERc 5S Digital Camera and Axion software 4.8 (all Carl Zeiss MicroImaging, Inc., Thornwood, NY). The images were then processed with Adobe Photoshop 7.0 software. The following monoclonal antibodies (MoAbs) were used for flow cytometric analysis: FITC-conjugated mouse antihuman CD41 MoAb, PE-conjugated mouse antihuman CD42b MoAb, and PE-conjugated mouse anti-human GPA MoAb (all from Dako; Milano, Italia;
Immunofluorescence staining
Cytospins were fixed with 4% paraformaldehyde (PFA) and permeabilized using 0.3% Triton X-100 in PBS for 20 min at room temperature. After blocking with 5% FBS, 2% BSA, and 0.1% Triton X-100 in PBS for 30 min at 37°C, slides were incubated with mouse anti-human CD41a (1:200 in blocking solution, BD Pharmingen™; BD Biosciences, San Jose, CA) or mouse anti-human Glycophorin A (clone JC159, 1:100 in blocking solution; DakoCytomation, Carpiteria, CA) overnight at 4°C. This was followed by incubation with horseradish peroxidase-conjugated goat anti-mouse (1:1,000 in blocking, catalog #sc2005; Santa Cruz Biotechnology, Inc., Heidelberg, Germany) for 30 min at 37°C. All incubations were followed by three washes with PBS solution. Nuclear counterstaining was performed with 4′,6-diamino-2-phenylindole (DAPI). The slides were mounted with the DakoCytomation fluorescent mounting medium (DAKO). Finally, fluorescence imaging was performed using the Zeiss LSM 510 Meta Confocal Microscope (Zeiss, Germany) and digital images of representative areas were taken. To ensure random sampling, 50 images/slide were captured and cells positive for GPA or CD41 were scored.
Western blot
CALR protein levels were assessed by means of western blot analysis in CD34+ cells. Briefly, cells were harvested 48 h after the last nucleofection or immediately after NGFR-positive cell isolation, washed twice with cold phosphate-buffered saline (PBS), and lysed in 50 mM Tris (tris(hydroxymethyl) aminomethane)-Cl (pH 7.4), 150 mM NaCl, 1% Nonidet P-40, 10 mM KCl, 1 mM EDTA, 20 mM NaF, 0.25% Na doexycholate, 5 mM dithiothreitol (DTT). Protease inhibitors (Roche, Indianapolis, IN; Complete, catalog #1697498) and phosphatase inhibitors (ThermoFisher Scientific) were added to the lysis buffer. Total cellular lysates (30 μg for each sample) were loaded and separated on 10% SDS-polyacrylamide gel and then transferred onto a nitrocellulose membrane. To visualize loading and transfer, Ponceau staining has been performed. Membranes were then preblocked in a blocking solution of 0.1% TBST containing 5% non-fat dry milk (NFDM) and then incubated with the following primary antibodies: rabbit policlonal anti-CALR antibody (Abcam; catalog# ab2907, 1:250 dilution at 4°C overnight) and rabbit polyclonal anti-β-actin primary antibody (Thermo Fisher Scientific Inc, catalog #PA1-16889; 1:2,000 dilution for 1 h at RT). The blots were washed three times with 0.1% TBST and then incubated with 1:1,000 dilution of HRP-conjugated goat anti-rabbit secondary antibody (Thermo Fisher Scientific, Inc.; catalog #32460) for 1 h at RT in secondary antibodies. After three successive washes with TBS, BM chemiluminescence blotting substrate (POD) (Roche) was used for protein detection.
RNA extraction
Total cellular RNA was harvested from 1 × 105 cells from each sample using the miRNeasy Micro RNA isolation kit (QIAGEN) according to the manufacturer's instructions. RNA sample concentration and purity (assessed as 260/280 nm and 260/230 nm ratios) were evaluated by NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE), while RNA integrity was assessed by using the Agilent 2100 Bioanalyzer (Agilent Technologies, Waldbrunn, Germany).
Quantitative reverse transcription–polymerase chain reaction
Total RNA (100 ng) was reverse transcribed to cDNA using a High Capacity cDNA Archive Kit (Life technologies; Carlsbad, CA). TaqMan PCR was carried out using the TaqMan Fast Advanced PCR master mix and TaqMan gene expression assays (all reagents from Life Technologies) by means of a 7900HT Fast Real-Time PCR System (Applied Biosystems). Assays were performed in triplicate. Gene expression profiling (GEP) was achieved using the comparative cycle threshold (CT) method of relative quantitation using Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as housekeeping genes. To normalize data, ΔΔCT was calculated for each sample using the mean of its ΔCT values subtracted from the mean ΔCT value measured in the control sample, set as a calibrator; relative quantitation (RQ) value was expressed as 2−ΔΔCT.
Gene expression profiling
GEP was performed on RNA samples isolated from NGFR-positive cells purified after retroviral transduction from the samples transduced with the empty control vector LXIDN (LXIDN), and from the samples transduced with the vector carrying CALR cDNA (LCALRIDN) (three independent experiments). GEP cDNA synthesis and biotin-labeled target synthesis were performed using the GeneChip® 3′ IVT PLUS Reagent Kit according to the protocol supplied by Affymetrix. The HG-U219 Array Strips (Affymetrix, Santa Clara, CA) hybridization, staining, and scanning were performed by using the GeneAtlas Platform.
Robust multiarray average procedure was used to perform probe-level normalization and conversion into expression values [19]. Differentially expressed genes (DEGs) were then selected following a supervised approach with the analysis of variance (ANOVA) module supplied by the Partek GS. 6.6 Software Package (
In the same way, GEP was performed on RNA samples isolated from CALR-silenced (CALRsiRNA sample) and non-targeting negative control siRNA-transfected (NTsiRNA sample) CD34+ cells 24 h after last nucleofection from three independent experiments. GEP data were analyzed by using the Partek GS 6.6 Software Package as reported above, by selecting the probesets with a fold change contrast ≥1.5 (P < 0.05) as DEGs in the pairwise comparison between CALR siRNA and NTsiRNA samples.
Raw and normalized GEP data have been submitted to the NCBI's Gene Expression Omnibus (GEO) public repository [20] (
Statistical analysis
The statistics used for data analysis was based on two-tailed Student t-tests for average comparisons in paired samples (equal variance). Data were analyzed with Microsoft Excel (Microsoft Office, 2011 release) and are reported as mean ± standard error of the mean (SEM). A P-value <0.05 was considered significant.
Results
CALR overexpression in CD34+ HSPCs
To characterize the role of CALR in the proliferation and differentiation of HPSCs, we investigated the effects induced by CALR overexpression in normal CB CD34+ cells. To this end, we constructed the LCALRIDN retroviral vector expressing the full-length CALR cDNA and DLNGFR as reporter gene, in the context of a bicistronic transcript driven by the viral LTR [14]. Freshly isolated CB CD34+ cells were transduced with either LCALRIDN vector or the empty control vector and cell viability was assessed by trypan blue exclusion assay. No significant differences among LXIDN and LCALRIDN after four cycles of infection were found (data not shown).
In a set of five independent experiments, gene transfer efficiency, assessed by flow cytometric analysis of DLNGFR positivity, ranged from 25% to 33%. Transduced cells were then purified by nerve growth factor receptor (NGFR) expression 36 h post-infection. Real-time qPCR and western blot analysis performed on transduced/NGFR-purified cells showed a significant increase in both mRNA (RQ ± SEM, 5.7 ± 1.4, P < 0.05) and protein levels in LCALRIDN-transduced cells compared to the control empty vector (Fig. 1A, B).

Effects of CALR overexpression on HSPC differentiation.
Biological Effects exerted by CALR overexpression in CD34+ HSPCs
To shed light on the biological role played by CALR during hematopoietic commitment and differentiation, CALR cDNA was overexpressed in CB-derived HSPCs and the biological effects exerted by its overexpression were assessed by means of in vitro differentiation assays. Flow cytometry analysis clearly shows that CALR overexpression did not affect granulocyte or mono/macrophage differentiation, as demonstrated by the unchanged expression of granulocyte (ie, CD66b, CD15, and MPO) or monocyte/macrophage markers (ie, CD14 and CD163) (Fig. 1C, D, panels).
Since the presence of HS inhibits erythroid and MK differentiation of HSPCs in vitro, the effects of CALR overexpression on erythroid and MK differentiation were evaluated in serum-free culture conditions by assessing the expression of the erythroid marker GPA and the MK markers CD41 and CD42b. Flow cytometry analysis showed a statistically significant increase in the percentage of GPA-positive cells associated with CALR overexpression (Fig. 1E). Moreover, our data also revealed a statistically significant increase in the fraction of CD41+ and CD42b+ MK cells in the sample transduced with LCALRIDN vector compared to the control sample (Fig. 1F, G).
In agreement with liquid culture results, methylcellulose-based clonogenic assay showed a significant increase in the percentage of erythroid colonies (BFU-E and CFU-E) upon CALR overexpression, while no statistically significant differences in myeloid colonies were noticed (CFU-G, CFU-M, and CFU-GM) (Fig. 1H). The collagen-based clonogenic assay demonstrated a significant increase in the percentage of CFU-MK colonies coupled to a decrease in the non-MK ones in the LCALRIDN sample compared to the control (Fig. 1I). Moreover, morphological analysis of May-Grunwald-Giemsa-stained cytospins and immunofluorescent analysis performed on days 9, 13, and 15 of liquid culture revealed that CALR overexpression lead to a strong enrichment in erythroid and MK precursors at different stages of maturation (Fig. 2A, B).
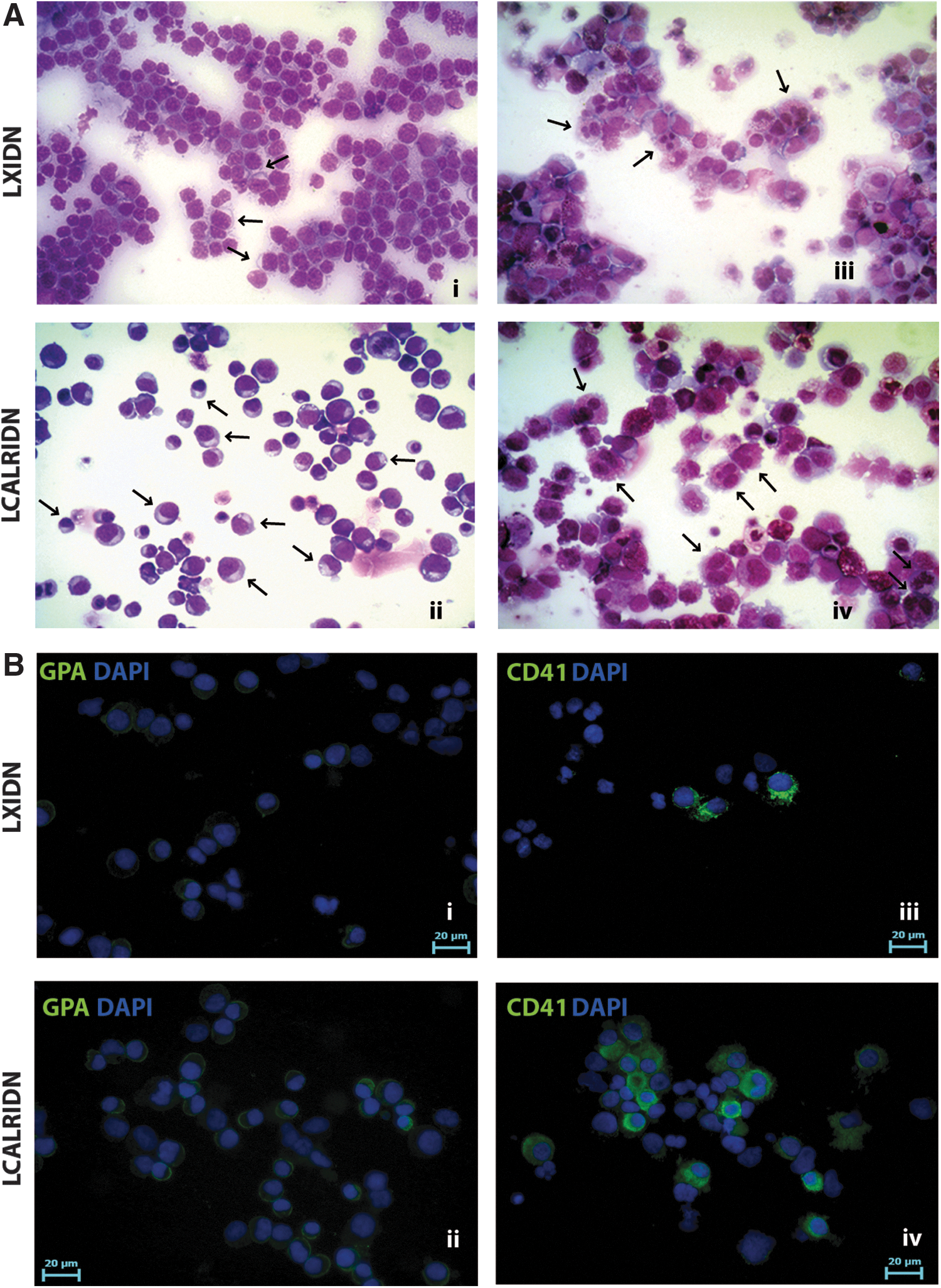
Morphological and immunofluorescent analyses of LXIDN and LCALRIDN samples.
Gene expression profile of CALR-overexpressing CD34+ cells
To better characterize the molecular mechanisms underlying the biological effects of CALR overexpression on erythroid and MK differentiation, we carried out a microarray-based gene expression analysis on NGFR-positive cells purified after retroviral transduction (Methods section) to compare CALR-overexpressing cells vs LXIDN-transduced cells. The list of 54 DEGs is shown in Supplementary Table S1 (Supplementary Data are available online at
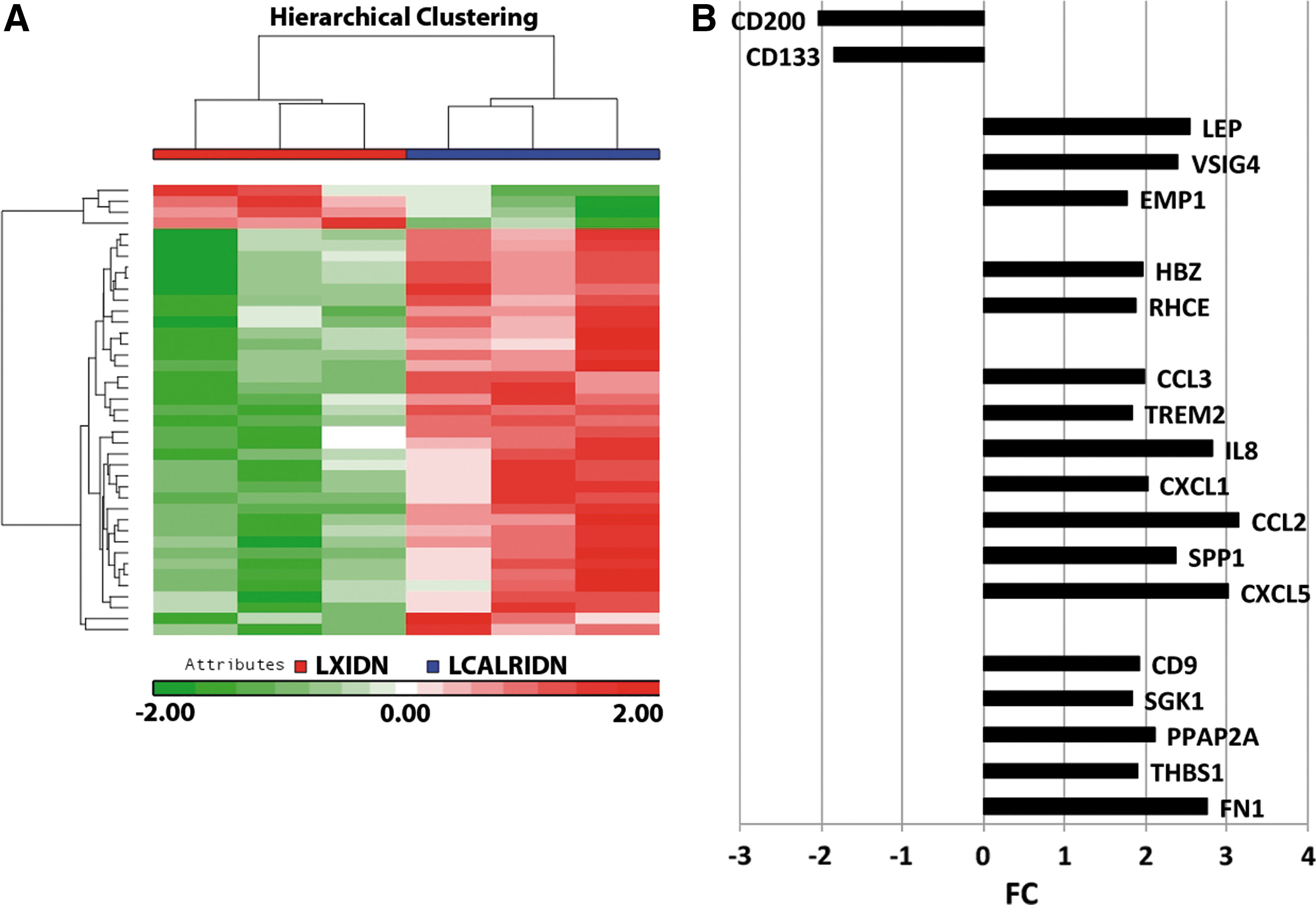
Effects of CALR overexpression on HSPC gene expression profile.
Functional analysis by means of Ingenuity Pathway analysis software (IPA, version 8.6; Ingenuity Systems, Redwood City, CA,
CALR silencing in CD34+ HSPCs
To confirm the role played by CALR in the regulation of megakaryocytopoiesis and erythropoiesis, CALR silencing experiments were performed. To this end, CB CD34+ cells were transfected with either CALR siRNA (CALR siRNA sample) or a non-targeting control siRNA (NT siRNA sample). Real-time quantitative reverse transcription–polymerase chain reaction (qRT-PCR) and western blot analysis, performed, respectively, 24 and 48 h after last nucleofection, confirmed CALR downregulation both at the mRNA (RQ ± SEM, 0.12 ± 0.014, P < 0.001) and protein level in the CALR siRNA sample compared to the control (Fig. 4A, B).
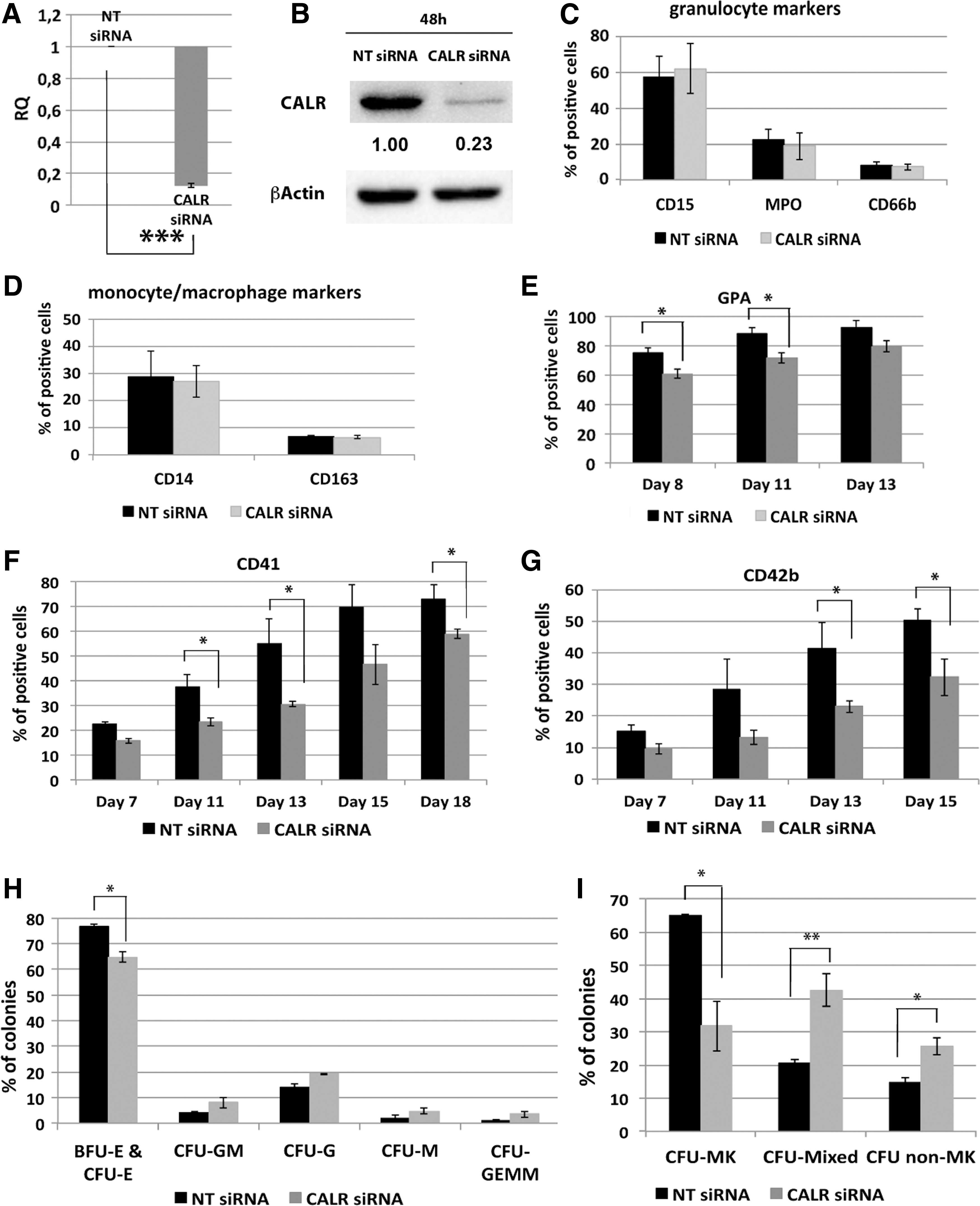
Effects of CALR silencing on HSPC differentiation.
Flow cytometric analysis of myeloid lineage differentiation markers showed no difference in the expression level of either granulocytic (ie, CD66b, CD15, and MPO) or mono/macrophage (ie, CD14 and CD163) markers in CALR siRNA sample compared to the control (Fig. 4C, D). On the other hand, results from multilineage and EPO-driven cultures, clearly demonstrated a strong effect induced by CALR silencing on both MK and erythroid commitment. As for the erythroid differentiation, we observed a steady decrease of the erythroid marker GPA in CALR-silenced cells compared to control cells (Fig. 4E). Strikingly, our data showed a significant decrease in the expression of the MK markers CD41 and CD42b in CALR-silenced cells compared to the negative control sample (Fig. 4F, G).
To better characterize the role of CALR in HSPC differentiation, cells were plated in methylcellulose-based medium 24 h after the last nucleofection. Methylcellulose clonogenic assay showed no statistically significant difference in the percentage of myeloid colonies (CFU-G, CFU-GM, CFU-M, and CFU-GEMM) in CALR-silenced cells compared to the negative control cells. On the other hand, CALR silencing was able to induce a statistically significant decrease in the percentage of erythroid colonies (BFU-E & CFU-E) compared to the control sample (Fig. 4H).
To assess the effect mediated by CALR silencing on MK differentiation, a collagen-based clonogenic assay supporting megakaryocyte progenitor growth in vitro was performed. Results showed a significant decrease in the percentage of MK colonies coupled to an increase in the percentage of non-MK and mixed colonies in samples transfected with CALR siRNA compared to the control (Fig. 4I). Morphological analysis of May-Grunwald-Giemsa-stained cytospins and immunofluorescent analysis confirmed that CALR silencing led to a decrease in the number of MK and erythroid precursors in unilineage MK and erythroid cultures, respectively (Fig. 5A, B). Altogether, our data demonstrated that CALR silencing is able to inhibit MK and erythroid differentiation of HSPCs, in agreement with the results obtained in CALR overexpression experiments.
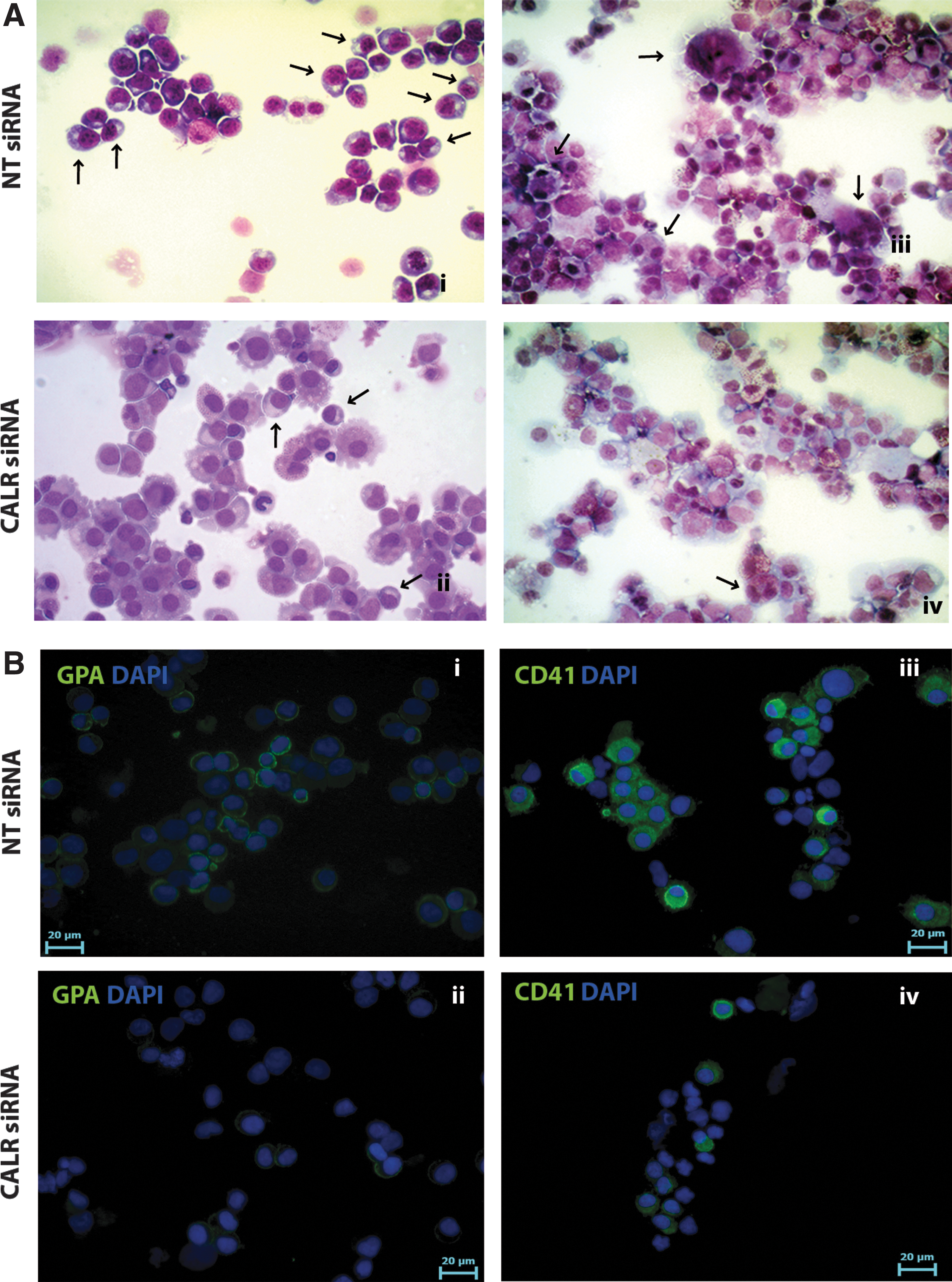
Morphological and immunofluorescent analyses of LXIDN and LCALRIDN samples.
Gene expression profile of CALR-silenced CD34+ cells
To shed light on the molecular mechanisms supporting the biological effects exerted by CALR silencing, GEP analysis was performed on CALR-silenced cells (CALRsiRNA sample) and non-targeting negative control siRNA-transfected cells (NTsiRNA sample). The list of 154 DEGs is shown in Supplementary Table S2. Figure 6A shows the hierarchical clustering of NTsiRNA and CALRsiRNA samples based on the list of 154 DEGs.
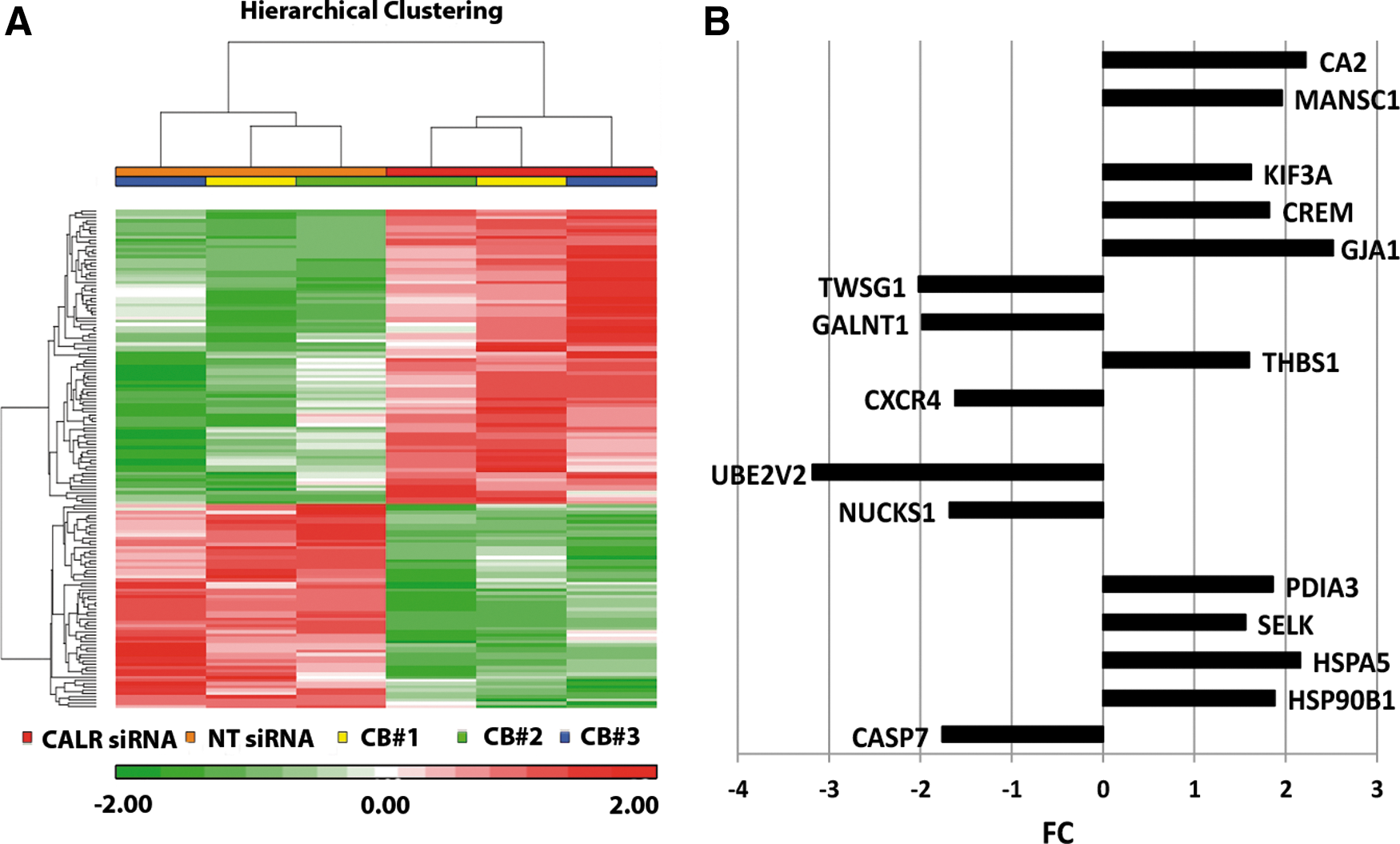
Effects of CALR silencing on HSPC differentiation on gene expression profile.
Functional analysis by means of Ingenuity Pathway analysis software identified several categories of DEGs, such as ER stress, UPR, proliferation of blood cells, quantity of myeloid cells, quantity of hematopoietic progenitor cells, and DNA repair. In particular, among upregulated genes (Fig. 6B and Supplementary Fig. S1B), we found genes involved in UPR and ER stress response, such as Heat Shock Protein 90 kDa Beta 1 (HSP90B1) [33], Heat Shock 70 kDa Protein 5 (HSPA5) [34], and selenoprotein K (SELK) [35] and in the regulation of self-renewal of hematopoietic stem cells (HSCs) such as cAMP responsive element modulator (CREM) [36] and kinesin family member 3A (KIF3A) [37], and hematological malignancies markers such as MANSC domain containing 1 (MANSC1) [38] and carbonic anhydrase II (CA2) [39].
On the other hand, among downregulated genes (Fig. 6B), we found genes involved in DNA repair such as nuclear casein kinase and cyclin-dependent kinase substrate 1 (NUCKS1) [40] and ubiquitin-conjugating enzyme E2 variant 2 (UBE2 V2) [41]. Altogether, GEP results suggest a role for CALR in several biological processes, some of which, such as ER stress response and UPR, might have been inferred from the role of chaperone that CALR plays in the ER, while some others were unexpected, such the involvement in DNA repair or in HSC self-renewal.
Discussion
Recently, the discovery of mutations affecting the CALR gene has filled a gap regarding the genomic abnormalities responsible for roughly 60–80% of the JAK2- and MPL-unmutated ETs and PMFs. CALR mutations have been demonstrated to induce the development of an ET-like phenotype, progressing to myelofibrosis in case of CALRdel52 in a retroviral mouse model [8]. Moreover, mutated CALR has been shown to bind to the THPO receptor MPL, causing its dimerization and activation and thus leading to the constitutive activation of JAK2 signaling pathway [10,11]. This has established an important link between CALR, MPL, and JAK2 in the pathogenesis of CALR-mutated MPNs.
So far, most of the research efforts have been focused on the understanding of the role played by mutated CALR in the development of MPNs. Before the discovery of CALR mutations in 2013, no involvement of CALR has ever been suggested in the context of physiological hematopoiesis, and currently no data are available on the function of CALR during the hematopoietic development.
To shed some light on CALR function during physiological hematopoiesis, we performed overexpression and gene silencing experiments in CD34+ HSPCs isolated from normal donors. CALR has been overexpressed in CD34+ cells by means of retroviral-mediated gene transfer, and its effects on hematopoietic progenitor cell differentiation have been assessed both in liquid and semisolid cultures.
Our results unravel a new and unexpected role for CALR in the differentiation of CD34+ cells. In particular, liquid culture differentiation assays showed that CALR overexpression in CD34+ cells enhances the expression of both erythroid and MK lineage markers. In agreement with liquid culture results, methylcellulose-based and collagen-based clonogenic assays showed a significant increase in the percentage of the erythroid and MK colonies, respectively, in CALR-overexpressing cells compared to the controls.
Moreover, GEP analysis unveiled the upregulation in CALR-overexpressing CD34+ cells of several genes involved in erythroid differentiation, such as RHCE [28] and HBZ [29], and in platelet activation, such as SGK1 [21], CD9 [22], FN1 [23], and THBS1 [24]. These results support the role of wild-type CALR in the regulation of physiological hematopoiesis enhancing differentiation toward the erythroid and MK lineages. Furthermore, our GEP data pointed out the upregulation of several proinflammatory genes, such as the chemokines CCL2 [25], CCL3 [26], and CXCL5 [27], and cytokines like IL8 [27], and cancer-related markers already described in hematological neoplasms, such as EMP1 [30], VSIG4 [31], and LEP [32].
These data suggest that CALR expression in HSPCs is able to induce the expression of several genes that have already been described to play a role in the development of MPNs, this in turn implies that CALR might contribute to the pathogenesis of the MPN disease not only through the above-mentioned interaction with THPO receptor but also by enhancing erythroid and MK differentiation together with inducing the expression of proinflammatory cytokines already described to be responsible for the development of BM fibrosis that characterizes PMF.
In agreement with the overexpression results, CALR silencing in CD34+ cells significantly inhibited both erythroid and MK differentiation. Our data showed that CALR silencing causes a significant decrease in the percentage of cells positive for the erythroid marker GPA as well as a significant decrease in the percentage of cells expressing the MK markers CD41 and CD42b. The inhibition of erythroid and MK differentiation was also confirmed by morphological analysis of May-Grunwald-Giemsa-stained cytospins, which evidenced in CALR-silenced cells a decrease in polychromatic and orthochromatic erythtoblasts in erythroid unilineage culture and a decrease in the number of megakaryoblasts in THPO-driven culture.
According with liquid culture data, methylcellulose-based and collagen-based clonogenic assays demonstrated a significant decrease in the percentage of erythroid and MK colonies, respectively, upon CALR silencing.
Moreover, GEP analysis unravels several signaling pathways modulated after CALR silencing, such as ER stress response, UPR, and DNA repair. Among upregulated genes upon CALR silencing, we found genes involved in UPR and ER stress response such as, HSP90B1 [33], HSPA5 [34], and SELK [35], genes involved in the regulation of self-renewal of HSCs such as CREM [36] and KIF3A [37]. Among downregulated genes, we found genes involved in DNA repair such as NUCKS1 [40] and UBE2 V2 [41]. GEP results after CALR silencing confirm a role of CALR in ER stress response and UPR, exemplified by the differential expression of HSP90B1, HSPA5, and SELK. In addition, GEP results disclose a possible role of CALR in DNA repair or in HSC self-renewal, which might have not been foreseen otherwise.
Altogether, our results suggest a role for CALR during physiological hematopoiesis: CALR expression may affect HSPC differentiation toward the erythroid and MK lineages. Moreover, GEP analysis suggested that CALR modulates the expression of several genes involved in ER stress response, UPR, and DNA repair, and of several genes that have already been described to play a role in the development of MPNs, such as proinflammatory chemokines and hematological neoplasm-related markers.
To our knowledge, this is the first report describing the role of WT CALR in hematopoiesis. Moreover, our data are strongly suggestive of an additional role for CALR in the development of the MPN disease. So far, the oncogenic role for CALR mutants has been linked only to the binding of mutated CALR to the THPO receptor and its consequent JAK2 pathway activation. Our data pointed out a role for WT CALR in the induction of MK and erythroid differentiation, which are the two main lineages affected in the Philadelphia-negative classical MPNs.
Moreover, we also showed a role for CALR in several biological processes, which might take part in the pathogenesis of MPNs such as the expression of proinflammatory cytokines, or the involvement in ER stress response, UPR, and DNA repair, processes that have been already described to be linked to cellular transformation and oncogenesis. This work sheds light on the physiological function of WT CALR during hematopoiesis and moreover suggests novel pathways that might be affected by CALR mutations that have not been investigated yet.
Footnotes
Acknowledgments
This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC), project number #10005 “Special Program Molecular Clinical Oncology 5 × 1000” to AGIMM (AIRC-Gruppo Italiano Malattie Mieloproliferative,
Author Disclosure Statement
The authors declare no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.