
Abstract
Producing hair cells of the inner ear is the major goal of ongoing research that combines advances in developmental and stem cell biology. The recent advent of an inner ear organoid protocol—resulting in three-dimensional stem cell-derived tissues resembling vestibular sensory epithelia—has sparked interest in applications such as regeneration, drug discovery, and disease modeling. In this study, we adapted this protocol for a novel mouse embryonic stem cell line with a fluorescent reporter for Pax2 expression. We used Pax2EGFP/+ organoid formation to model otic induction, the pivotal developmental event when preplacodal tissue adopts otic fate. We found upregulation of Pax2 and activation of ERK downstream of fibroblast growth factor signaling in organoid formation as in embryonic inner ear development. Pax2 expression was evident from the EGFP reporter beginning at the vesicle formation stage and persisting through generation of the sensory epithelium. The native ventralizing signal sonic hedgehog was largely absent from the cell aggregates as otic vesicles began to form, confirming the dorsal vestibular organoid fate. Nonetheless, cochlear- or vestibular-like neurons appeared to delaminate from the derived otic vesicles and formed synaptic contacts with hair cells in the organoids. Cell lines with transcriptional reporters such as Pax2EGFP/+ facilitate direct evaluation of morphological changes during organoid production, a major asset when establishing and validating the culture protocol.
Introduction
H
Stem cells are heralded as the key to replacing damaged tissues as well as modeling disease states in the laboratory. To realize this potential, we must establish reliable routes from naive pluripotency to mature cell fates. One approach is to follow cues from developmental literature, supplying signaling molecules as switches guiding differentiation along known pathways. During embryonic development, cochlear and vestibular organs and cochleovestibular neurons develop from thickened regions of surface ectoderm between the neural plate/crest and epidermis. These regions, the otic placodes, are influenced by morphogens secreted from surrounding tissues, including the underlying mesoderm [fibroblast growth factors (FGFs)], neural plate (FGFs), dorsal neural tube [Wnts and bone morphogenetic proteins (BMPs)], and ventral neural tube [sonic hedgehog (SHH)] (reviewed in Groves and Fekete [2]).
Koehler et al. recently pioneered a method for generation of inner ear sensory epithelia using a three-dimensional stem cell culture paradigm [3,4]. Their protocol was built on methods for making retinal and cerebral organoids from aggregates of mouse embryonic stem cells (mESCs) in serum-free culture (ie, serum-free floating culture of embryoid body-like aggregates with quick reaggregation, or SFEBq) [5 –7]. The initial SFEBq methods were remarkably simple, belying the complexity of the tissues they produced and exploiting the tendency of dissociated mESCs to default to neural fates under serum-free conditions [8,9]. Two key modifications based on developmental mechanisms resulted in otic tissue. Differentiation was guided toward non-neural ectoderm via regulation of transforming growth factor (TGF)β/BMP signaling [10,11] and subsequently toward otic placode via regulation of BMP and FGF signaling [12 –15].
During formation of the otic placode, the specialized ectoderm from which all inner ear structures develop, FGF signaling upregulates the transcription factor Pax2 [15 –19]. Although Pax2 is expressed in multiple embryonic tissues [20 –22], it is commonly regarded as indicative of otic lineage in hair cell regeneration studies [4,23 –26]. In this study, we produced inner ear organoids using mESCs with a reporter for expression of Pax2. We demonstrated the utility of such a reporter system in identifying vesicle formation in live cultures and the maintenance of marker expression through terminal hair cell differentiation. This allowed a direct relationship to be established between exogenous FGF2 dose and formation of otic vesicle-like structures. The dose of FGF2 also corresponded directly with ERK phosphorylation, suggesting that ERK mediates otic induction in this model system as in avian and zebrafish embryos [27,28]. Finally, we compared neurons that arise in these organoids with embryonic neuroblasts and identified opportunities for future investigation of neurogenesis.
Overall, we draw parallels between several features of our organoids and developing inner ears. The efficiency of self-patterning in organoid cultures fluctuates, however, with multiple determinants in need of clarification. Therefore, since our evidence supports the use of inner ear organoids as a developmental model, we present insights into troubleshooting in hopes of advancing the field toward achieving the promise of stem cell technology.
Materials and Methods
Mice
All mice were housed as per institutional standards and used according to experimental protocols approved by the University of Michigan Institutional Animal Care and Use Committee.
We used mice carrying the Pax2EGFP allele, in which EGFP is inserted upstream of the Pax2 translational start site, and wild-type (WT) controls on the same genetic background [29]. Breeding cages with Pax2EGFP/+ mice were set up to obtain embryos of all 3 genotypes; postnatal studies were performed only with WT and Pax2EGFP/+ genotypes as the Pax2EGFP/EGFP genotype is perinatal lethal due to renal agenesis [29]. Genotyping was performed using the following primers: EGFP-F (5′-CTCGTGACCACCCTGACCTA-3′) and EGFP-R (5′-GTCCATGCCGAGAGTGATCC-3′); WT-F (5′-ACCGTATTACCGCCATGCAT-3′) and WT-R (5′-ACCTCTACAAATGTGGTATGGCT-3′). Amplification with EGFP primers results in a 525-bp PCR product, and amplification with WT results in a 230-bp PCR product. Presence of the 525-bp product represents the Pax2EGFP allele.
To prepare images of marker expression at roughly equivalent developmental stages in comparison with differentiating mESC aggregates, timed pregnant C57BL/6 mice at E11.5 and E15.5 were obtained from The Jackson Laboratory. Embryos were collected at E11.5 and E15.5, fixed in 4% paraformaldehyde (PFA), cryoprotected with sucrose, and cryosectioned in optimal cutting temperature compound (OCT) in transverse and parasagittal planes at 12 μm thickness. Staining of cryosections was performed according to the method described for mESC aggregates below.
Auditory brainstem response recording
Mice were anesthetized with 65–120 mg/kg ketamine and 7 mg/kg xylazine, with or without 2 mg/kg acepromazine, administered intraperitoneally before the procedure. Mice were then placed on a heating pad inside the recording booth. Electrodes were then attached at the vertex of the head, beneath the test ear, and beneath the contralateral ear. Acoustic stimuli consisting of 4 ms tone bursts with 1 ms rise and fall times were delivered from a speaker at 30 bursts per second via a tube placed just outside the ear canal. Auditory-evoked potentials were recorded at 3 tone frequencies: 8, 16, and 32 kHz. Data were acquired using the Tucker Davis Technologies System III, with up to 1,024 responses averaged for each stimulus. Recordings began at 80 dB sound pressure level, which was sufficient to elicit a response. Stimulus level was reduced systematically in 5–10 dB decrements. The minimum level eliciting a reproducible waveform was determined to be the response threshold. The recording system was routinely calibrated in a closed system using a reference microphone and lock-in amplifier.
Derivation of Pax2EGFP mESCs
Blastocysts were flushed from the uterine horns of Pax2EGFP/+ females 3.5 days after mating to Pax2EGFP/+ males. Individual blastocysts were placed in wells of a 96-well culture plate seeded with irradiated mouse embryonic feeder (MEF) cells in Dulbecco's modified Eagle's medium (DMEM) (high glucose; Gibco) supplemented with 15% fetal bovine serum (FBS; Harlan), 0.1 mM β-mercaptoethanol (Sigma), 50 IU/mL penicillin and 50 μg/mL streptomycin (Gibco), 1,000 U/mL leukemia inhibitory factor (LIF; Chemicon), and 12.5 μM PD98059 (Sigma). Inner cell mass outgrowths were trypsinized and passaged sequentially until mESC lines were established in 35-mm cell culture dishes. For genotyping, mESCs were passaged on gelatin-coated dishes twice to eliminate feeder cells before being genotyped as described [29]. For expanding cultures, mESCs were maintained on irradiated MEF cells with media consisting of DMEM (Gibco), 15% FBS (Atlas Biologicals), 1× sodium pyruvate, 1× nonessential amino acids, 2× glutamax, and 0.1 mM β-mercaptoethanol (all from Gibco), and 1,000 U/mL LIF (ESGRO). Cells used in this study were frozen at passage 8, thawed, and expanded in feeder-free culture conditions before organoid formation.
mESC cultures
ESCs from the Pax2EGFP/+ mouse line were used to generate organoids between passages 13 and 17. Pluripotency staining was performed at passages 10 and 21. Colonies were maintained in feeder-free conditions on a 0.1% gelatin substrate with maintenance medium consisting of DMEM (high glucose; Gibco), 10% FBS (Atlas Biologicals), 1.5 mM
Differentiation protocol
Spherical aggregates of mESCs were differentiated according to the inner ear organoid protocol modified from Koehler et al. [3,4]. In brief, mESC colonies were dissociated to single cells and aggregated at 3,000 cells per well in round-bottomed 96-well Nunclon Sphera Microplates (Thermo Scientific) in 100 μL ectodermal differentiation medium. Medium consisted of Glasgow's minimum essential medium, 10% KnockOut serum replacement (KSR), 15 mM HEPES, 1× nonessential amino acids, 1 mM sodium pyruvate (all from Gibco), and 0.1 mM β-mercaptoethanol (Sigma). Y-27632 (Calbiochem) was included in the medium at 10 μM on day 0 only.
On day 1, half of the volume was replaced with medium containing growth factor-reduced (GFR) Matrigel (Corning) to achieve 2% final concentration. On day 3, 25 μL of medium containing SB431542 (1 μM; Stemgent) and BMP4 (10 ng/mL; Stemgent) was added to each well. On day 4.5, 25 μL of medium containing LDN193189 (1 μM; Stemgent) and FGF2 (0, 5, 25, or 100 ng/mL; Sigma) was added to each well. Aggregates were transferred into a new 96-well plate in 200 μL maturation medium containing 1% GFR Matrigel on day 8, with half of the medium exchanged daily for the remainder of the protocol. Maturation medium consisted of advanced DMEM/F12, 1× N-2 supplement, 15 mM HEPES, and 1 × glutamax (all from Gibco). CHIR99021 (3 μM; Stemcell Technologies) was optionally added on day 8 as noted.
Modifications to the Koehler et al. [4] protocol included use of 10% KSR in the ectodermal differentiation medium instead of 1.5%, our variation of FGF2 dose, and our use of nonenzymatic dissociation buffer for passaging and forming aggregates from mESCs.
Immunostaining
Aggregates were collected, fixed 20 min overnight in 4% PFA, and rinsed in phosphate-buffered saline (PBS) before further processing.
For staining of cryosections, fixed aggregates were cryoprotected via 30-min incubations with increasing concentrations of sucrose up to 30% in PBS. Following overnight incubation in 30% sucrose at 4°C, aggregates were incubated 4–5 h in a 1:1 mixture of 30% sucrose and OCT at room temperature, incubated 1 h in OCT, and frozen in cryomolds on dry ice. A Leica 3050S cryostat was used to section tissue at 10-μm thickness. Slides were dried overnight, rehydrated in PBS for 15 min, and then transferred to humid chambers for the remainder of the staining procedure. Sections were blocked and permeabilized 15 min with 10% normal donkey serum (NDS) and 0.1% Triton X-100. Primary antibodies were applied overnight at 4°C in a 1:1 mixture of blocking/permeabilization solution with PBS. Alexa Fluor secondary antibodies were applied at 1:500 in PBS for 1 h at room temperature with slides covered to prevent photobleaching. Nuclei were counterstained by 5-min incubation with Hoechst 33242. Coverslips were added with ProLong Gold Antifade Mountant (Molecular Probes). For a list of antibodies used, refer to Supplementary Table S1 (Supplementary Data are available online at
For staining of whole-aggregate tissue, fixed aggregates were processed as described previously [3]. To prepare a custom imaging chamber, Sylgard 184 (1:10) prepared at ∼1-mm thickness was cut to fit glass slides, and a metal punch was used to create a well to hold ScaleA2 solution containing a stained aggregate. A coverslip was placed across the well, and the imaging chamber was sealed with clear nail polish.
For staining of isolated organoids, fixed aggregates were transferred to PBS for microdissection of the hair cell-containing regions. Minutien pins were used to stabilize aggregates against 35-mm dishes of Sylgard, while hair cell-containing cysts were dissected using iris scissors and fine forceps. The regions of protruding cysts distal to the aggregate bodies were cut open using scissors to expose the apical surfaces of hair cells, and the aggregate bodies were cut or teased away using scissors or forceps and discarded. The hair cell-containing epithelia were retained. Custom-made microwells were used to process single tissues in small solution volumes. Organoids were blocked and permeabilized in PBS with 5% NDS and 0.1% Triton X-100 for 15 min and incubated with primary antibodies in blocking/permeabilization solution overnight at 4°C. Alexa Fluor secondary antibodies (1:500) and phalloidin conjugates (1:100) were added in PBS for 1 h at room temperature, and Hoechst 33242 was applied for 5 min at 1:2,500 in PBS. Organoids were mounted using ProLong Gold.
For staining of mouse organs of Corti, 4-week-old WT and Pax2EGFP/+ mice were decapitated under anesthesia (80 mg/kg ketamine and 20 mg/kg xylazine). Each temporal bone was removed and inner ear placed in 4% PFA. The cochlear duct was then perfused with 4% PFA by perforating the cochlear windows and apex. Following dissection, surface preparations of the apical turn were blocked and permeabilized in 5% NDS and 0.1% Triton X-100 in PBS for 1 h at room temperature and incubated with primary antibody overnight at 4°C. After extensive washes, the preparations were stained with Alexa Fluor secondary antibodies (1:500) and Alexa Fluor 488 phalloidin (1:200) for 2 h and counterstained by a 5-min application of Hoechst 33242 (1:2,500). After a final series of washes in PBS, preparations were mounted using ProLong Gold.
For pluripotency staining, colonies grown on gelatin-coated coverslips or in 6-well culture plates were washed with PBS and fixed with 4% PFA for 15 min. They were then blocked and permeabilized in PBS with 5%–10% NDS and 0.1% Triton X-100 for 15 min. Primary antibodies were diluted in PBS or a 1:1 mixture of blocking/permeabilization solution for incubation at 4°C overnight. Alexa Fluor secondary antibodies (1:500) were diluted in PBS for incubation at room temperature for 1–2 h. Hoechst 33242 was then applied for 5 min at 1:2500 in PBS.
Brightfield and epifluorescence images were obtained using Leica DM IL and Olympus BX51WI microscopes. Confocal images were obtained using Olympus FluoView 1000, Leica TCS SP5, and Leica TCS SP8 confocal microscopes.
FM 4-64FX labeling
For live imaging of FM 4-64FX dye labeling, a day 20 organoid (with distal portion of cyst removed to expose hair cells) was dissected away from the aggregate body, affixed to a collagen droplet in a 35-mm dish, and maintained through day 33. After dissection, the culture was maintained in serum-free medium consisting of 1× basal medium Eagle (Sigma), 2.2 g/L sodium bicarbonate, 1× insulin–transferrin–selenium–ethanolamine (Gibco), 1% bovine serum albumin, 5 mg/mL glucose, and 8.8 U/mL penicillin G (Sigma). A stock solution of 2 mM FM 4-64FX in distilled water was prepared and stored at −20°C. The 5 μM working solution was prepared immediately before use by 1:400 dilution with low-calcium Hank's balanced salt solution (LC-HBSS), prepared by adding 0.1 mM CaCl2 to HBSS (Gibco) and filter sterilizing. The organoid tissue to be labeled was rinsed in LC-HBSS, incubated 10 s in FM 4-64FX working solution at room temperature, and washed 3 times with LC-HBSS for 1 min per wash. The time needed to add and remove the dye, taking care not to dislodge the tissue, and add the first wash totaled <30 s. Organoids were immediately imaged live in LC-HBSS.
Aminoglycoside treatment
Organoids were dissected and maintained on collagen droplets as described above. On day 33 of culture, gentamicin (Sigma) was applied to the media at 6 μM final concentration. After 72 h, organoids were fixed in 4% PFA for 30 min at room temperature and assayed for presence of stereocilia bundles via staining with rhodamine phalloidin (1:100; Thermo Fisher Scientific).
FGF assay
Solutions containing 1 μM LDN193189 and varying concentrations of FGF2 in ectodermal differentiation medium were prepared and added at day 4.5 of the differentiation protocol. After brief incubation at 37°C in a humidified culture incubator with 5% CO2, aggregates were collected and rinsed twice with prechilled PBS with 1× Halt phosphatase inhibitor cocktail (phosphatase inhibitor [PI]; Thermo Scientific). Aggregates were lysed in RIPA lysis and extraction buffer (Thermo Scientific) with 1× PI. For lysis, aggregates were incubated 15 min on ice and sonicated on ice in three 10-s pulses at 50% power. Lysates were centrifuged at 14,000 g for 15 min and supernatants retained. Total protein concentration was assayed using the BCA Protein Assay Kit (Thermo Scientific).
Western blotting
Lysates were diluted 1:1 with 2× Laemmli buffer from Bio-Rad (1610737) and denatured at 95°C for 5 min. Bio-Rad Mini Protean TGX gels (4%–15%) were loaded with equal amounts of total protein per well. Molecular weight standards from Bio-Rad (1610317) and Cell Signaling Technology (7720) were used. Proteins were transferred to nitrocellulose membranes, and membranes were blocked with 5% nonfat dry milk in TBS +0.1% Tween (TBST) for 1 h. Primary antibody incubations were performed overnight in TBST at 4°C. Horseradish peroxidase-conjugated secondary antibody incubations were performed for 1 h in TBST at room temperature. For a list of antibodies used, refer to Supplementary Table S1. Enhanced chemiluminescent substrates included SuperSignal West Pico (Thermo Scientific) and Amersham ECL Select (GE Healthcare). Images were obtained using a FluorChem SP system (Alpha Innotech). Densitometry was performed using Fiji software (version 2.0) [30].
Vesicle and organoid quantification
The percentage of aggregates with at least 1 organoid, indicated by EGFP signal at the base of a protruding cyst, was quantified at day 20. The percentage of aggregates with at least 1 visible EGFP+ otic vesicle-like structure was estimated at day 12 using epifluorescence. This analysis excluded vesicles not yet formed at day 12 or not visible due to orientation away from the camera. Therefore, since all organoids arose from vesicles, quantifying the percentage of aggregates with vesicles identified at day 12 or organoids at day 20 (referred to as “% vesicle- or organoid-positive”) more accurately reflected the total number of vesicle-positive aggregates. Note that this percentage does not double-count aggregates in which both structures were identified.
Aggregate size measurements
Average long-axis diameter of 4 aggregates per time point was measured from brightfield images in Fiji software (version 2.0) [30]. Data were averaged across multiple cultures and plotted to track changes in aggregate size over time.
Statistical analysis
Statistical comparisons between 2 groups were performed using unpaired t-tests in Microsoft Excel. Comparisons among more than 2 groups were performed by one-way analysis of variance with Tukey's post-hoc test using the XLSTAT plug-in.
Results
Pax2EGFP/+ mice develop normal inner ears
To develop a reporter system for monitoring otic induction during inner ear organoid formation, we derived mESCs from mice encoding EGFP between 5′ regulatory elements and the endogenous Pax2 transcription start site in 1 allele (Fig. 1A). Mice with a single copy of the allele (Pax2EGFP/+) develop normal kidneys and midbrain/hindbrain tissues [29]. Moreover, they appear to form normal otic vesicles, with EGFP in the ventromedial domain where Pax2 is expressed (Fig. 1B–D′) [22], as do the majority of mouse lines carrying Pax2 loss-of-function alleles [29,31 –33]. No apparent difference was seen in the gross morphology of cochlear and vestibular structures in Pax2EGFP/+ mice compared to WT animals (data not shown).
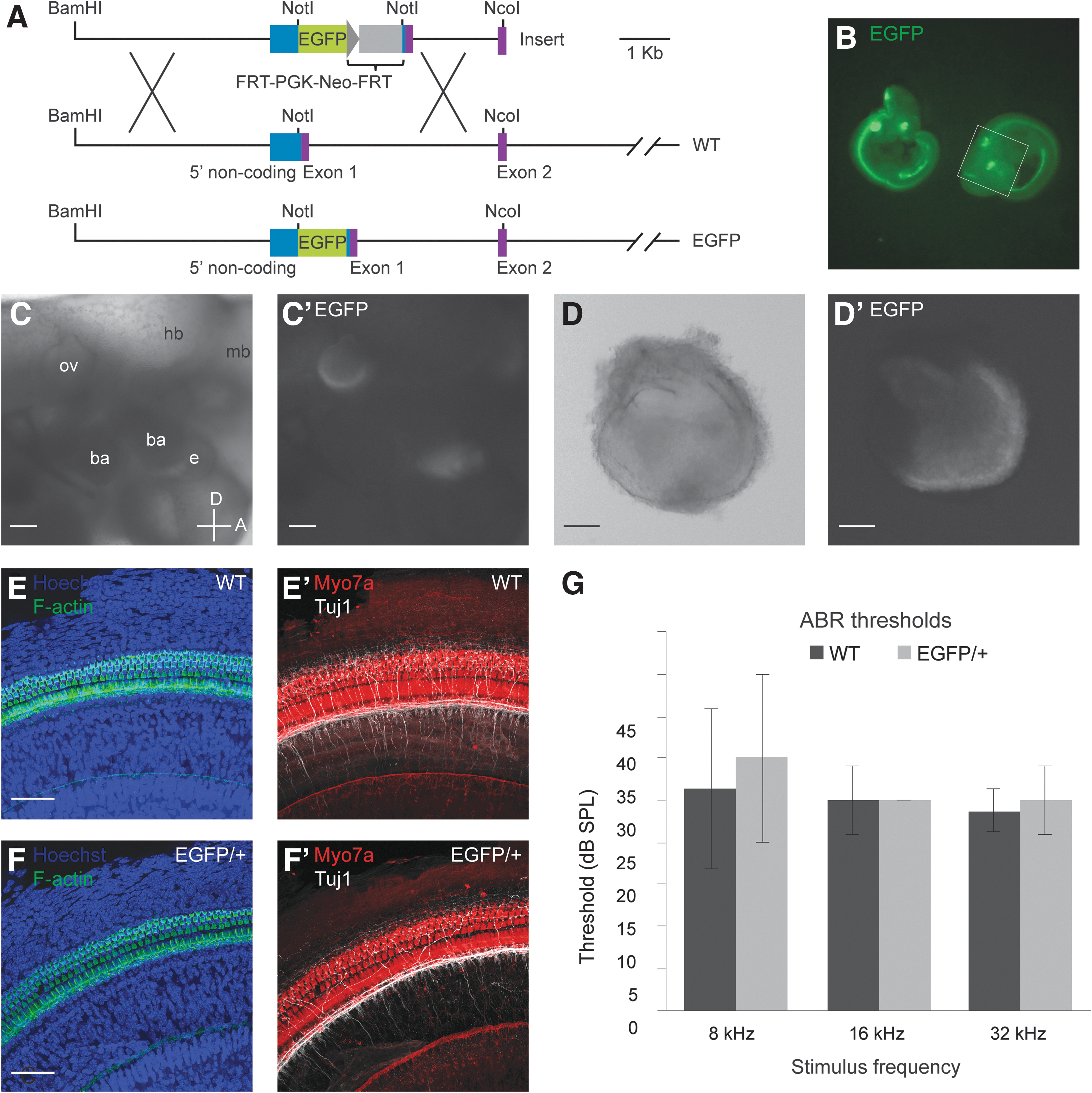
Features of Pax2EGFP reporter system and characterization of Pax2EGFP/+ mice and WT controls at 4 weeks postnatal.
To investigate more closely, we stained whole-mount preparations of the organ of Corti from adult Pax2EGFP/+ and WT littermates for markers of hair cells and neurons. Staining for myosin VIIa (Myo7a), an unconventional myosin characteristic of hair cells, and phalloidin labeling of F-actin-rich hair bundles revealed the characteristic pattern of 3 rows of outer hair cells and 1 row of inner hair cells in both WT and Pax2EGFP/+ mice (Fig. 1E–F′). Tuj1 staining revealed robust peripheral projections extending toward the hair cells, with some continuing past the inner hair cells and into the outer hair cell region. In 1 of 3 Pax2EGFP/+ embryos, we detected an aberrant pattern of innervation. Instead of extending radially toward the outer hair cells, multiple neurites traversed the tunnel of Corti at acute angles, occasionally parallel to the curvature of the organ of Corti.
Auditory brainstem response (ABR) testing was performed on postnatal mice at 4 weeks of age to measure thresholds for response to auditory stimuli. Three frequencies, processed by different regions of the mouse cochlea, were presented: 8, 16, and 32 kHz. Average thresholds from WT and Pax2EGFP/+ mice were not significantly different at any frequency (unpaired t-test, P > 0.5, n = 4 mice per genotype) (Fig. 1G).
Overall, our investigation of Pax2EGFP/+ mice revealed normal auditory and vestibular function based on ABR testing for auditory-evoked potentials and absence of behavioral signs of vestibular dysfunction (eg, circling, head bobbing, or torticollis). The Pax2EGFP allele, therefore, reports a normal pattern of Pax2 expression and permits development of the inner ear from the otic vesicle to mature auditory and vestibular organs in heterozygotes. This supports the use of Pax2EGFP/+ ESCs to produce inner ear organoids.
Pax2EGFP/+ mESCs form inner ear organoids
Clonal mESCs were obtained from Pax2EGFP/+ blastocysts (clone C5, Fig. 2A). We transitioned these cells from MEF dependence to feeder-free maintenance on gelatin. At passage 10, Pax2EGFP/+ cells were screened for the pluripotency marker Oct3/4, which was uniformly expressed throughout colonies (Fig. 2B–B′′′). To assay maintenance of pluripotency, cells were stained at passage 21 for a panel of pluripotency markers (Fig. 2C–E′′′). Oct3/4, Sox2, and Rex1 were uniformly expressed, but Klf4 and Nanog were variable with regard to staining intensity and the percentage of positive cells, suggesting that some cells in these populations may be switching between naive and epiblast-like states.
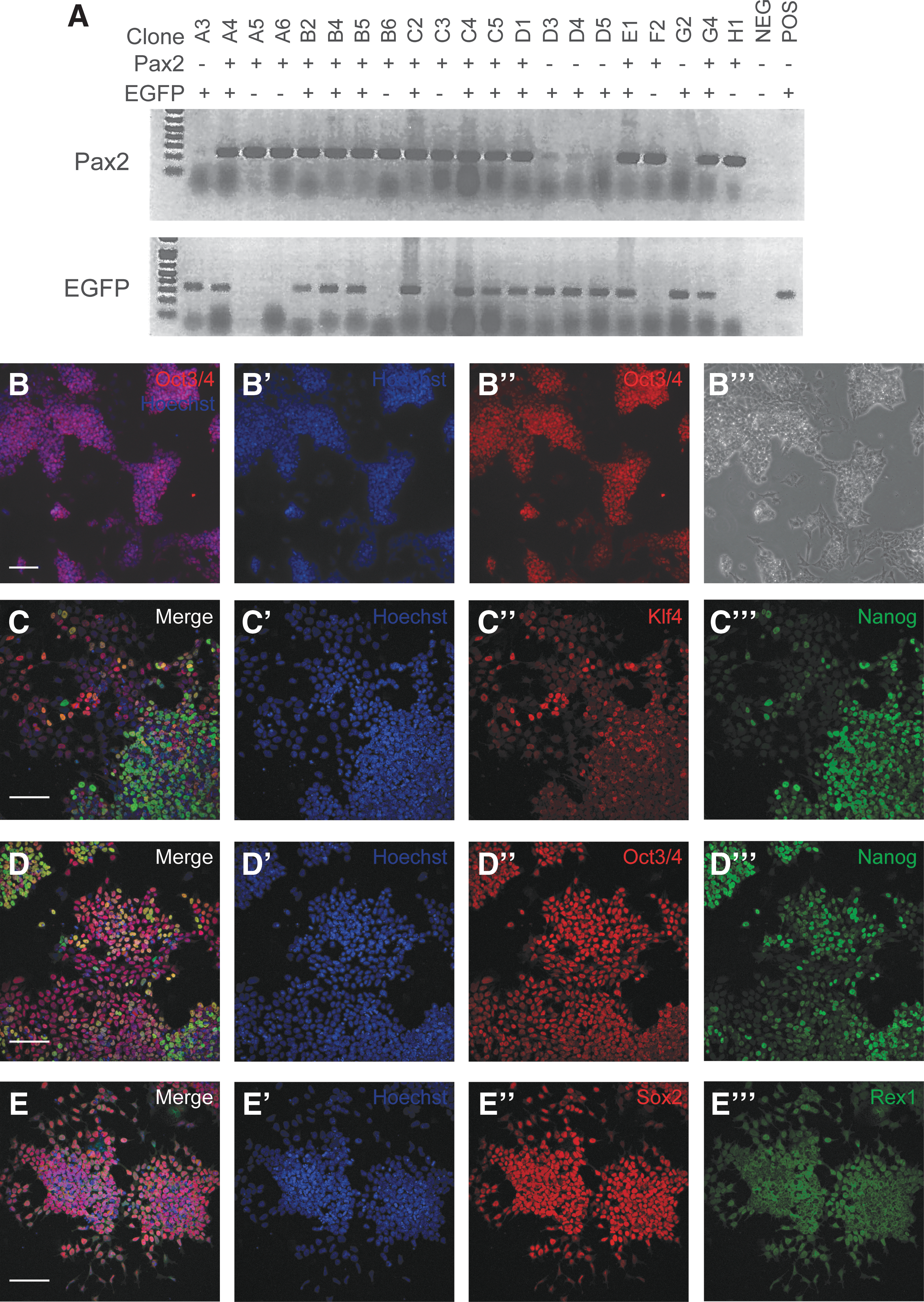
Establishment of Pax2EGFP/+ mESC line.
Pax2EGFP/+ cells at passages 13–17 were evaluated for capacity to generate inner ear organoids (Fig. 3A). At day 0, mESC aggregates were formed, and their size expansion was tracked over the course of the protocol (Fig. 3B and Supplementary Fig. S1). When 1.5% KSR was included in the ectodermal differentiation medium as per the original protocol, aggregates expanded at a slower rate than expected and never reached the 1.5–2 mm diameter by day 12 described by Koehler and Hashino [3]. When 10% KSR was used, aggregates began at an average diameter of 320.3 ± 21.6 μm (n = 21) at day 1 and expanded to 1054.3 ± 36.0 μm by day 8 (n = 11) after 100 ng/mL FGF2 was applied at day 4.5. By day 20, aggregates treated in this way reached a final diameter of 1449.9 ± 81.7 μm (n = 6). The final diameter of aggregates treated with 25 ng/mL FGF2 at day 4.5 (n = 5) was not significantly different (P = 0.201). In addition, the inclusion of the Wnt signaling agonist CHIR99021 during early maturation did not appear to affect aggregate size. For all experiments going forward, the ectodermal differentiation medium contained 10% KSR, a departure from the original inner ear organoid protocol, which may reflect a unique requirement of this cell line.
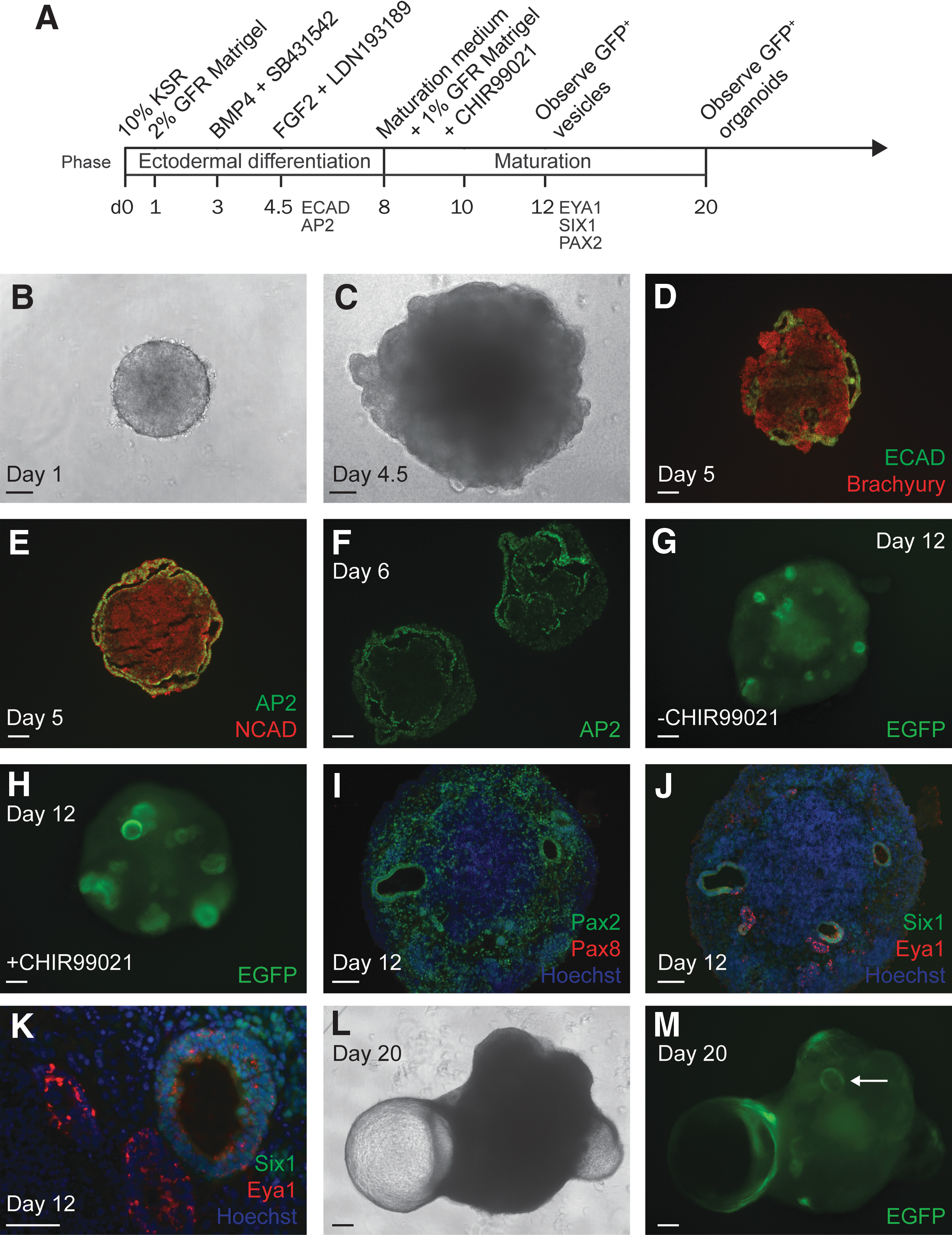
Process of forming Pax2EGFP/+ organoids.
Guided differentiation along the otic lineage was assessed via immunofluorescence staining of cryosections. Following treatment with BMP4 and an inhibitor of TGFβ signaling SB431542 (B/S) at day 3 to encourage differentiation of non-neural ectoderm and reduce mesoderm and endoderm, the aggregates formed a layer positive for E-cadherin (ECAD) and Tfap2a (AP2) (Fig. 3C–F). This layer, detected at day 5–6, excluded staining for brachyury and N-cadherin (NCAD), markers of mesendoderm and neural ectoderm, respectively.
The addition of FGF2 and an inhibitor of BMP signaling LDN193189 (F/L) at day 4.5 guided non-neural ectoderm toward preplacodal and, specifically, otic placodal fate. By day 12, otic vesicle-like structures were easily visualized due to the upregulation of EGFP driven by the Pax2 promoter (Fig. 3G, H). In trials of 1.5% KSR, we observed markedly weaker EGFP epifluorescence than in trials of 10% KSR (not shown). In 10% KSR trials, EGFP signal increased gradually throughout the aggregates beginning on day 9 but was brighter in the vesicles compared to surrounding tissue. Staining for Pax2 confirmed the uniform expression of Pax2 in vesicle epithelia as well as scattered expression throughout the aggregates, whereas Pax8 expression was absent at this same time point (Fig. 3I). Preplacodal markers Eya1 and Six1 were detected at the vesicle epithelia at day 12 (Fig. 3J, K). Inclusion of CHIR99021 early in the maturation phase (days 8–10) appeared to increase the size and number of vesicles at day 12 (Fig. 3H) [34]; however, this condition was not necessary for vesicle formation using Pax2EGFP/+ cells and was not included unless otherwise noted.
Aggregates were observed via epifluorescence microscopy until day 20, by which time the progressive expansion of vesicles into protruding cysts described by Koehler et al. was replicated in our cultures (Fig. 3L, M) [4]. The EGFP signal became concentrated at the base of each protrusion, bordering the aggregate body, marking the “organoid” region where hair cells were expected. Overall, the Pax2EGFP/+ mESC aggregates cultured with 10% KSR met major checkpoints for otic differentiation and reported on formation of Pax2+ vesicles and organoids.
Otic induction during organoid protocol supports FGF-ERK-Pax2 mechanism
FGF signaling is a necessary step in the adoption of inner ear fate. Therefore, we investigated whether a relationship existed between FGF2 dose and the efficiency of organoid production. Using a range of FGF2 doses (0, 5, 25, and 100 ng/mL) at day 4.5, we maintained cultures through day 20 and quantified the percentage of aggregates featuring at least 1 organoid as defined by an EGFP+ border region at the base of a protruding cyst. A clear pattern of dose dependency was observed: FGF2 was required for organoid formation, and increasing doses resulted in more aggregates forming organoids (Fig. 4A–A′′). The efficiency of vesicle production also corresponded with FGF2 dose, as expected, based on our finding that all organoids arose from EGFP+ vesicles (Fig. 4A′, B). Using 25 ng/mL FGF2 resulted in a similar percentage of organoid-positive aggregates (24.1% ± 10.0) compared to the 10%–20% reported previously [3]. The maximum dose tested, 100 ng/mL, resulted in an even higher percentage of organoid-positive aggregates (41.8% ± 19.8) (Fig. 4B).

Evidence of ERK mediating FGF-driven otic induction in mouse organoid model.
FGF receptors signal through multiple downstream pathways, including those mediated by ERK, AKT, and PLCγ. Recent reports have demonstrated the necessity of ERK as a mediator of otic induction in chickens and zebrafish [27,28]. Furthermore, the zebrafish study provided evidence that ERK is needed for hair cell development, whereas AKT is involved in otic neurogenesis. Given the correlation between vesicle formation and FGF2 dose, we decided to investigate whether ERK activation would demonstrate the same relationship. We hypothesized that the process of otic induction in organoids would replicate the in vivo developmental mechanism, supporting the use of inner ear organoids as a model to study signaling mechanisms involved in early mammalian otic development.
To examine the activation of ERK downstream of FGF2, aggregates were harvested at 1 h after application of LDN193189 and varying doses of FGF2 (0, 5, 25, and 100 ng/mL) at day 4.5. Lysates were prepared and screened for the activated, phosphorylated form of ERK (pERK). ERK phosphorylation increased in an FGF2 dose-dependent manner (Fig. 4C). Interestingly, the strongest ERK activation was induced by 100 ng/mL FGF2, a 4-fold higher dose than prescribed by the original organoid protocol [3,4]. These data reveal correlations between FGF2 and both organoid production and ERK phosphorylation, in agreement with developmental literature on otic induction [27,28].
Pax2EGFP/+ organoids model several features of developing ear
Organoids were screened for Myo7a+ cells with F-actin+ apical structures, indicative of hair cells with stereocilia bundles. Myo7a+/Pax2+ cells were found in the organoid regions where EGFP was observed (Fig. 5A). Cryosectioning revealed the presence of internal organoids containing Myo7a+ hair cells arrayed in an epithelial layer with F-actin+ stereocilia-like bundles oriented inward toward the central lumen of each cyst (Fig. 5B, B′). Protruding organoids were dissected and stained as isolated surface preparations. Imaging the apical face of the preparation revealed a variety of bundle morphologies, with some splayed and some tightly bundled stereocilia (Fig. 5C). A network of thick F-actin-rich belts similar to those formed by supporting cells in vivo invariably marked the organoid regions where hair cells were found; this distinctive feature served as a useful marker by which to screen tissues for hair cells efficiently.
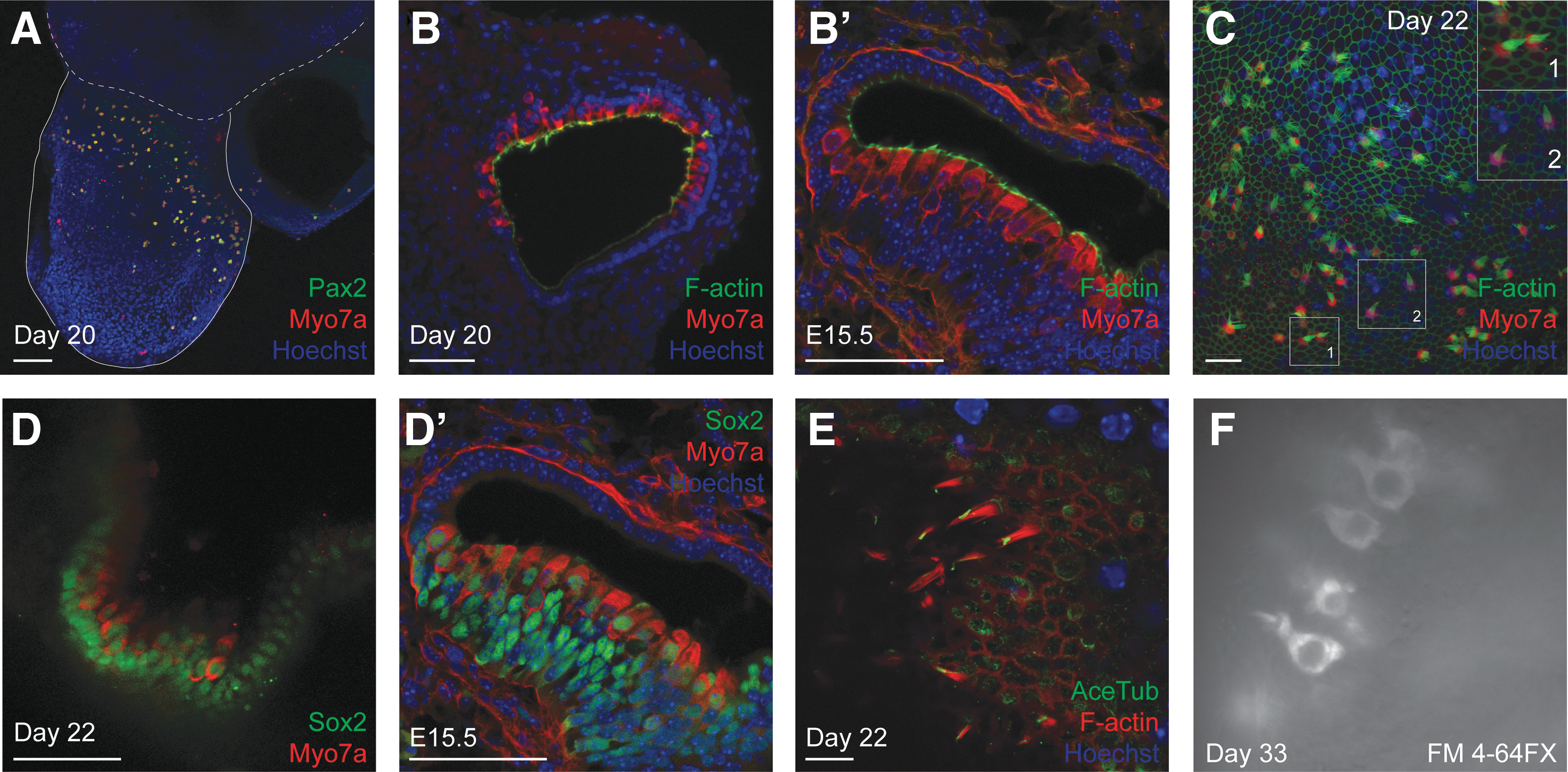
Characterization of Pax2+ inner ear organoids.
We screened for additional characteristics of auditory and vestibular organs to demonstrate the suitability of the Pax2EGFP/+ cell line for inner ear organoid formation. The organoid hair cells were lined basally by a layer of Sox2+ cells similar to supporting cells of the inner ear. The hair cells themselves were positive for Sox2 as well (Fig. 5D). This may be evidence of arrest at an immature stage as Sox2 is downregulated in auditory hair cells of neonatal mice [35,36]. Alternatively, it may point to a vestibular phenotype as particular vestibular hair cells express Sox2 into adulthood [37]. The presence of a single kinocilium, marked by expression of acetylated tubulin (AceTub), at each apical bundle supports characterization as either vestibular or immature auditory hair cells (Fig. 5E).
To test for functional mechanotransduction channels, we applied the styryl dye FM4-64FX to an organoid that had been dissected at day 20 and cultured until day 33 adhered to a collagen droplet. Following 10-s application, the dye was washed out, and the organoid was imaged live. Positive labeling indicative of hair cells was observed (Fig. 5F). Because the dye application was brief, entry presumably occurred through functional mechanotransduction channels rather than via endocytosis or uptake through other large pores such as those from P2X receptors [38].
To test for susceptibility to aminoglycoside-induced ototoxicity, additional organoids cultured on collagen to day 33 were treated with 6 μM gentamicin for 72 h. Rhodamine phalloidin was used to screen for stereocilia bundles. Bundles were present in 3 out of 4 organoids in both treated and untreated conditions, indicating that the hair cells were not severely affected by aminoglycoside treatment (not shown).
Vesicle-associated neurons model features of embryonic inner ear neurogenesis
The incidence of neurons in inner ear organoids has been reported [4]. This prompted us to further investigate the source and synaptogenic potential of these neurons. We first examined whether the neurons in our organoids arose from neuroblasts associated with vesicles at day 12. Staining revealed cells expressing islet-1/2 (Isl1/2) immediately adjacent to vesicles positive for Pax2 (Fig. 6A). This pattern closely resembled Isl1/2+/Sox2+ neuroblasts delaminating from otocysts in C57BL/6 mouse control tissue at E11.5 (Fig. 6A′, A′′) [39,40].
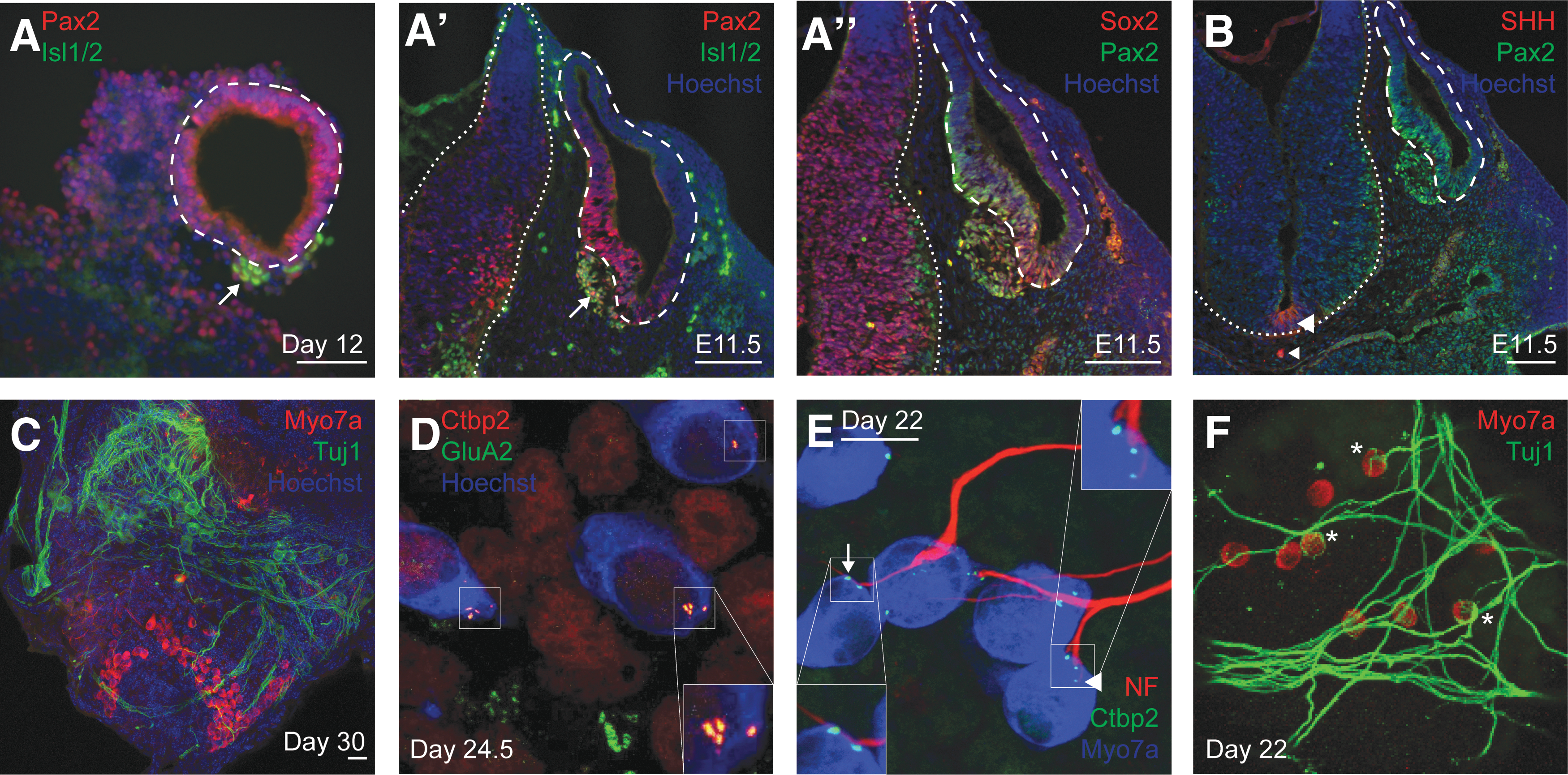
Comparison of immunofluorescence staining for Isl1 and SHH in day 12 aggregates and E11.5 embryos and evidence of organoid synapses.
In development, SHH signaling extrinsic to otic tissue impacts the specification of cochleovestibular ganglion (cvg) neuroblasts indirectly through neurogenin 1, and SHH signaling intrinsic to otic tissue is necessary for cvg neuroblast proliferation and survival [41]. Given these relationships between SHH and inner ear neurogenesis, we therefore examined SHH expression in our day 12 aggregates through immunofluorescence staining. SHH signal was faint and nonspecific (not shown). In contrast, SHH is readily detected at the ventral neural tube and notochord in E11.5 mice (Fig. 6B).
Whether or not organoid-associated neurons are specified by SHH, their ability to form synaptic contacts with hair cells is important to evaluate as this could be exploited in disease modeling or regeneration studies. Tuj1+ neurons were found in clusters near regions of Myo7a+ hair cells within the isolated day 30 organoids (Fig. 6C). These cells extended projections traversing the hair cell epithelia. Furthermore, these cells formed putative synaptic connections on hair cells as indicated by the colocalization of the presynaptic marker Ctbp2 and postsynaptic marker GluA2 in puncta at the basal aspect of hair cells (Fig. 6D). This direct apposition of presynaptic and postsynaptic markers mimics the one-to-one pairing of these markers in the mature inner ear [42,43].
To characterize the interaction between neurons and hair cells in our organoids, we stained for neurofilament and Tuj1 and found projections with terminal boutons and calyces as well as en passant synapses (Fig. 6E, F). Altogether, these results support the potential use of inner ear organoids as a model not only of hair cell development but also inner ear neurogenesis.
Discussion
The generation of inner ear organoids from mouse and human pluripotent stem cells could transform in vitro disease modeling, drug discovery, and regenerative studies in auditory and vestibular research [4,44]. Adoption of the methodology is, however, lacking; to the best of our knowledge, all prior publications report efforts of a single laboratory. For our study, we applied the method to mESCs derived from blastocysts of a heterozygous Pax2 reporter mouse line. The results reflect a dependence of otic vesicle induction on FGF concentration and support the vestibular-like characterization of the organoids. Variability in the efficiency of organoid production over time and passaging of cells suggests that further optimization is needed to make this protocol more robust and translatable to a broad range of pluripotent stem cell lines.
Here we demonstrated the utility of transcriptional reporter ESCs from an established mutant mouse line. Direct comparison between in vivo development and in vitro differentiation in the same genetic background is a prototype for organoid cultures as model systems. However, verifying that the reporter system recapitulates embryonic expression patterns and does not interfere with development is critical.
We used mESCs with an EGFP reporter driven by the endogenous Pax2 promoter sequence. In general, transcriptional regulatory elements are proximal to start sites, but some tissue-specific enhancer elements can be located far upstream. For example, enhancers within a 6.9-kb region upstream of the Pax2 transcription start site are sufficient to drive expression in mid-hindbrain domains, but a 30-kb 5′ region is required for expression in the ear, kidney, and spinal cord [19]. Therefore, confirming that transcriptional reporter lines reproduce in vivo expression patterns is a necessary consideration when modeling development of specific tissues in vitro. Our results showed otic expression of the EGFP reporter in vivo, normal phenotypic hearing thresholds of Pax2EGFP/+ mice compared with WT mice, and sustained expression of Pax2 from ESC-derived otic vesicles to sensory hair cell stages. Since Pax2 is a marker for otic vesicles and terminally differentiated sensory cells in mouse development [22,32,45], the Pax2EGFP allele is a potential asset for future studies of molecular events leading from progenitors to mature hair cells.
Our results were consistent with evidence that Pax2 heterozygosity on C57BL/6 background does not adversely affect formation of inner ear tissue [32]. Although the C57BL/6 background—previously untested with the inner ear organoid protocol—and Pax2 heterozygosity did not prevent organoid formation, modification of the original protocol was necessary. A higher concentration of KSR was required to induce aggregate growth to the expected size [3], raising the possibility that KSR influences proliferation rate. In 2 different mESC lines based on the 129 background strains, EB size did not change systematically with increasing KSR concentration [46]. Whether KSR affects proliferation during early stages of the inner ear organoid protocol and whether this is cell line-specific remain to be determined.
Alternatively, KSR may affect differentiation or competence to adopt otic lineage. We found that higher KSR concentration was associated with stronger EGFP—and therefore likely higher Pax2—expression within the vesicles and throughout the aggregates. From this we infer that some components of KSR influenced differentiation potential in addition to or independent of aggregate growth. Differential requirements of organoid cultures for KSR concentrations ranging from 1.5% to 20% have been described and an unidentified caudalizing factor implicated [36]. One candidate is insulin, a weakly caudalizing factor in SFEBq cultures [47]. The uncharacterized influence of each component of KSR is an important consideration in optimizing protocols for independent cell lines. Although a higher concentration of KSR was advantageous for producing inner ear organoids with the Pax2EGFP/+ cell line, its components could impact the efficiency of organoid formation in conflicting ways when added indiscriminately.
Inner ear organoid formation is largely dependent on self-organization with limited input of exogenous patterning molecules during the maturation phase. In development, secreted factors Wnt (from the dorsal neural tube) and SHH (from the ventral neural tube and notochord) influence dorsal–ventral patterning of otic vesicles; their opposing gradients establish vestibular and auditory/neurogenic domains [41,48 –51]. Day 12 aggregates lacked a particular source of SHH analogous to the ventral neural tube and notochord. In keeping with the absence of this ventralizing morphogen, the organoids expressed various features of vestibular-like end organs. For instance, the presence of calyceal terminals was indicative of type I vestibular hair cells [45]. Given the vestibular hair cell characteristics and the need for auditory hair cells in clinical applications, future inner ear organoid studies should evaluate directed patterning of vesicles through exogenous SHH.
In addition to its role in ventralization and cochleogenesis, SHH signaling is required for maturation of the cvg, which becomes a source of SHH later in development [52 –56]. In day 12 aggregates, Isl1/2+ neurons did not themselves express SHH. Future studies should establish definitively whether Isl1/2+ cells originate from vesicles and give rise to the mature neurons found within the organoids. Resolving this issue is important if inner ear organoids are to be used as a model system for disease and developmental studies. Moreover, if these neurons are indeed otic in nature, then the protocol may ultimately provide sensory, nonsensory, and neural elements for inner ear repair and regeneration.
Additional differences between mESC-derived and embryonic inner ear tissues represent opportunities for optimization. For instance, our gentamicin treatment paradigm resulted in no obvious effect on derived hair cells. In contrast, treatment of neonatal mouse cochlea explants with 3.5 μM gentamicin for 72 h results in a gradient of outer hair cell death, with nearly complete loss at the base of the cochlea [57]. We used 6 μM gentamicin based on experiments demonstrating severe loss of hair cells throughout cochlear explants (data not shown). Therefore, while the inner ear organoid protocol resulted in a close approximation of native hair cells with regard to molecular, structural, and functional characteristics, refinements are still needed to fully replicate hair cell physiology.
Another notable difference from embryonic tissue was the absence of Pax8 staining in the newly formed Pax2+ otic vesicles at day 12 of the protocol. Pax2 and Pax8 show overlapping expression in the ventral domains of otic vesicles of E9.5–10.5 mice [58], and Pax8 expression has been demonstrated previously using the organoid culture protocol [4]. The lack of Pax8 may indicate a unique feature of the cell line and may prompt experiments on the role of Pax genes in organoid formation. Pax2, Pax5, and Pax8 can compensate for one another in development, but Pax5 is not expressed in the embryonic inner ear and was absent from our aggregates [31,59]. Therefore, the development of otic tissue in vitro relied solely on heterozygous Pax2 expression.
Tracking protocol efficiency is necessary for optimization
As mentioned, the adaptability of the inner ear organoid protocol to several distinct cell lines has been demonstrated. DeJonge et al. optimized FGF2/LDN193189 treatment timing for each cell line within a window of just 6 h [34]. This showed that careful consideration of the relative efficiencies of different cell lines is crucial. Our experience suggested that cultures vary in efficiency not only with parameters defined by the protocol (eg, timing or dose of drug treatments) but also with additional parameters yet to be elucidated. We found that mESC passage number, reagent lots, and seasonal environmental fluctuations may influence outcomes. In addition, the reliance on Matrigel, an animal-derived product that varies unpredictably in composition from lot to lot, is not ideal, although no reliable alternative has been established.
The data presented in this study were produced during a single period of cultures meeting criteria for “success” in generating otic cultures. At least 10% of aggregates were vesicle positive, with some expanding into large, protruding cysts. During that time, nonotic cultures (lacking one or both criteria for success) corresponded to higher mESC passage numbers. It is important to note that subsequently, without changing materials or methods, our success rate dropped suddenly and without relationship to passage number for a prolonged interval. In contrast, hair cells were reliably identified in the organoid region at the base of each protruding cyst in our previous and subsequent cultures.
We present a summary of our culture attempts categorized by major variations to the original protocol tested (Supplementary Fig. S2) as well as a strategy we recommend for adopting the inner ear organoid protocol (Table 1). The summary corresponds to our trials with Pax2EGFP/+ cells only, although we tested other cell lines (R1, R1/E, and E14Tg2a). As noted, our Pax2EGFP/+ cell line required 10% KSR in the ectodermal differentiation medium. During the period when our cultures were consistently producing organoids, we performed drug treatments within ±3 h from the timing prescribed by the original protocol with no adverse effects. The combinations of additional variables predicted to influence the success of inner ear organoid cultures are myriad; this presents a challenge even when using cell lines known to be amenable to the protocol.
KSR, KnockOut serum replacement.
The collective understanding of stem cell differentiation and pluripotency is still evolving, despite the fact that mESCs were first derived several decades ago [60,61]. Naive and primed states of pluripotency have recently been distinguished, and pharmacological means of reverting primed cells to a naive state are being discovered [62 –64]. Although our outcomes did not correlate with passage number, maintenance of naive pluripotency is a key consideration for stem cell cultures. Interestingly, human ESCs are more comparable to mouse epiblast stem cells (primed) than to mESCs (naive), perhaps underlying the modifications that have been necessary to adapt mouse organoid protocols for human cells [44,65,66]. As our study demonstrated, the influence of cells surrounding target tissue in three-dimensional stem cell culture paradigms—in terms of both secreted factors and physical cues—is relatively uncharacterized. In addition, the possibility that selecting for reporter cells may bias outcomes toward higher efficiency has been suggested in retinal organoid literature [67]. Until we gain control over pluripotency states, environmental cues, and distinctions between cell lines, these remain necessary caveats for organoid researchers.
Efforts to understand, refine, and apply inner ear organoid technology are still in early stages. However, the potential value of reporter lines able to recapitulate endogenous expression patterns and restore function in vivo cannot be overstated. The many parallels between the Pax2EGFP/+ organoids and true organs of hearing and balance are a strong impetus for continued investment in what may be a highly lucrative area of research.
Footnotes
Acknowledgments
We are grateful to Liqian Liu for technical assistance with cell cultures and to Lisa L. Kabara and Mark A. Crumling for assistance with auditory brainstem response recordings. We acknowledge Elizabeth Hughes, Galina Gavrilina, and Thom Saunders for the preparation of transgenic mice and ESCs and the Transgenic Animal Model Core of the University of Michigan's Biomedical Research Core Facilities. This work was supported by the Department of Defense (W81XWH-12-1-0492 to R.K.D.), a NIH-NIDCD core grant (P30-DC005188), and NIH institutional training grants (T32-DC00011 and T32-NS07640 to S.A.S.).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.