
Abstract
Cardiac stem cell (CSC) therapy is a promising approach to treat ischemic heart disease. However, the poor survival of transplanted stem cells in the ischemic myocardium has been a major impediment in achieving an effective cell-based therapy against myocardial infarction. Inhibiting mitochondrial fission has been shown to promote survival of several cell types. However, the role of mitochondrial morphology in survival of human CSC remains unknown. In this study, we investigated whether mitochondrial division inhibitor-1 (Mdivi-1), an inhibitor of mitochondrial fission protein dynamin-related protein-1 (Drp1), can improve survival of a novel population of human W8B2+ CSCs in hydrogen peroxide (H2O2)-induced oxidative stress and simulated ischemia-reperfusion injury models. Mdivi-1 significantly reduced H2O2-induced cell death in a dose-dependent manner. This cytoprotective effect was accompanied by an increased proportion of cells with tubular mitochondria, but independent of mitochondrial membrane potential recovery and reduction of mitochondrial superoxide production. In simulated ischemia-reperfusion injury model, Mdivi-1 given as a pretreatment or throughout ischemia-reperfusion injury significantly reduced cell death. However, the cytoprotective effect of Mdivi-1 was not observed when given at reperfusion. Moreover, the cytoprotective effect of Mdivi-1 in the simulated ischemia-reperfusion injury model was not accompanied by changes in mitochondrial morphology, mitochondrial membrane potential, or mitochondrial reactive oxygen species production. Mdivi-1 also did not affect mitochondrial bioenergetics of intact W8B2+ CSCs. Taken together, these experiments demonstrated that Mdivi-1 treatment of human W8B2+ CSCs enhances their survival and can be employed to improve therapeutic efficacy of CSCs for ischemic heart disease.
Introduction
I
Mitochondria play a central role in cell death and have been the main organelles being targeted for various cytoprotective strategies [8]. Mitochondria are morphologically dynamic organelles that are constantly undergoing fusion and fission, processes essential to maintain mitochondrial fidelity and cell survival. These two opposing processes are regulated by a group of evolutionarily conserved mitochondrial fusion proteins (mitofusin-1, mitofusin-2, and optic atrophy-1) as well as mitochondrial fission proteins [dynamin-related protein-1 (Drp1), mitochondrial fission 1 protein, mitochondrial fission factor, mitochondrial dynamics proteins of 49 and 51 kDa]. Current literature supports the close link between mitochondrial morphology and the bioenergetic health of mitochondria [9]. Studies have shown that mitochondrial fission occurs during apoptosis and necrosis, and shifting the balance of mitochondrial morphology toward fission increases susceptibility to death of various cell types [10,11]. In contrast, elongated mitochondria are energetically active and can better withstand oxidative stress [12,13]. Inhibition of Drp1 by expression of a Drp1-dominant negative mutant, by siRNA-mediated Drp1 gene silencing, or with mitochondrial division inhibitor-1 (Mdivi-1) have been shown to prevent mitochondrial fission and exert cytoprotective effects [10,14]. Mdivi-1 is a cell-permeable small molecule that inhibits GTPase activity and self-assembly of pro-fission protein Drp1 [15]. Although previous studies have demonstrated that Mdivi-1 can promote survival of different types of mammalian cells [10], the cytoprotective effect of Mdivi-1 has not been examined in human adult stem cells. In this study, we investigated whether inhibiting mitochondrial fission with Mdivi-1 promotes survival of human W8B2+ CSCs using two different in vitro injury models, hydrogen peroxide (H2O2)-induced oxidative stress, and simulated ischemia-reperfusion injury.
Materials and Methods
W8B2+ CSCs
W8B2+ CSCs were isolated from human atrial appendages from consented adult patients as previously described [4]. Human tissue sample collection was approved by the Human Research Ethics Committee of St Vincent's Hospital (Melbourne, Australia) (HREC-A 07/08). Briefly, atrial appendages were minced into small fragments and subjected to brief collagenase digestion. Tissue fragments were plated onto fibronectin-coated dishes and cultured in an explant medium containing IMDM (Thermo Fisher Scientific, MA) supplemented with 20% fetal bovine serum (FBS; Sigma-Aldrich, MO), 0.1 mM mercaptoethanol (Thermo Fisher Scientific), 100 U/mL penicillin (Lonza, MD), 100 μg/mL streptomycin (Lonza), and 0.25 μg/mL amphotericin B (Lonza). The resulting monolayer of outgrowth cells was collected and expanded in a growth medium containing an endothelial growth medium (EGM-2) and M199 medium (Lonza), in 1:3 ratio, supplemented with 10% FBS, 0.1 mM nonessential amino acids (Thermo Fisher Scientific), 100 U/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin B. Once confluent, cells were enriched for W8B2+ cells by staining with the W8B2 PE-conjugated antibody (mouse anti-human IgG1; Biolegends, CA), sorted using a Flow cytometer and sorter (FACSAria; BD Biosciences), and expanded in growth medium.
H2O2-induced oxidative stress
Cells were plated at 5 × 103 cells/well in a 96-well plate precoated with 0.1% gelatin (Sigma-Aldrich) and cultured in the growth medium. After overnight incubation, cells were randomized to one of the following treatment groups: (i) 0.1% dimethylsulfoxide (DMSO; Sigma-Aldrich), (ii) Mdivi-1 (1–50 μM; Enzo Life Sciences), (iii) H2O2+0.1% DMSO, and (iv) H2O2+Mdivi-1 (1–100 μM). After 3 h of incubation at 37°C in a humidified 5% CO2 incubator, cells were stained with 3 μg/mL of Hoechst 33258 (Sigma-Aldrich) and 5 μg/mL of propidium iodide (PI; Thermo Fisher Scientific). Images were captured at 200× magnification with an inverted microscope (Olympus IX71, Tokyo, Japan). The number of dead cells (PI+) was counted and expressed as a percentage over total number of cells (Hoechst 33258 positive). At least 100 cells were counted per group for each independent experiment.
Simulated ischemia-reperfusion injury
Cells were plated at 4 × 104 cells/well in a 24-well plate precoated with 0.1% gelatin and cultured in the growth medium. To simulate ischemia, cells were washed twice with phosphate-buffered saline (PBS) and cultured in glucose- and serum-free DMEM medium (Thermo Fisher Scientific). Hypoxia (<0.1% O2) was induced in an airtight GENbox anaerobic jar containing a Genbox Anaer generator sachet (BioMérieux, Marcy-l'Étoile, France) at 37°C. After 18 h of simulated ischemia, reoxygenation was performed in the growth medium for 1 h at 37°C in a humidified 5% CO2 incubator to simulate reperfusion. Cells were treated with 0.1% DMSO or Mdivi-1 (1–50 μM) in three treatment protocols: (i) 2-h pretreatment before simulated ischemia, (ii) throughout simulated ischemia and reperfusion, and (iii) during simulated reperfusion. In normoxic cohorts, cells were cultured in the growth medium at 37°C in a humidified 5% CO2 incubator. At the end of experiment, cells were stained with Hoechst 33258 and PI. The number of dead cells (PI+) was counted and expressed as a percentage over total number of cells (Hoechst 33258 positive). At least 100 cells were counted per group for each independent experiment.
Apoptosis analysis by flow cytometry
Cells were trypsinized, centrifuged, and resuspended in an annexin-binding buffer (10 mM HEPES, 140 mM NaCl, and 2.5 mM CaCl2, pH 7.4) containing Alexa Fluor-488-conjugated Annexin V (Thermo Fisher Scientific) and PI. After 15 min of incubation at room temperature, the percentage of apoptotic cells were analyzed using LSRFortessa Flow cytometer (BD Bioscience) and 10,000 cells were analyzed.
Mitochondrial morphology
The cell-permeant mitochondrial-selective fluorescent dye MitoTracker Red (Thermo Fisher Scientific) was used to stain mitochondria in live cells. In H2O2-induced oxidative stress model, cells were stained with 100 nM of MitoTracker Red for 30 min at 37°C in a humidified 5% CO2 incubator, washed once with PBS, and subjected to oxidative stress. In the simulated ischemia-reperfusion injury model, MitoTracker staining was performed during simulated reperfusion. At the end of treatment period, cells were fixed with 4% paraformaldehyde for 15 min followed by ice-cold acetone for 1–5 min. Images were captured at 600× magnification with a fluorescence microscope (Olympus BX60) and analyzed with ImageJ software. Cells were categorized into either having tubular or fragmented mitochondria. At least 100 cells were counted per group for each independent experiment.
Mitochondrial membrane potential
The cationic fluorescent dye 5,5′,6,6′-tetrachloro-1,1′3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1; Thermo Fisher Scientific) was used for mitochondrial membrane potential measurement. Low concentration of JC-1 exists as monomers inside the cytoplasm and yield green fluorescence. The highly negative mitochondrial membrane potential attracts large uptake of the dye into the mitochondria, enabling formation of J-aggregates, which yield orange to red fluorescence. These characteristics enable assessment of mitochondrial membrane potential in single cells by quantifying the ratio of red and green fluorescence intensity. Cells were stained with 1 μM of JC-1 dye for 10 min (in H2O2-induced oxidative stress model) or 30 min at reperfusion (in simulated ischemia-reperfusion injury model) at 37°C in a humidified 5% CO2 incubator. Cells treated with 50 μM of carbonyl cyanide m-chlorophenyl hydrazine (CCCP; Sigma-Aldrich), a mitochondrial respiratory uncoupler, were served as a positive control for the assay. Images were captured at 200× magnification with a fluorescence microscope (Olympus IX71). Fluorescence intensity was quantified using ImageJ software and a background-subtraction method [16]. Briefly, the color images were split into their respective color channel images in grayscale. The corresponding images of the same field (ie, green channel of JC-1 monomers and red channel of JC-1 aggregates) were stacked into an image frame to ensure selection of the same cells. An outline was drawn around each cell, and area and integrated density were measured. For background intensity, five random areas remote from the cells were selected and measured for their average mean gray value. The total corrected cellular fluorescence was calculated as “integrated density”—(“area” × “background intensity”). At least 100 cells were randomly selected and analyzed for individual red to green fluorescence ratio.
Mitochondrial superoxide production
The fluorescent dye MitoSOX Red (Thermo Fisher Scientific) was used to assess mitochondrial superoxide production. Cells were stained with 1 μM of MitoSOX Red for 10 min (in H2O2-induced oxidative stress model) or for 15 min at reperfusion (in simulated ischemia-reperfusion injury model) at 37°C in a humidified 5% CO2 incubator. Cells treated with antioxidant were served as a positive control. In H2O2-induced oxidative stress model, cells were treated with 100 μM of EUK-134 (Cayman Chemical). In simulated ischemia-reperfusion injury model, cells were treated with 5 mM of N-acetyl-
Intracellular total reactive oxygen species production
The cell-permeant fluorescein 2′,7′-dichlorodihydrofluorescein diacetate (DCFH2-DA; Thermo Fisher Scientific) was used to assess intracellular total reactive oxygen species (ROS) production. Cells were prestained with 3 μM of DCFH2-DA in PBS for 60 min at 37°C in a humidified 5% CO2 incubator before being subjected to H2O2-induced oxidative stress. Fluorescence was then measured with excitation and emission at 490 and 530 nm, respectively, with a POLARstar OPTIMA microplate reader (BMG Labtech, Ortenberg, Germany) at 37°C.
Recombinant his6-Drp1 production and purification
For bacterial expression of recombinant human Drp1 (N-term his6) protein, cDNA for human Drp1 (Isoform2, Uniprot ID: O00429-3, kindly provided by Prof Michael Ryan) was cloned into pQE-30 vector and transformed in Rosetta (DE3) competent cells (Novagen, Merck Millipore). Transformed cells were propagated in 2 L Luria-Bertani broth, in the presence of 100 μg/mL ampicillin at 37°C, 120 rpm to A600 = 0.6. Protein expression was induced by the addition of 0.5 mM IPTG, after which cultures were incubated at 16°C for 18 h. Cells were harvested by centrifugation at 3,500 rpm, 20 min, and the pellet resuspended in lysis buffer containing 50 mM Tris.HCl (pH 7.3), 0.5 M NaCl, 50 mM imidazole, 5% glycerol, 2 mM β-mercaptoethanol, 0.1 mM LEUPEP, 0.1 mM AEBSF, and 1 mM benzaminidium chloride. Cell lysates were prepared by sonication on ice and clarified by centrifugation at 20,000 rpm for 30 min. Clarified cell lysate was loaded onto a 5 mL Nickel Chelating Sepharose Fast Flow column (GE healthcare, Buckinghamshire, United Kingdom). The column was washed with 20 column volumes of a wash buffer (50 mM Tris.HCl (pH 7.6), 150 mM NaCl, 10% glycerol, and 2 mM β-mercaptoethanol), and Drp1 protein was eluted with the wash buffer supplemented with 400 mM imidazole. Eluted proteins were equilibrated with 50 mM Tris.HCl (pH 7.6), 150 mM NaCl, 10% glycerol, and 2 mM TCEP using a PD-10 desalting column (GE Healthcare) and aliquots stored at −80°C.
Western blot analysis of Drp1
Cells were lysed using RIPA lysis buffer (Sigma-Aldrich) supplemented with protease inhibitor cocktail (Sigma-Aldrich), and extracted proteins were concentrated using Amicon Ultra centrifugal filters (30K MWCO; Millipore, County Cork, Ireland). Protein samples were electrophoresed on 12% sodium dodecyl sulfate–polyacrylamide gels and transferred to Immobilon FL polyvinylidene-fluoride membranes (Millipore). The membranes were blocked with 2.5% nonfat dry milk in PBS containing 0.1% Tween-20 (PBS-T) for 1 h at room temperature. Membranes were incubated with mouse monoclonal Drp1 (1 μg/mL, 4°C, overnight; Abcam) and α-Tubulin (1:2,000, room temperature, 2 h; Cell Signaling) primary antibodies, followed by incubation with anti-mouse IgG secondary antibody (1:10,000) fluorescently labeled with IR680 for 1 h at room temperature. Immunoreactive bands were visualized on an Odyssey membrane imaging system (LI-COR Biosciences). A 75 ng of recombinant his6-Drp1 was included as a positive control.
Analysis of oxidative phosphorylation and glycolysis
All extracellular flux analyses were performed using the Seahorse XFe96 Extracellular Flux Analyzer (Agilent, Santa Clara, CA). Cells were plated at 5 × 104 cells/well in a 96-well Seahorse V3-PS plate precoated with 0.1% gelatin. After overnight incubation and before assay, cells were washed with assay media [Seahorse XF base medium supplemented with 25 mM glucose, 1 mM glutamine, and 1 mM sodium pyruvate for oxygen consumption rate (OCR) assay, or Seahorse XF base medium supplemented with 1 mM glutamine for extracellular acidification rate (ECAR) assay] and equilibrated in 175 μL of respective assay media per well at 37°C with no CO2 for 30 min. The assay protocol consisted of repeated cycles of 3-min mix and 3-min measurement periods, with OCR and ECAR measured simultaneously. Basal energetics was established after four of these initial cycles, followed by injection of either DMSO or Mdivi-1 (5, 10, or 50 μM) for three cycles. This was followed by performing a mitochondrial stress test that includes sequential injection of the following compounds and subsequent measurement of OCR after each: the proton ionophore carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP, 125 nM) together with sodium pyruvate (1 mM), and the mitochondrial complex III and complex I inhibitors antimycin A/rotenone (1 μM). The glycolysis stress test includes sequential injection of the following compounds and subsequent measurement of ECAR after each: glucose (10 mM), oligomycin (1 μM), and 2-deoxy-glucose (50 mM). All treatment conditions were analyzed as three to four replicates for each independent experiment. At the completion of each assay, cells were lysed and protein concentration was determined using the Bradford dye-binding method (Bio-Rad, Sydney, Australia) according to manufacturer's instructions.
Statistical analysis
Data sets were expressed as mean ± standard error of the mean. Statistical analysis was performed using GraphPad Prism software (version 6). The significance of the differences was evaluated using one-way analysis of variance followed by Bonferroni post hoc analysis. Values of P < 0.05 were considered statistically significant.
Results
Mdivi-1 protects W8B2+ CSCs against H2O2-induced cell death
Treatment with 3 mM of H2O2 for 3 h induced significant cell death in W8B2+ CSCs (62.0% ± 8.2% in DMSO + H2O2 vs. 0.8% ± 0.4% in DMSO, P < 0.01) and the presence of Mdivi-1 at 50 μM significantly reduced the percentage of cell death (Fig. 1A). To assess the antiapoptotic effect of Mdivi-1, cells were stained with Annexin V and PI. Treatment of Mdivi-1 at 50 μM significantly reduced the percentage of early (Annexin V+/PI−) and late (Annexin V+/PI+) apoptotic cells (Supplementary Fig. S1; Supplementary Data are available online at
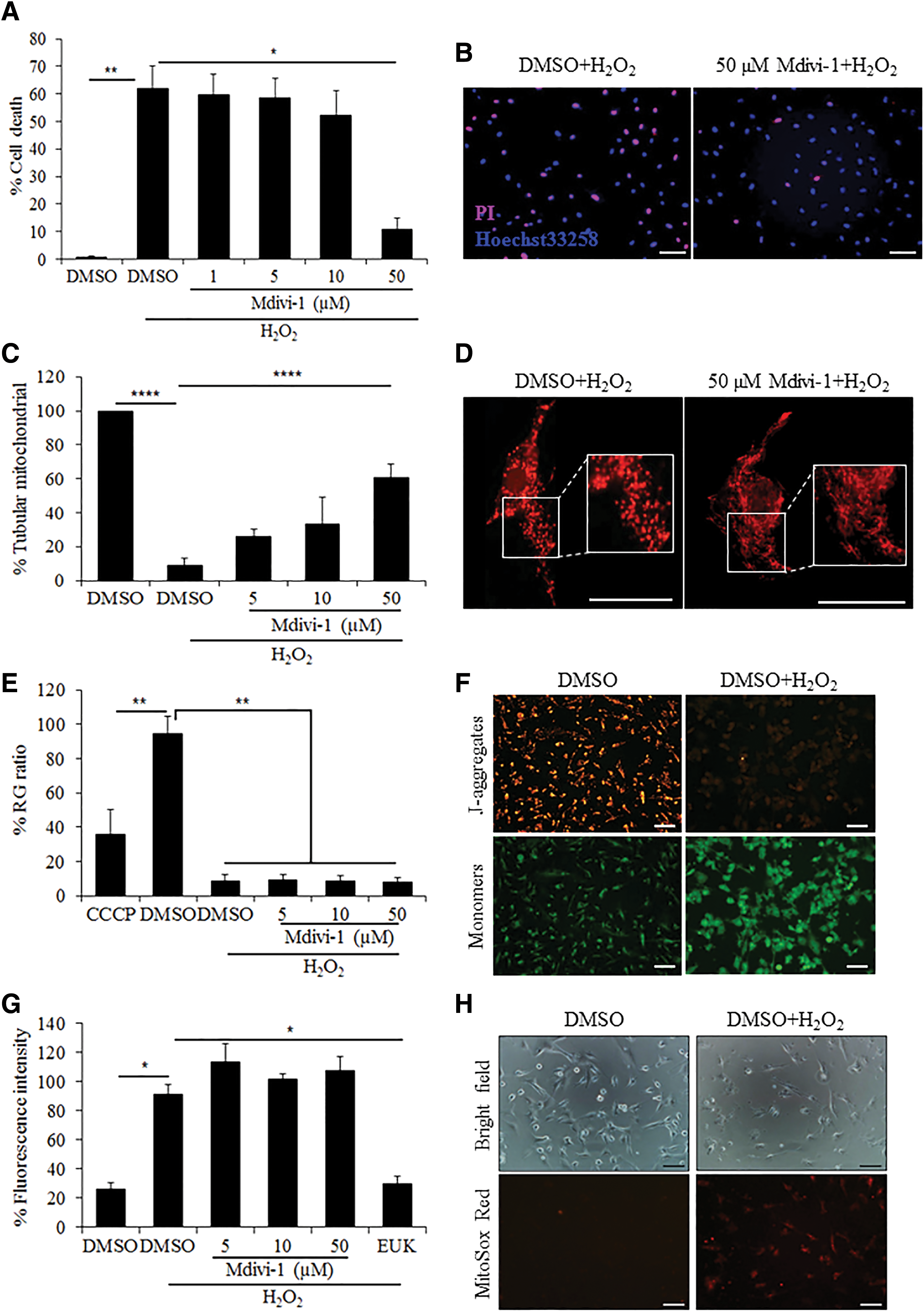
Mdivi-1 protects W8B2+ CSCs against H2O2-induced oxidative stress. The effect of Mdivi-1 on survival (n = 6)
To assess mitochondrial morphology, cells were stained with MitoTracker Red. Under basal conditions (ie, no H2O2 treatment), the majority of W8B2+ CSCs displayed predominantly tubular mitochondria. Exposure to 3 mM of H2O2 markedly reduced the percentage of cells with tubular mitochondria (9.1% ± 4.2% in DMSO + H2O2 vs. 99.8% ± 0.1% in DMSO, P < 0.0001). The presence of Mdivi-1 at 50 μM significantly increased the percentage of cells with tubular mitochondria (60.9% ± 7.5% in Mdivi-1+H2O2 vs. 9.1% ± 4.2% in DMSO + H2O2, P < 0.0001) (Fig. 1C).
Mitochondrial membrane potential was assessed with JC-1 staining. Under basal condition, CCCP, but not Mdivi-1, induced a significant reduction in mitochondrial membrane potential in W8B2+ CSCs. Treatment with H2O2 markedly reduced mitochondrial membrane potential compared to vehicle control (8.9% ± 3.5% in DMSO + H2O2 vs. 94.7% ± 10.0% in DMSO, P < 0.01). However, Mdivi-1 did not rescue H2O2-induced mitochondrial membrane potential dissipation (Fig. 1E).
Treatment with H2O2 significantly increased the production of mitochondrial superoxide in W8B2+ CSCs (90.9% ± 6.8% in DMSO + H2O2 vs. 25.8% ± 4.4% in DMSO, P < 0.05). While the production of mitochondrial superoxide can be effectively reduced by antioxidant EUK-134, treatment with Mdivi-1 did not show a significant reduction of mitochondrial superoxide compared to vehicle control (Fig. 1G). Similarly, treatment with H2O2 significantly increased the intracellular total ROS in W8B2+ CSCs and Mdivi-1 did not influence H2O2-induced ROS production (Supplementary Fig. S2).
Mdivi-1 protects W8B2+ CSCs against simulated ischemia-reperfusion injury-induced cell death when given as a preconditioning agent or throughout ischemia-reperfusion injury
W8B2+ CSCs pretreated with 5 μM of Mdivi-1 modestly, but significantly, reduced the percentage of cell death after simulated ischemia-reperfusion injury (46.6% ± 5.1% in 5 μM of Mdivi-1 vs. 55.4% ± 3.3% in DMSO, P < 0.05). However, pretreatment with higher doses of Mdivi-1 at 10 and 50 μM did not significantly reduce the percentage of cell death after simulated ischemia-reperfusion injury (Fig. 2A). When given throughout ischemia and reperfusion, Mdivi-1 significantly reduced the percentage of cell death (42.8% ± 7.3% in 5 μM of Mdivi-1 and 41.3% ± 5.5% in 10 μM of Mdivi-1 vs. 61.3% ± 5.9% in DMSO, P < 0.05) (Fig. 2B), as well as the percentage of early (Annexin V+/PI−) and late (Annexin V+/PI+) apoptotic cells (Supplementary Fig. S3) after simulated ischemia-reperfusion injury. However, treatment with 1–50 μM of Mdivi-1 during simulated reperfusion did not protect W8B2+ CSCs against simulated ischemia-reperfusion injury-induced cell death (Fig. 2C).
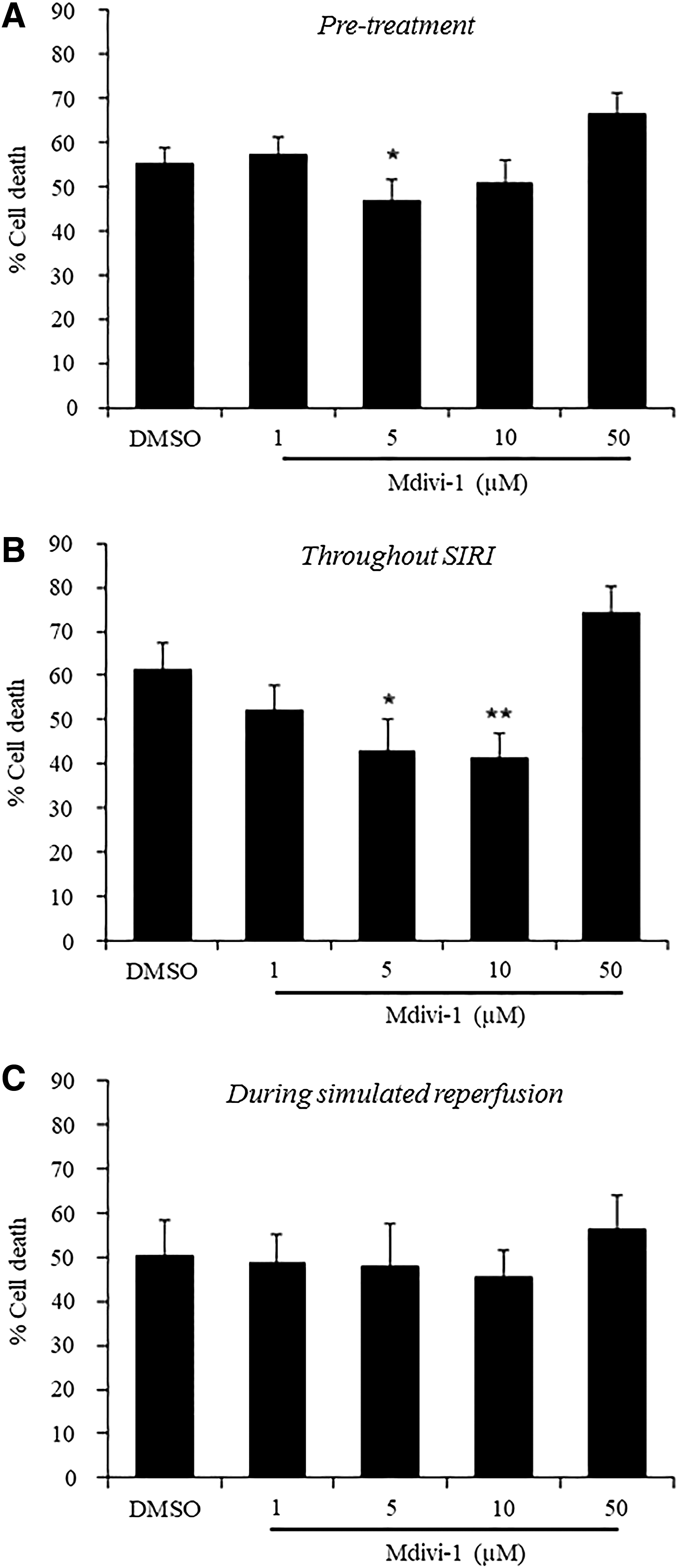
Mdivi-1 protects W8B2+ CSCs against SIRI.
Given that the cytoprotective effect of Mdivi-1 was observed only in the pretreatment cohort and when given throughout simulated ischemia-reperfusion injury, these two treatment regimens were selected for subsequent mechanistic studies. Compared to cells cultured under normoxic conditions, W8B2+ CSCs subjected to simulated ischemia-reperfusion injury have a lower percentage of cells with tubular mitochondria (Fig. 3A), lower mitochondrial membrane potential (Fig. 3B), and higher levels of mitochondrial superoxide (Fig. 3C). However, treatment with Mdivi-1, either as pretreatment or throughout simulated ischemia-reperfusion injury, did not significantly revert these changes to normoxic levels. Phosphorylation and mitochondrial translocation of Drp1 were not assessed because protein expression of Drp1 cannot be detected in the whole cell lysate of human W8B2+ CSCs after being subjected to simulated ischemia-reperfusion injury (Supplementary Fig. S4).

The cytoprotective effect of Mdivi-1 in SIRI model is not associated with changes in mitochondrial morphology, mitochondrial membrane potential, and mitochondrial superoxide production. The effect of Mdivi-1 on mitochondrial morphology (n = 4–6)
Acute treatment of Mdivi-1 does not affect the mitochondrial bioenergetics of W8B2+ CSCs
A recent study has reported that Mdivi-1 can inhibit complex I-dependent reverse electron transfer-mediated ROS production in rat neurons [17]. To determine whether this could be the explanation for the cytoprotective effect of Mdivi-1 observed in this study, the effect of acute treatment of Mdivi-1 on mitochondrial respiration and glycolytic flux of intact W8B2+ CSCs was evaluated. Mdivi-1 (5–50 μM) did not significantly affect the basal cellular respiration (measured before Mdivi-1 injection), Mdivi-1 respiration (basal cellular respiration after Mdivi-1 injection), maximal respiration (measured after addition of FCCP), nonmitochondrial respiration, and spare respiratory capacity of W8B2+ CSCs (Fig. 4A). Treatment with Mdivi-1 (5–50 μM) did not significantly affect the glycolytic metabolism of W8B2+ CSCs, as determined by the ECAR (Fig. 4B).
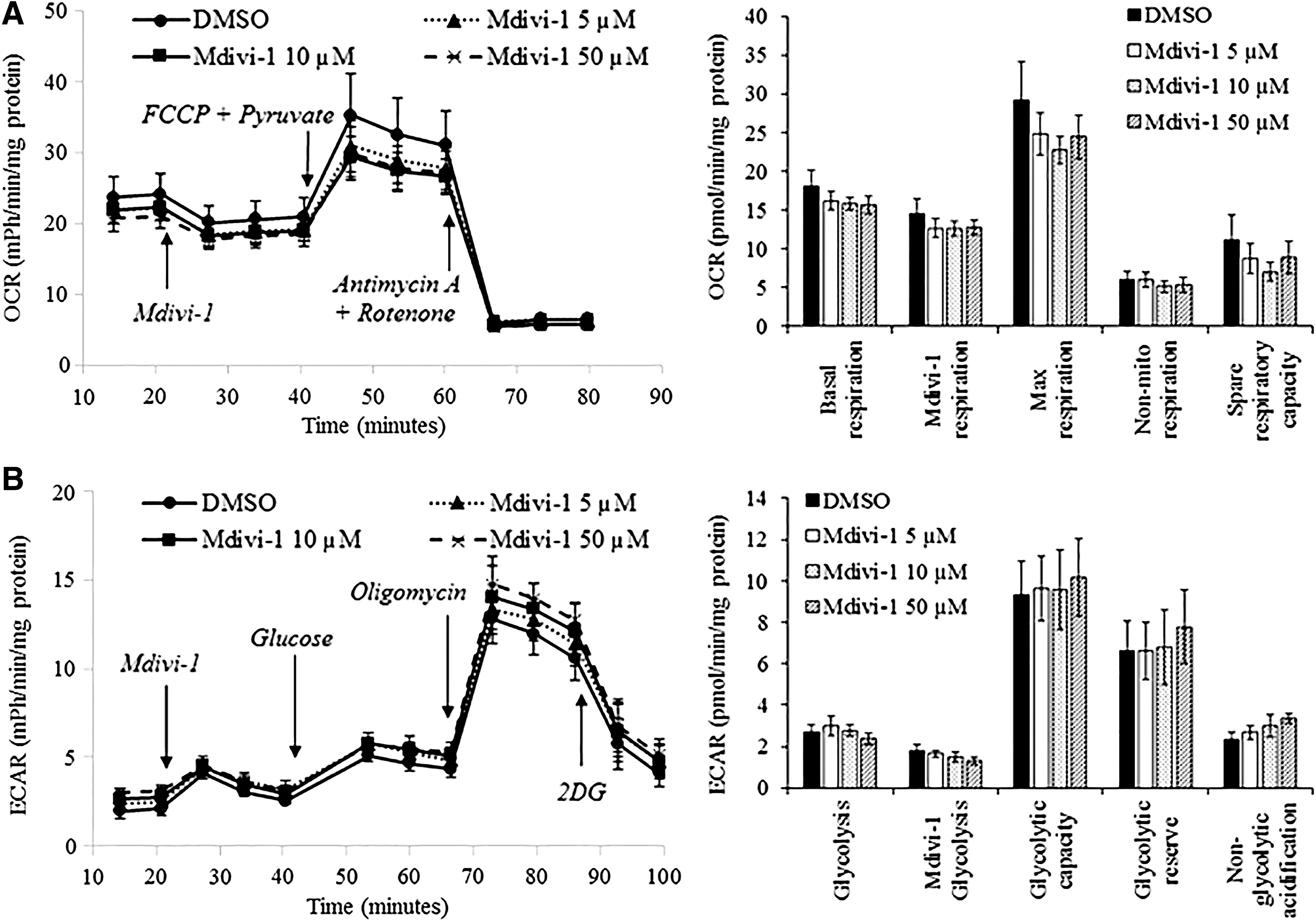
Mdivi-1 does not affect the oxidative phosphorylation and glycolysis of W8B2+ CSCs. The effect of Mdivi-1 on OCR
Discussion
This study demonstrates for the first time that Mdivi-1 can promote the survival of human W8B2+ CSCs using two different clinically relevant in vitro cell stress models. The H2O2 model simulates the detrimental burst of ROS occurring at the onset of reperfusion, mimicking the oxidative stress experienced by cells following percutaneous coronary intervention delivery. The simulated ischemia-reperfusion injury model mimics the situation where cells are delivered intramyocardially into the already ischemic myocardium, as well as the formation of avascular cell clumps following delivery, with reperfusion ensuing thereafter either through physical diffusion of nutrient and oxygen or as a result of neoangiogenesis over time. Furthermore, the prosurvival effect of Mdivi-1 in human W8B2+ CSCs appeared to be independent of effects on recovery of mitochondrial membrane potential, reduction in ROS production, and changes in mitochondrial bioenergetics.
In the simulated ischemia-reperfusion injury model, Mdivi-1 was cytoprotective as a preconditioning agent and when given throughout ischemia and reperfusion, but was not effective when given during reperfusion. This suggests that the window of protection conferred by Mdivi-1 is limited to before and during ischemia. This is in line with previous studies that demonstrated the cytoprotective effect of Mdivi-1 is dependent on the timing of treatment [18,19]. Using the HL-1 cells, a cardiac cell line derived from mouse atrial tumor lineage, and a lethal hypoxia-reoxygenation injury model, Dong et al. showed that inhibition of Drp1 mitochondrial translocation with Mdivi-1 before hypoxia was cytoprotective, but exacerbated cell death when cells were treated with Mdivi-1 at the time of reoxygenation [18]. Similarly, Zhang et al. has reported aggravation of neuronal injury by Mdivi-1 when administered at reperfusion in both in vitro and in vivo settings [19]. They also showed that the detrimental effect of Mdivi-1 on cell survival was due to the suppression of the protective mitophagic process during reperfusion, where inhibition of mitochondrial fission could interfere with the mitophagic process in removing dysfunctional mitochondria from the cell, leading to accumulation of damaged mitochondria and finally cell death [19 –21].
Mdivi-1 has been shown to confer cytoprotection by preventing increases in mitochondrial fission under cell stress conditions [10,22]. While we have shown that the cytoprotective effects of Mdivi-1 in W8B2+ CSCs was accompanied by less mitochondrial fission in the H2O2-induced oxidative stress model, this was not the case in the simulated ischemia-reperfusion injury model where the percentage of cells with tubular mitochondria was equally reduced in both the Mdivi-1 and vehicle control groups. Moreover, protein expression of Drp1 was barely detected in human W8B2+ CSCs after being subjected to simulated ischemia-reperfusion injury (Supplementary Fig. S4). These observations may suggest that the cytoprotective effect of Mdivi-1 could be independent from its inhibitory effect on Drp1-mediated mitochondrial fission. In fact, a recent study by Bordt et al. has demonstrated that Mdivi-1 can also inhibit complex I-dependent reverse electron transfer-mediated ROS production in rat primary cortical neurons, immortalized mouse embryonic fibroblasts, and COS-7 cells [17]. Zhang et al. [23] also showed that both genetic and pharmacologic inhibition of Drp1 with Mdivi-1 could suppress mitochondrial respiration of adult rat and cardiomyocytes. However, we could not reproduce this inhibitory effect of Mdivi-1 on mitochondrial oxygen consumption in human W8B2+ CSCs (Fig. 4). A likely explanation could be the differences in mitochondrial characteristics between cell types, but this warrants further investigation. Supporting this conjecture are previous studies, which have reported that mitochondrial subtypes within the same cell have distinct ultrastructure [24] and respiratory preference, which affects its susceptibility toward pathological stimuli [25 –27].
The rapid burst of ROS during reperfusion following an episode of ischemia can induce opening of the mitochondrial permeability transition pore, resulting in a collapsed mitochondrial membrane potential and release of proapoptotic factors into the cytosol leading to cell death [28,29]. Previous studies in neurons, cardiovascular cells, and other cell types have shown that the cytoprotective effect of Mdivi-1 is accompanied by recovery of mitochondrial membrane potential and attenuation of ROS production [10,11,30 –33]. However, under normal conditions, treatment of Mdivi-1 has been shown to induce mitochondrial membrane potential depolarization and increased ROS production in human umbilical vein endothelial cells [21] and porcine embryos [34]. Moreover, accumulation of oxidative stress could still occur in elongated tubular mitochondria [20,35], suggesting a complex relationship between mitochondrial morphology and the health status of mitochondria. In this study, we showed that the cytoprotective effect of Mdivi-1 in human W8B2+ CSCs is independent of improvement of mitochondrial membrane potential and reduction of mitochondrial superoxide generation. This may indicate the existence of other protective mechanisms that may or may not be dependent on Drp1. These include reduced autophagic flux [19,36 –38], increased adenosine signaling [39], reduced endoplasmic reticulum stress [40], and upregulation of mitochondrial large-conductance calcium and voltage-activated potassium channel [31], which require further investigation.
Conclusion and Future Perspective
In summary, this study has demonstrated that treatment with Mdivi-1 can promote the survival of human W8B2+ CSCs. This pharmacological approach can be employed to render human stem cells more resistant to cell death after transplantation into the ischemic myocardium, thereby prolonging their benefits, either by differentiating into cardiovascular cell types or by evoking intrinsic cardiac repair and regenerative mechanisms through paracrine mechanisms.
Footnotes
Acknowledgments
This work was carried out with support from the St Vincent's Hospital (Melbourne) Research Endowment Fund, CASS foundation, and Stafford Fox Medical Research Foundation. Ayeshah Rosdah is supported by Australia Awards Scholarship. The O'Brien Institute Department and St Vincent's Institute of Medical Research receive Operational Infrastructure Support from the Victorian State Government of Innovation, Industry and Regional Development.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.