
Abstract
Wolfram syndrome (WFS) is a rare autosomal premature aging syndrome that shows signs of diabetes mellitus, optic atrophy, and deafness in addition to central nervous system and endocrine complications. The frequent form of WFS type 1 (WFS1) harbors causative mutations in the WFS1 gene, whereas the rare form or WFS type 2 (WFS2) involves CISD2. Mutations in these two genes are recognized by a subset of variable clinical symptoms and a set of overlapping features. In this study, we report on the generation of stable human-induced pluripotent stem cells (hiPSCs) derived from primary fibroblasts of a previously reported Italian family with CISD2 mutation (c.103 + 1G>A), occurring in the consensus intron 1 splicing site in two sisters, deleting the first exon of the transcript. The generated hiPSCs provide a cell model system to study the mutation's role in the multisystemic clinical disorders previously described and test eventual drug effects on the specific and associated clinical phenotype.
Introduction
W
The two types of WFS are recognized with a subset of variable clinical symptoms and a set of overlapping features [12,13], although some are disease specific. WFS shows clinical and genetic heterogeneity affecting the function of several organs with clinical signs apparent in early childhood [14,15], in fact, pancreatic β-cells and neuronal cells are selectively destroyed [4,8,16,17]. Although reported cases of CISD2-dependent WFS did not present diabetes insipidus (DI) and psychiatric disorders [8,18,19], they can be present in patients faced with pituitary gland dysfunction and urinary tract problems. CISD2 is a well-conserved, ER and mitochondria-resident protein and the gene resides on the long arm of chromosome 4q24 within a region harboring genetic components for human longevity [20,21].
The exact function of the CISD2 protein is unknown but is thought to keep mitochondria functional [4] and control lifespan. A CISD2 knockout model has shown that the deficiency drives premature aging in mice since it is an essential gene that regulates lifespan [22]. Three different CISD2 deleterious mutations have been reported, the most recent homozygous mutation (c.215A>G; p.Asn72Ser) was found in a Moroccan patient associated with overlapping WFS1 and WFS2 [23]. The first CISD2 mutation was first described in a large consanguineous Jordanian family [8,24,25]; two further homozygous intragenic mutations were reported by Mozzillo et al. [18] and a third affects the consensus splicing site of the gene in two sisters of an Italian family [19]. Recently, two siblings demonstrated clinical characteristics of WFS2, whereas previously were misdiagnosed with type 1 DM and diabetic retinopathy-related blindness [26]. Although wolframin, encoded by WSF1gene, has been studied extensively, knowledge on the biological function of the CISD2 proteins is still in its infancy [10,27]. Both wolframin and CISD2 proteins share overlapping roles in intracellular Ca2+ homeostasis, ER-stress response, and autophagy [11,28,29]. Insights into the functions of CISD2 will not only provide knowledge regarding the etiology of WFS, but will also shed light on important regulatory proteins that link metabolic disease with aging.
In this study, we report on the generation and characterization of human-induced pluripotent stem cells (hiPSCs) using primary fibroblasts of homozygous and heterozygous c.103 + 1G>A CISD2 individuals [19], to provide an in vitro cell model system to study the effects, of this rare mutation, on the development of the multisystemic clinical manifestations observed in the patients. The family under investigation consists of two homozygous sisters and heterozygous parents for the c.103 + 1G>A mutation. The hiPSCs can serve as a valuable tool for testing drug effects on the specific clinical phenotype associated with this particular mutation.
Materials and Methods
Clinical cases
Skin biopsies were obtained from the WFS2 Italian family (WFS2_1, WFS2_2, WFS2_3, and WFS2_4) at MultiMedica Hospital (Milan, Italy) using a 3 mm AcuPunch biopsy kit and were approved by the IRCCS MultiMedica Ethical Committee. The family consists of two homozygous sisters and heterozygous parents for the c.103 + 1G>A CISD2 mutation. The sisters (patients) carried the mutation mapping within the donor splice site of intron 1 and clinically developed clinical and biochemical signs of nonautoimmune insulin-requiring DM, polynuria, peptic ulcer, and hematological alterations [19].
Molecular characterization of the c.103 + 1G>A mutation
Total RNA was extracted from homozygous patient's fibroblasts (WFS2_1 and WFS2_2), heterozygous parents (WFS2_3 and WFS2_4), generated hiPSCs (WFS2_1#2, WFS2_1#3, WFS2_1#24, WFS2_3#1, and WFS2_3#6), and fibroblasts MGM18004E from a healthy control subject (CTR) using Trizol, and amplification of the complementary DNA (cDNA) was obtained using primers indicated in Table 1. Amplified products were fractionated on 1.5% agarose and observed with Imagequant (GE).
F, forward; R, reverse; UTR, untranslated region.
Generation of hiPSCs
Primary fibroblasts were established using standard procedures and cultured in 4.5 g/L Dulbecco's modified Eagle's medium (DMEM) high-glucose (Gibco), 20% fetal bovine serum (Gibco), 1%
To validate the electroporation efficiency, 1.27 μg/μL of eGFP C2 was used as control and incorporation was tested 24 h postnucleofection using Amaxa Human Dermal Fibroblast Nucleofector Kit (Lonza). Cells at passage 4 were transfected through U-020 program (Amaxa) and 2 μg of a plasmid encoding eGFP. A total of 5^105 transfected cells were seeded on a six-well matrigel-coated plate and incubated in a humidified 37°C/5% CO2 incubator for 48 h with antibiotic-free fibroblast medium. From the third day after nucleofection, the fibroblast medium (days 1 and 2) was replaced daily (days 3 to 25+) with human embryonic stem (hES) cell medium during the induction phase of reprogramming.
hES medium is a xeno-free and defined reprogramming culture medium optimized for the generation of hiPS: 4.5 g/L DMEM high-glucose (Gibco), 2 mM
The healthy control line BF15#2 (passage 12) was obtained from Dr. Delia (National Cancer Institute, Milan, Italy and stored in ISENET Biobank) and generated by donor fibroblasts reprogrammed simultaneously using the same vectors and technology.
Characterization of fibroblasts and hiPSCs
Real-time PCR and immunofluorescence analysis
RNAs were isolated with Trizol according to the manufacturer's recommendations (Sigma-Aldrich) and quantified with Nanodrop ND-1000 (Thermo Fisher Scientific). RNA was retrotranscribed to cDNA with the RevertAid™ H Minus First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). quantitative reverse transcription polymerase chain reaction (qRT-PCR) was assessed in triplicate on at least two independent biological replicates by the ΔΔCt method on Rotor-Gene Q (Qiagen) using the Maxima SYBR Green qPCR Master Mix (Thermo Fisher Scientific). GAPDH was selected as housekeeping gene and data were normalized to its expression. Statistical analysis was performed using REST (relative expression software tool) software. Primer sequences are described in Table 2.
For immunofluorescence analysis, cells, after thawing, were seeded in four-well plates and fixed in 4% paraformaldehyde, incubated in 0.1 M glycine and then in blocking solution consisting of 5% goat serum and 0.6% Triton in phosphate-buffered saline (PBS). Cells were immunostained at 4°C overnight in blocking solution with primary antibodies for pluripotency markers: anti-Sox2 (1:300; Millipore), anti-Oct4A (1:400; Cell Signaling), anti-SSEA-4 (1:50; Millipore), anti-TRA-1-81 (1:50; Millipore), and anti-Vimentin (30 μg/mL; Sigma-Aldrich). For the immunofluorescence characterization, samples were incubated with appropriate secondary antibodies (Rhodamine-Red antimouse IgM and antirabbit immunoglobulin G (IgG) and Alexa Fluor 488 antimouse IgG, (Jackson ImmunoResearch, distributed by Li StarFish, Milan, Italy), and nuclei were counterstained with Hoechst 33258.
Cells were mounted with GelMount aqueous mounting medium (Sigma-Aldrich). The images were acquired using a Leica DMI4000B inverted microscope linked to a DFC360FX or to a DFC280 camera (Leica Microsystems).
Genomic stability
Fibroblasts from the entire family and from WFS2_1#3 and WFS2_3#1 iPS clones in addition to BF15#2 were assayed for genomic stability by karyotyping and by array comparative genomic hybridization (aCGH). Metaphase spreads were prepared from 80% confluent cultures according to standard procedures. Actively dividing cells were treated with 10 ng/mL colcemid (Gibco KaryoMAX Colcemid; Thermo Fisher Scientific) for 16 h (overnight) at 37°C. Cells were combined in 0.56% KCl for hiPS cells, and a 1:1 mixture of 0.56% KC1 and 1% sodium citrate for 20 min at 37°C, and were fixed with methanol/acetic acid (3:1 v/v). Chromosomes were evaluated by Q-banding using quinacrine following routine procedures and guidelines of the International System for Chromosome Nomenclature 2009 [30].
Karyotyping was performed using Leica DMI4000B inverted microscope linked to a DFC360FX or to a DFC280 camera (Leica Microsystems), equipped with the acquisition and analysis Cytovision software (Leica Biosystems). On average, 25 metaphases were evaluated.
aCGH analysis was performed using Agilent Human Genome CGH Microarray 44K Kit (Agilent Technologies, Palo Alto, CA), following the manufacturer's instructions. Sex-matched commercial DNA sample (male, Promega) was used as reference DNA. Hybridization signals were analyzed by means of Feature Extraction software (v10.7) and DNA Analytics software (v5.0; Agilent Technologies). Aberration detection method 2 algorithm (threshold 5.0) was used to identify DNA copy number aberrations. We applied a filtering option of minimum three aberrant consecutive probes [31] and absolute average log 2 ratio of 0.30. University of California Santa Cruz human genome assembly hg18 was used as a reference and copy number variations (CNVs) were identified with a database integrated into the Agilent Genomic Workbench analytic software. Log 2 ratios lower than −0.30 were classified as losses, those >0.3 as gains.
Embryoid body and trilineage differentiation
WFS2_1#3, WFS2_3#1 iPS clones, and BF15#2 hiPSCs were detached as cell clumps, plated in six-well low-attachment plates, and cultured in Essential 8 medium (Thermo Fisher Scientific) supplemented with 4 mg/mL PVA (polyvinyl alcohol; Sigma-Aldrich) and 10 μg/mL ROCK inhibitor.
Two days after, cell aggregates were nourished with Essential 8 medium (Thermo Fisher Scientific) and E6 medium (1:1 mixture) supplemented with 4 mg/mL PVA. At day 6, embryoid bodies (EBs) were collected, plated on matrigel-coated wells, and allowed to differentiate for further 8 days with daily medium changes. For immunofluorescence analysis, at day 14, cells were seeded in four-well plates and fixed in 4% paraformaldehyde, incubated in 0.1 M glycine, and blocked in a solution consisting of 5% goat serum and 0.6% Triton in PBS.
Cells were immunostained in blocking solution with primary antibodies: for ectoderm anti-βIII-Tubulin (1:100; Sigma-Aldrich), mesoderm antismooth muscle actin (SMA) (1:200; Sigma-Aldrich), and endoderm anti-alpha-fetoprotein (AFP) (1:50; R&D Systems). For the immunofluorescence characterization, samples were incubated with appropriate secondary antibodies (Alexa Fluor 488 antimouse IgG and Rhodamine-Red antirabbit IgG; Jackson ImmunoResearch, distributed by Li StarFish, Milan, Italy) and nuclei were counterstained with Hoechst 33258. The images were acquired using a Leica DMI4000B inverted microscope linked to a DFC360FX or to a DFC280 camera (Leica Microsystems).
Statistical analysis
Three independent experiments were performed for each experimental condition tested and each experiment was performed at least in triplicate. Experimental data are expressed as mean ± standard error. Statistical analysis was performed by two-tailed Student's t-test, A p-value <0.05 was considered statistically significant (*).
Results
Molecular characterization of the CISD2 mutation in WFS2 subjects
The gene CISD2 consists of three exons encoding for a protein of 15 kDa. The homozygous CISD2 (c.103 + 1G>A, NM_001008388) mutation was predicted to eliminate the first exon of the gene since it occurs in the donor splice site in intron 1. To molecularly characterize the CISD2 mutation, RNA was extracted from the patient's fibroblasts (homozygous WFS2_1 and WFS2_2), parents (heterozygous WFS2_3 and WFS2_4), and fibroblasts MGM18004E healthy control subject (CTR). Overlapping primers were designed against each exon and upstream regulatory region. PCR amplification consisted in coupling each primer pair with one designed on the 3′ untranslated region (UTR), generating three fragments (1128nt, 1031nt, and 967nt) consisting of progressive 5′ deletions spanning the entire CISD2. As predicted, the patients' transcript lacked exon 1 and upstream regulatory region but retained exons 2 and 3, since the primers designed against exon 2 amplified the expected PCR product of 967 nt in size, and no amplification was observed using primers designed on exons 1 and 5′ regulatory region. MGM18004E (CTR), WFS2_3, and WFS2_4 samples retained the entire CISD2 transcript (Fig. 1A). Quantitative PCR analysis revealed that the level of CISD2 expressed in the homozygous and heterozygous samples was reduced by 98% and 65%, respectively, compared with those obtained from the healthy control (Fig. 1B), suggesting a further alteration in the promoter region of the siblings, this is presently being explored. The mentioned data confirm total loss of exon 1 and partial modifications of the 5′ UTR in the siblings.
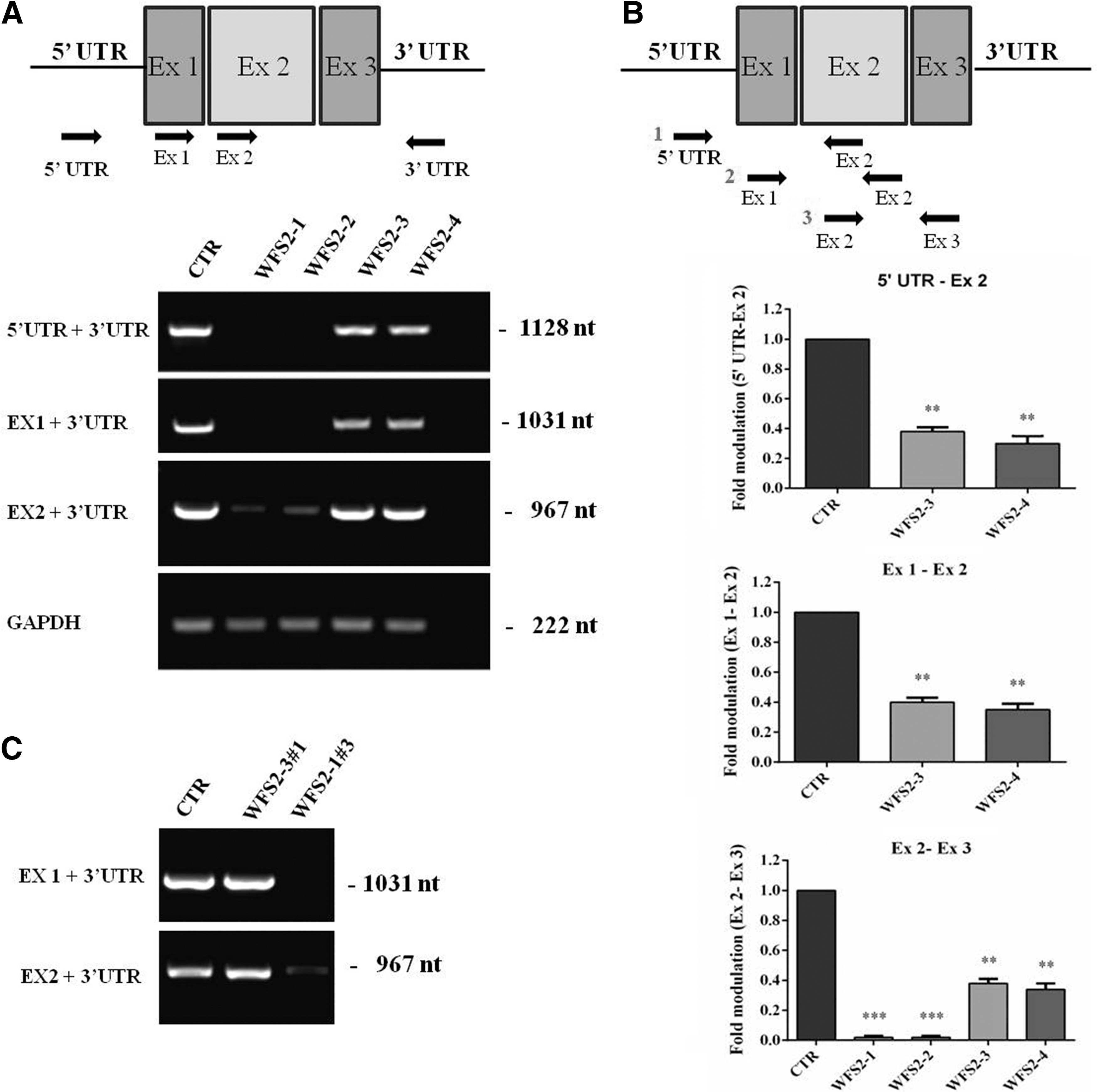
Molecular characterization of the c.103 + 1G>A mutation.
Phenotypical and genomic characterization of c.103 + 1G>A CISD2 WFS2 primary fibroblast cell lines
The primary fibroblast cell lines from homozygous and heterozygous CISD2 c.103 + 1G>A subjects showed normal growth patterns as indicated by bright field morphology and vimentin staining. Patient WFS2_2 showed consistent reduction in proliferation (Fig. 2A, B). Although no gross chromosomal alterations were observed by Q-banding (Fig. 2C), aCGH revealed CNVs on chromosomes 2, 3 (WFS2-2) and 4, 6 (WFS2-3). In WFS2-2, >10 CNVs, at chromosome 2, consisting of 120 kb loss were detected, these are described as benign in Database of Genomic Variants and International Standards for Cytogenomic Arrays. The CNVs on chromosome 3 are smaller involving the CTD small phosphatase-like a tumor suppressor gene. CNV losses in WFS2_3 consist of 527 and 77 kb on chromosomes 4 and 6, respectively, involving the RAP guanine nucleotide exchange factor 2 (RAPGEF2), p53 effector related to PMP22 (PERP), and tumor protein p53 (TP53) genes with no significant associated clinical phenotype. Neither WFS2-1 nor WFS2-4 showed any genomic alterations (Fig. 2D).
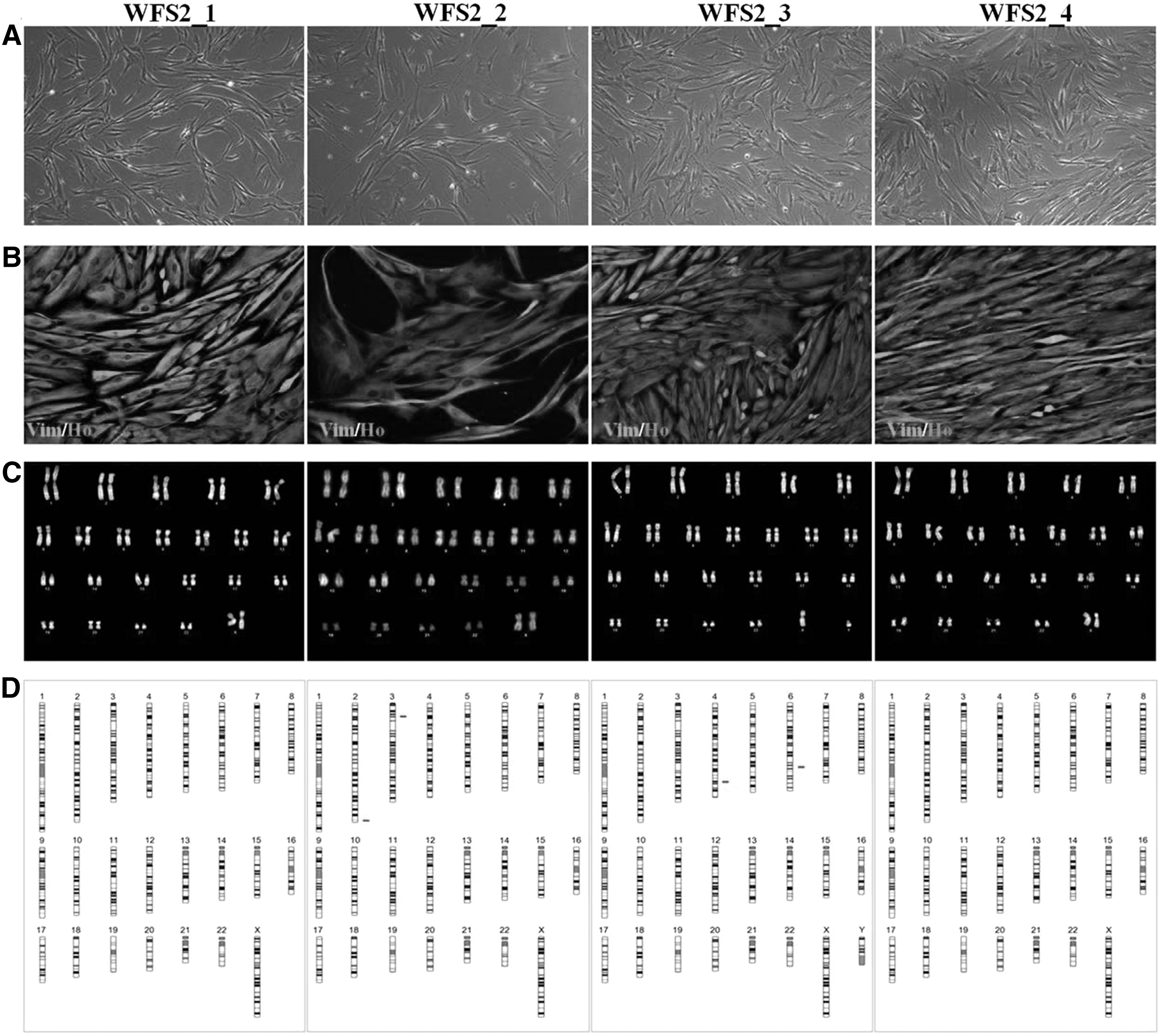
Phenotypical and genomic characterization of c.103 + 1G>A CISD2 in WFS2 primary fibroblast cell lines.
Generation and characterization of hiPSCs from primary CISD2 mutated fibroblast cell lines
Dermal fibroblasts from homozygous CISD2 proband with c.103 + 1G>A mutation (WFS2_1) and heterozygous parent (WFS2_3) were transduced with the nonintegrating episomal reprogramming factors OCT3/4, I-Myc, KLF4, SOX2, and a short hairpin RNA against p53 [32], recovered in fibroblast medium and cultured in hES media, following, essentially, the protocol described in materials and methods. Supplementary Fig. S1A schematizes the reprogramming process and indicates cell morphology changes as they turn from fibroblasts to established iPSC lines. Five clones from WFS2_1 (WFS2_1#2, WFS2_1#3, WFS2_1#10, WFS2_1#11, and WFS2_1#24) and two from WFS2_3 (WFS2_3#1 and WFS2_3#6) subjects were expanded and cryostored after pluripotency testing. All clones were found positive for NANOG and SOX2 by qRT-PCR analysis compared with human embryonic stem cells RC17 and hiPSCs BF15#2. Vimentin messenger RNA expression of each generated hiPSC line was absent compared with the initial fibroblast cell lines (human fibroblasts WFS2_1 and WFS2_3) (Fig. 3A).
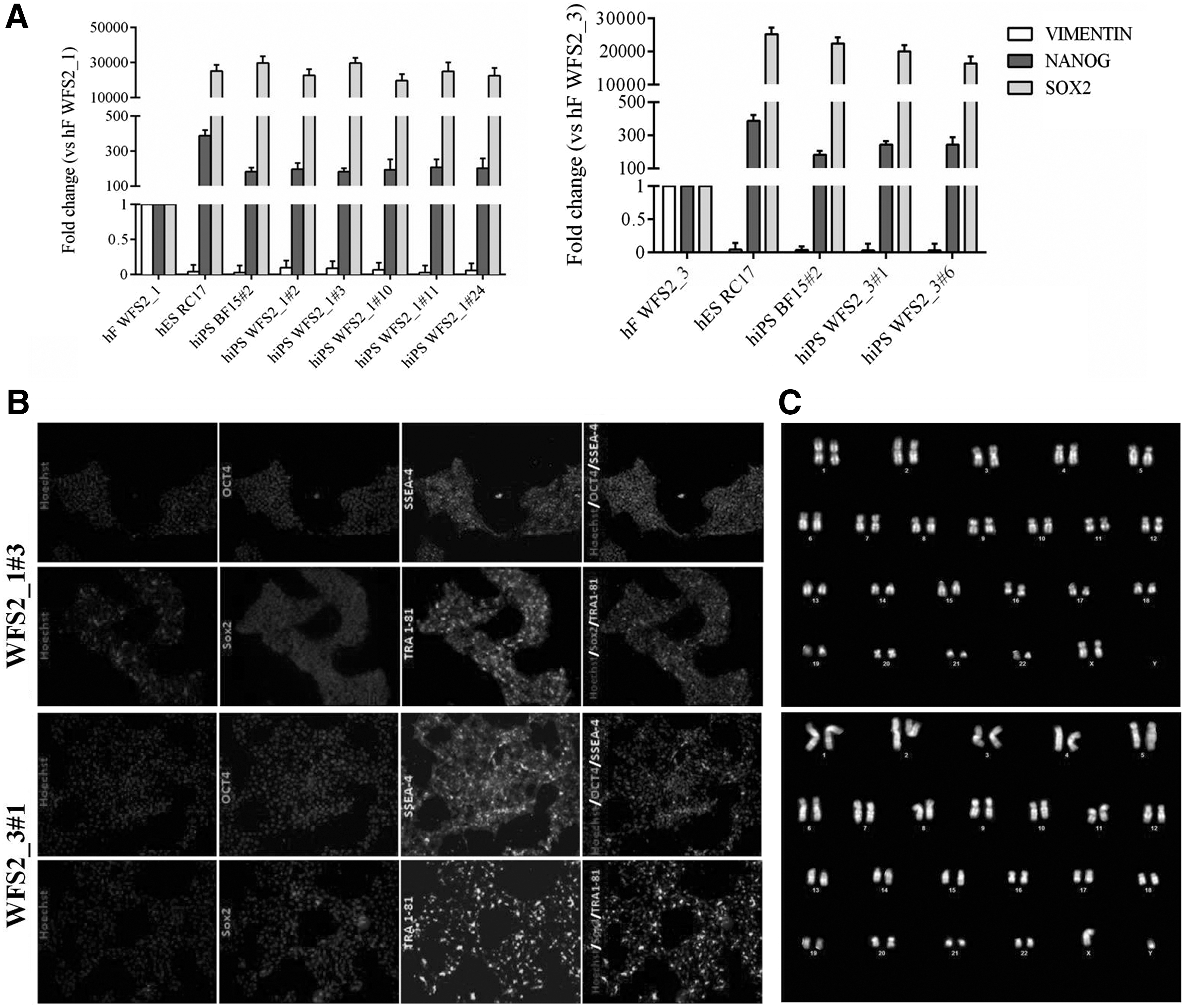
Characterization of hiPSCs from primary CISD2 mutated fibroblast cell lines.
Two iPS clones (WFS2_1#3 and WFS2_3#1) were selected and pluripotency using SOX2, OCT4, TRA 1-81, and SSEA4 markers was confirmed by immunofluorescence analysis (Fig. 3B). These hiPSC clones and BF15#2 hiPSCs line (46XY, data not shown) retained the normal karyotype as the primary fibroblast cells (Fig. 3C). Moreover, the pluripotent character of WFS2_1#3, WFS2_3#1, and BF15#2 hiPSCs was confirmed by their multilineage differentiation potential. All iPS clones were spontaneously differentiated into EBs and examined for immunopositivity of markers specific for all 3 g layers in vitro. Immunofluorescence analysis at day 14 showed that resulting cells expressed markers of ectoderm: βIII-Tubulin, mesoderm: α-SMA, and endoderm: α-FP (Fig. 4). The architecture of CISD2 gene was maintained in the mentioned hiPSC lines as revealed by PCR analysis using the same pair of primers (Fig. 1C).
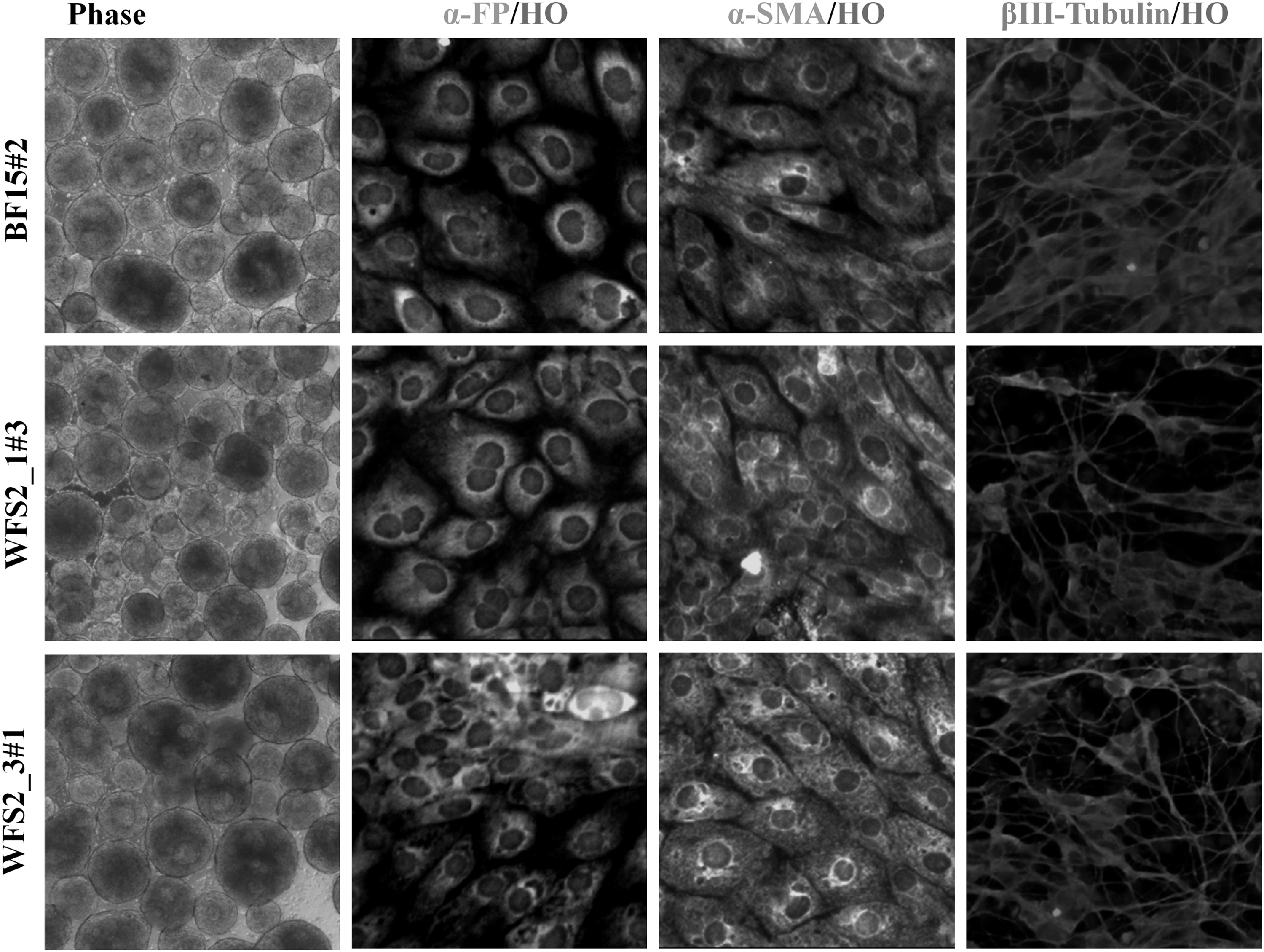
EB and trilineage differentiation of BF15#2 hiPSC line, WFS2_1#3, and WFS2_3#1 iPS clones. Phase: day 4 EBs culture (10 × ). After 14 days, differentiated cultures exhibited the presence of cells immunopositive for endodermal (α-FP), mesodermal (α-SMA), and ectodermal (βIII-Tubulin), germ-layer markers. Magnification: 10 × . EBs, embryoid bodies.
Discussion
A promising technique able to recapitulate pathological processes, in vitro, involves the reprogramming of differentiated human somatic cells into iPSCs [33]. Among the WFS patients, 10% carry biallelic mutations in the WFS1 gene; however, a second causative gene regards CISD2 [34] mutations occur only rarely. To date, recessive CISD2 mutations have been described in five families living in Jordan and in Italy [8,18,19]. The patients with the c.103 + 1 CISD2 mutation described by Rondinelli et al. [19] exhibited clinical and biochemical signs of DI and endocrine malfunctions; furthermore, the homozygous bearing patients developed peptic ulcer and hematological abnormalities, phenotypes usually associated with WFS2.
In this study, we report a stem cell-based model consisting of hiPSCs generated from subjects affected with the c.103 + 1 CISD2 mutation, allowing further studies of the mutation's consequences on Wolfram patients. Although hiPSC lines were previously generated from WFS1 affected subjects [16], to our knowledge, this is the first report using CISD2 mutated individuals, at least for the mutation under investigation that causes drastic modifications of the encoded protein. It was predicted that the CISD2 mutation c.103 + 1G>A in the WSF2 family occurred in the donor splice site, resulting in the elimination of the first exon of the gene containing the signal peptide.
To molecularly characterize the specific mutation, a series of primer sets were designed and coupled to the 3′ UTR region generating progressive 5′ deletions. Indeed, as predicted, the patients lacked exon 1 and upstream regulatory region but retained exons 2 and 3, since primers designed against these two exons amplified the expected PCR products, whereas no amplification was observed using primers designed on exon 1 and on the 5′ regulatory region. Moreover, quantitative PCR analysis revealed that the level of CISD2 expression differed between homozygous (98%) and heterozygous (65%) samples, in both cases expression was significantly reduced compared with the healthy control subject. This suggested that further alterations occur in the promoter region of the siblings, but further analysis is required to explore this aspect in greater detail. The generated iPSC clones maintained the same gene structure as the parental fibroblasts.
We hypothesize that the CISD2 mutation encodes for a protein similar to native CISD2 but not identical and likely differs in function and subcellular location. Tsai et al. [10] reported on the generation of murine iPSC from CISD2 deficient mice, showing that protein deficiency impaired the Wnt/β-catening signaling pathway, a phenomenon contributing to the pathogenesis of osteopenia and lordokyphosis in WFS2 patients. Mouse embryonic stem cells derived from loss of Miner 1 [11] showed Ca2+ depletion in the ER and an overload in the mitochondria.
These cells also showed activation of ER stress and unfolded protein responses, phenomena associated with the incapacity to produce insulin, vasopressin, and neurotransmitters, ultimately leading to DI and DM, neuronal dysfunction, and cell death. Overall the mutation did not affect the reprogramming capacity of the fibroblasts into PSCs. A number of individual clones were generated, expanded, cryopreserved (available through the biobank ISENET,
Footnotes
Acknowledgments
The authors thank Dr. D. Delia (National Cancer Institute, Milan, Italy) for his guidance in reprogramming the primary fibroblasts and for fruitful discussion. A.L.S. was supported by a fellowship of National Research Council; A.N. was supported by a fellowship from Integrated Systems Engineering S.r.l.; P.D.B. financially supported the work. This work was supported by MIUR Regione Lombardia Network Lombardo iPS (NetLiPS, project ID 30190629-2011), Progetto Quadro Regione Lombardia-CNR (RSPPTECH 2013–2015), and InterOmics Flagship Project (2015).
Author Disclosure Statement
The authors declare that they have no competing interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.