
Abstract
Human endometrial mesenchymal stem cells (eMSCs) are a well-characterized adult stem cell type with potential for use in regenerative medicine or cell therapy. As a proof of principle, we demonstrated that eMSCs promoted wound healing by reducing the inflammatory response through a paracrine action in a subcutaneous rat model of wound repair. However, an efficient protocol for culturing eMSCs in the undifferentiated state and a reliable method of labeling them for cell tracking were lacking. In this study, we investigated the use of a lentiviral vector containing the mCherry fluorescent reporter gene to transduce and label eMSCs following in vitro culturing in A83-01 containing medium, and different methods of tracing the labeled cells following transplantation under the kidney capsule of immunocompromised NSG mice. Perivascular SUSD2+ eMSCs were isolated from human endometrium. Passage 1 eMSCs were transduced by lentiviruses with mCherry fluorescent reporter gene; mCherry+ cells were isolated by fluorescence-activated cell sorting and cultured until passage 6 in 5% O2 in serum-free medium with fibroblast growth factor 2 (FGF2) and epidermal growth factor (EGF). The cells were subsequently divided into two flasks and treated with either dimethyl sulfoxide (0.01%) or A83-01 (1 μM) for 7 days. 5 × 105 control or A83-01 pretreated cells were encapsulated into a fibrin gel and transplanted under the subrenal capsules of NSG mice. Tissues were analyzed at 7, 14, and 30 days posttransplantation. Human eMSCs were efficiently transduced with mCherry gene. They proliferated and maintained high mCherry expression over five passages. Analyzing transplanted kidneys using polymerase chain reaction, flow cytometry, and immunofluorescence showed that both cell types survived at least 30 days. Efficient labeling of eMSCs using a lentiviral vector and culturing them in an environment maintaining them in an undifferentiated state enable reliable detection in preclinical animal models and highlight the need for generating a pure population of undifferentiated MSCs for long-term survival in vivo to prolong their treatment effect.
Introduction
C
MSCs are present in low quantities in almost all postnatal tissues, including endometrium. Most cell therapies use a heterogeneous plastic-adherent population of MSCs, which have been expanded in culture. Enriching these cells using specific surface markers that purify perivascular cells such as CD146 [7] and SUSD2 [8,9] has improved the purity of isolated MSC. However, the extensive culture expansion of these rare cells is required to generate the large numbers required for cell-based therapies. The expansion of MSC typically leads to their spontaneous differentiation into fibroblasts and generation of heterogeneous cell populations with variable and often reduced stem cell properties than their parent cells [10 –12].
Recently we demonstrated that culturing endometrial mesenchymal stem cells (eMSCs) in serum-free medium (SFM) in a physiological oxygen concentration of 5% retained MSC properties in culture [13]. Furthermore, addition of a small molecule A83-01, a transforming growth factor (TGF) beta receptor inhibitor, prevented spontaneous differentiation of late passage SUSD2+ human endometrial MSC during culture expansion, maintaining stemness and promoting proliferation by blocking senescence and apoptosis in late passage/expanded cultures of SUSD2+ human eMSCs [11].
In a subcutaneous immunodeficient rat model of wound repair, eMSCs seeded on a polyamide mesh/gelatin composite construct were tested for wound healing properties [5]. In this study, the SUSD2+ sorted eMSCs were cultured in 10% fetal bovine serum (FBS) in a normoxic environment, and following extensive cell culture, only 10% of the expanded cells were SUSD2+ eMSCs [5] indicating that the eMSCs had differentiated into SUSD2− fibroblasts. Vybrant™ DiO-labeled cells were 95% positive at the time of implantation. Very few DiO+ cells were detected at 14 days and none at later time points [5]. However, these DiO+ cells could have been rat macrophages having engulfed dying human cells. Nonetheless, the bioengineered construct exerted angiogenic and anti-inflammatory effects and promoted wound healing with minimal fibrosis well beyond the persistence of eMSC. Deficiencies of this study were the lack of a method to maintain an undifferentiated SUSD2+ eMSC phenotype during expansion before implantation and the fact that a transient labeling technique was used to detect eMSCs ex vivo, which does not discriminate between the transplanted human cells, dye spillage into the surrounding cells, or rat macrophages which have engulfed the dye-labeled human cells.
In the present study, we used a lentiviral system to label SUSD2+ eMSCs with an mCherry fluorescent reporter gene and cultured them in a SFM and A83-01 in a 5% O2 hypoxic environment to maintain them in an undifferentiated state as previously published [11]. Late passage (P6) eMSC cultures were used as they contain a significant proportion of differentiated fibroblasts [5,14]. The aims were to establish a reliable approach to detect eMSCs in vivo and to determine whether A83-01 treatment of culture-expanded eMSCs maintained phenotype and promoted their in vivo survival in an immunocompromised mouse model. We hypothesized that pretreatment of eMSCs with A83-01 during culture expansion would increase their survival in vivo compared to untreated control eMSCs. We used several methods to detect mCherry positive human cells and found that they survived for at least 30 days when transplanted under the kidney capsule of NSG mice. Pretreatment of eMSCs with A83-01 had no adverse effect on their survival in vivo.
Materials and Methods
Human endometrial tissue samples
Human ethics approval was obtained from the Monash Health Human Research Ethics Committees (09270B). Human endometrial tissue samples were collected from women undergoing endometrial curette or hysterectomy for nonendometrial pathologies and who were not on any hormones for at least 3 months before surgery. Informed written consent was obtained from each woman. Six donors provided human endometrium for this study, and each donor's cells were used in replicate experiments for each time point for each of the two conditions.
Animals
The animal experimental procedure was approved by the Monash Medical Centre Animal Ethics Committee A (MMCA, 2015/29). NOD.Cg-Prkdcscid Il2rgtm1Wjl /SzJ (NSG) mice were housed in the animal house at Monash Animal Service facilities in compliance with the National Health and Medical Research Council guidelines for the care and use of laboratory animals. NSG is a strain of inbred mice, which lacks mature T cells, B cells (adaptive immunity), and natural killer cells [15].
Isolation of SUSD2+ human endometrial MSCs and cell culture
eMSCs were isolated using magnetic beads according to our previously published protocol [8,11]. Briefly, endometrial tissue was mechanically minced and dissociated into single cells with 0.5% collagenase type I (Worthington Biochemical Corporation) and 40 μg/mL DNase type I (Worthington Biochemical Corporation) in Dulbecco's modified Eagle's medium (DMEM)/F-12 in a MACSmix™ for 1 h at 37°C. Stromal cells were collected by filtering through a 40 μm cell strainer, and red blood cells were removed by density gradient centrifugation using Ficoll-Paque (GE Healthcare Bio-Science). Stromal cells, at the interphase, were incubated with SUSD2-PE antibody (10 μg/mL; BioLegend), followed by anti-PE magnetic beads [20 μL in 80 μL 2% FBS/phosphate-buffered saline (PBS); Miltenyi Biotec] for an hour then 30 min, respectively, at 4°C in the dark. The SUSD2+ eMSCs were eluted from a MS column (Miltenyi Biotec) in a magnetic field and then cultured in DMEM/F12-containing 10% FBS, 1% antibiotics-antimycotic, and 2 mM glutamine [stromal medium (SM)]. All cultures were maintained in 5% O2, 5% CO2, and 90% N2 in a Tri-Gas incubator at 37°C.
Transduction of eMSCs with mCherry lentivirus and culture
We used second-generation HIV-1 based lentiviral vector. The plasmids, plvx-IRES-mCherry (mCherry transfer plasmid, #6312237; Clontech), pMD2.G (envelope plasmid, #12259; Addgene), and psPAX2 (packaging plasmid, #12260; Addgene), were kindly donated by Dr. Daniel Gough, Centre for Cancer Research, Hudson Institute of Medical Research. To generate virus particles for transduction, 2–3 million 293T cells were seeded in a 100 mm dish with SM. The plasmid complex was made by gently mixing 1.5 mL Opti-MEM Reduced-Serum Medium (Sigma), 20 μg plasmid DNA comprising 10 μg plvx-IRES-mCherry, 1 μg pMD2.G, and 9 μg psPAX2 plasmids, 40 μL TransIT-X2® Dynamic Delivery system (MIR 6003; Mirus), and Chloroquine (25 μM) and incubated for 30 min at room temperature to promote complex formation. The 293T cells were transfected at ∼80% confluency by adding TransIT-X2 DNA complexes for 6 h followed by a media change to SM. After 48 h, the virus-containing supernatant was collected and centrifuged at 1,400 rpm for 4 min, passed through a 0.45 μm filter and added to passage 1 (P1) eMSCs together with polybrene (3 μg/mL hexadimethrine bromide, #107689; Sigma), and repeated after 6 h. eMSCs were transduced for 48 h to ensure subsequent inactivation of the lentivirus. mCherry positive eMSCs were collected by sorting on a MoFlo flow cytometer using Cyclops SUMMIT software (Version 5.2; Beckman Coulter) and cultured in SM until ∼70% confluence. The medium was changed to 5% and 1% FBS SM over 48 h and then to SFM [11] on fibronectin-coated flasks. At passage 6 (P6), eMSCs were divided into two flasks, one treated with 1 μm A83-01 and the second with vehicle control (0.01% dimethyl sulfoxide) for 7 days with media changed every 48 h (Fig. 1A).

Validation of mCherry expression in transduced eMSCs in vitro.
Casting fibrin gel constructs for delivering eMSCs
5 × 105 P6 A83-01-treated or control mCherry positive eMSCs were mixed with 10 μL warm fibrinogen (50 mg/mL, #F4753; Sigma) and dispensed into 2 μL droplets of thrombin (100 units/mL, #T9549; Sigma) on a Petri-dish lid to initiate fibrin gel formation (Fig. 1A). The lid was inverted and incubated at 37°C for ∼1 h to fully polymerize the gel. Cells were not kept in parallel culture while they were implanted in vivo because we wanted to see the in vivo effect of the cells following with and without treatment with A83-01.
Mouse subrenal capsule surgery
A total of 108 NSG mice older than 8 weeks were randomly divided into 2 groups (54 mice/group), which received control or A83-01 treated P6 eMSCs. A total of six patient cell lines were used for this study. eMSCs from 1 donor were transplanted into 18 mice in total (9 mice for control cells and 9 mice for A83-01 treated cells, with 3 mice/time point/each group. Three mice were harvested at each time point for each group for the three independent tests performed). Mice were anesthetized using intraperitoneal Ketamine/Xylazine (100 mg/kg and 10 mg/mL) and received subcutaneous Carprofen (0.3–0.5 mg/100 g body weight) and subcutaneous bupivacaine at the site of incision. The left kidney was exteriorized following mediolateral incision. A small hole was made in the kidney capsule, which was lifted from the underlying cortex. Using a dissecting microscope, the eMSC-fibrin gel was inserted under the capsule. The kidney was returned into the body and the incision closed with sutures. The left kidney, spleen, uterus, liver, and incision site skin/muscle were harvested 7, 14, or 30 days after grafting.
Polymerase chain reaction for detection of human mCherry cells
Freshly harvested tissues were digested in lysis buffer [100 mM Tris pH 7.4, 5 mM EDTA, 0.5% sodium dodecyl sulfate (SDS), and 200 mM NaCl] and proteinase K (100 μg/mL, #P2308; Sigma) at 50°C overnight, then centrifuged at 14,000 rpm for 10 min, and an equal volume of isopropanol was added to the supernatant to precipitate genomic DNA (gDNA). The gDNA was pelleted by centrifugation at 14,000 rpm for 10 min and washed with 70% ethanol. The supernatant was discarded, and the pellet was allowed to dry. gDNA was reconstituted in Milli-Q water, and the quality and quantity assessed using NanoDrop spectrophotometer. Undiluted gDNA was used to assess the mCherry sequence while 100 ng/μL gDNA was used for detecting Alu sequences. Polymerase chain reaction (PCR) was performed in 20 μL volumes consisting of 10 μL MyTaq (Bioline), 1 μL each reverse and forward primers (10 μM; Bioneer) (Table 1), gDNA, and Milli-Q water. The reaction consisted of initial denaturation of 95°C for 2 min, followed by 30 cycles of denaturation for 30 s, annealing at 55°C for 1 min, and extension at 72°C for 1 min. The PCR products were separated by 1.5% agarose gel electrophoresis with gel safe DNA ladder and PCR products visualized in ChemiDoc™ XRS+ System (Bio-Rad).
Fluorescence microscopy to detect mCherry labeled eMSCs in tissues
Freshly harvested kidneys were fixed in 4% paraformaldehyde (PFA) overnight at 4°C, washed in PBS then cryoprotected in 30% sucrose for 24 h at 4°C, and embedded in Optimal Cutting Temperature compound on dry ice. Eight-micron Cryo-sections were washed with PBS and nuclei stained with Hoechst 33258 (1:2,000; Molecular Probes) for 5 min. Images were visualized and photographed using a Nikon C1 microscope and analyzed using FIJI software [16].
Flow cytometry
The harvested kidneys were minced and digested as described above for endometrium to generate single cells. Cells were washed and incubated with APC-conjugated SUSD2 (1:20, #327408; eBioscience) antibody for 1 h in the dark on ice. Matched-isotype control IgG-APC was used at the same concentration to set the electronic negative control gate on the flow cytometer. Human mCherry positive and negative cells and cells from noninjected kidneys were used as controls. The cells were washed and fixed with 4% PFA in 2% FBS/PBS. Samples were analyzed using a MoFlo Flow Cytometer and Summit software (version 5.2.; Beckman Coulter).
Statistics
Results are reported as mean ± standard error of the mean for each experimental group. A Friedman test with Bonferroni and Hochberg correction at different time points between the groups was done using GraphPad Prism 7.02. Differences between the groups with P-values <0.05 were considered statistically significant.
Results
Validation of mCherry transduction and retention following extensive culture
eMSCs (P1) were cultured for 48 h following lentiviral mCherry vector transduction to promote plasmid integration and viral inactivation. Flow cytometric analysis revealed that the transduction efficiency of the lentivector ranged between 20% and 40% (n = 6). Of the positive cells, we opted to sort the cells with highest mean fluorescence intensity (20%) (Fig. 1B). The sorted cells were cultured in 5% O2 environment in SFM. Fluorescence microscopy and flow cytometric analysis demonstrated that eMSCs expressed mCherry, and the intensity was maintained following extensive (P6) culture (Fig. 1C, D). At P6 ∼ 99% of control and A83-01 treated eMSCs were mCherry positive (Fig. 1D). Propagation of eMSCs was not affected suggesting that lentiviral transduction did not adversely affect cell proliferation.
mCherry-labeled eMSCs persist in vivo
All mice had a normal postoperative recovery. Human cells were well tolerated and none of the mice developed any visual or palpable mass at any time point. Human mCherry eMSCs were tracked in NSG mice using three different ex vivo methods: PCR, immunofluorescence, and flow cytometry. The cells treated with and without A83-01 were transplanted under the left kidney capsule of NSG mice using fibrin gel as a carrier.
Human gDNA detected in transplanted mice
To detect human cells in the mouse kidney, we analyzed gDNA isolated from mouse tissues by PCR for human Alu sequences (Table 1). Human and mouse gDNA were used as positive and negative references for Alu sequences, respectively. Human cells were detected in the kidneys of mice transplanted with and without A83-01 treatment at 7, 14, and 30 days (Fig. 2A). To determine if the transplanted human cells had migrated from the site of delivery, gDNA from tissue at the incision site (skin), spleen, uterus, and liver was also tested. Human cells were detected at the incision site and spleen in some mice, although at lower abundance and frequency (Fig. 2B). To confirm that the detected human cells were from the transplanted mCherry positive cells, PCR with mCherry sequence was performed (Table 1) using undiluted gDNA. mCherry signal was detected at 7 days in control cells and at 7 and 14 days in the A83-01 treated groups (Fig. 2C), further demonstrating that the human cells were from the transduced eMSCs. However, mCherry DNA was not detected at day 30.
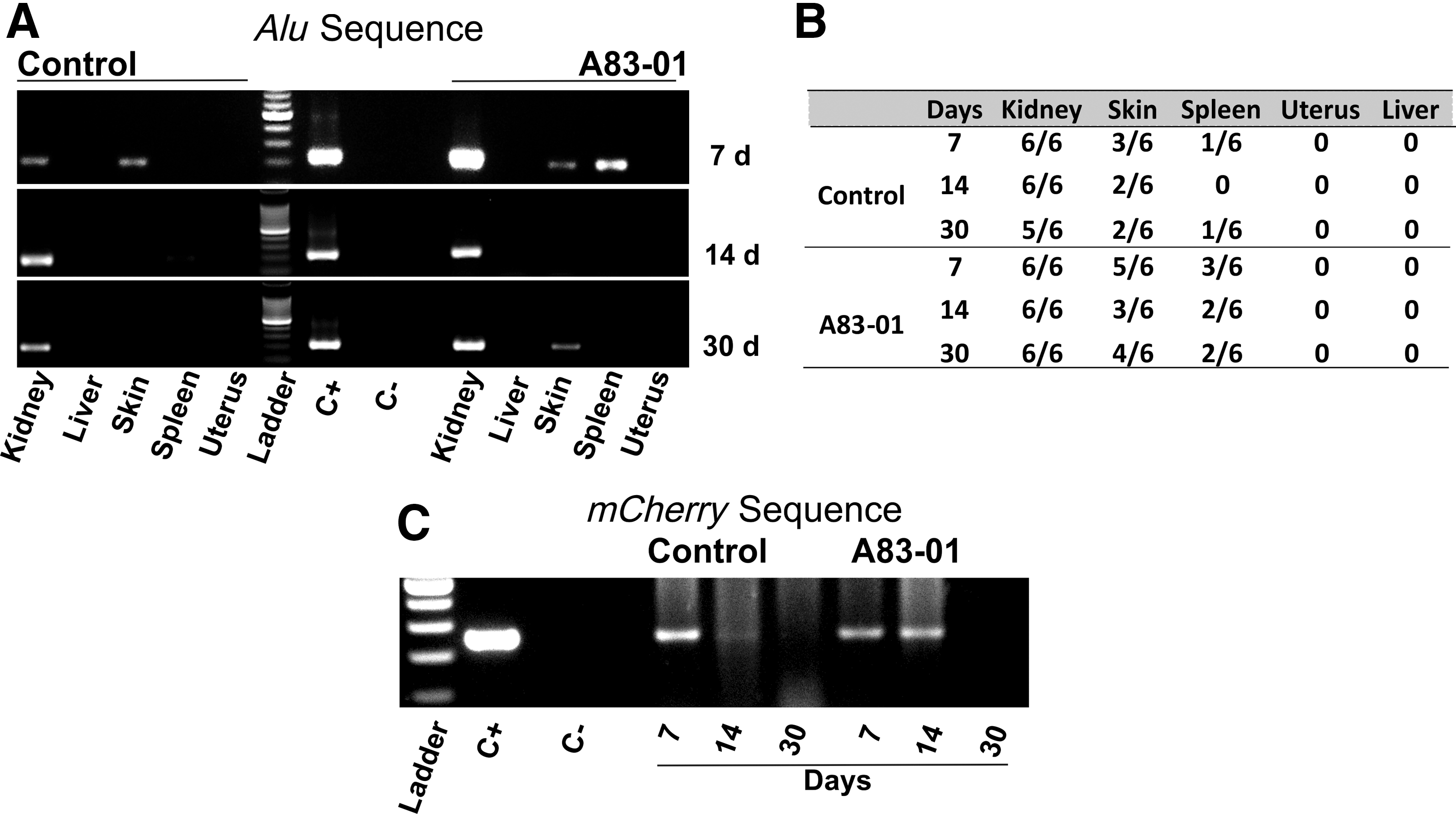
PCR amplification products from mouse tissues harvested posttransplantation by electrophoresis on 1.5% agarose gels.
Flow cytometric detection of mCherry+ SUSD2+ cells in transplanted mice
The identity of transplanted eMSCs was further confirmed by flow cytometry. mCherry positive and negative human eMSCs and cells from untreated mice kidneys were used as positive and negative controls to gate the flow cytometry plots. Inoculated cells from the harvested kidneys at different time points indicated mixtures of human mCherry positive cells and mouse cells. mCherry positive cells were detected at all time points in both treatment groups of mice (Fig. 3A). However, the total number of mCherry cells detected at 7, 14, and 30 days was <20%, 10%, and 1% of the initial cells transplanted. To assess if the transplanted mCherry positive eMSCs still expressed the SUSD2 surface marker, cells were stained with APC-SUSD2 antibody. Approximately, 2.5%, 6%, and 0.3% in the control group and 6%, 11.15%, and 0.7% of the mCherry positive cells in the treated group were SUSD2 positive at 7, 14, and 30 days after transplantation (Fig. 3B). Along with the decreasing number of mCherry eMSCs, there was also a significant decrease in the percentage of SUSD2 expressing cells from 14 to 30 days in the treatment group.
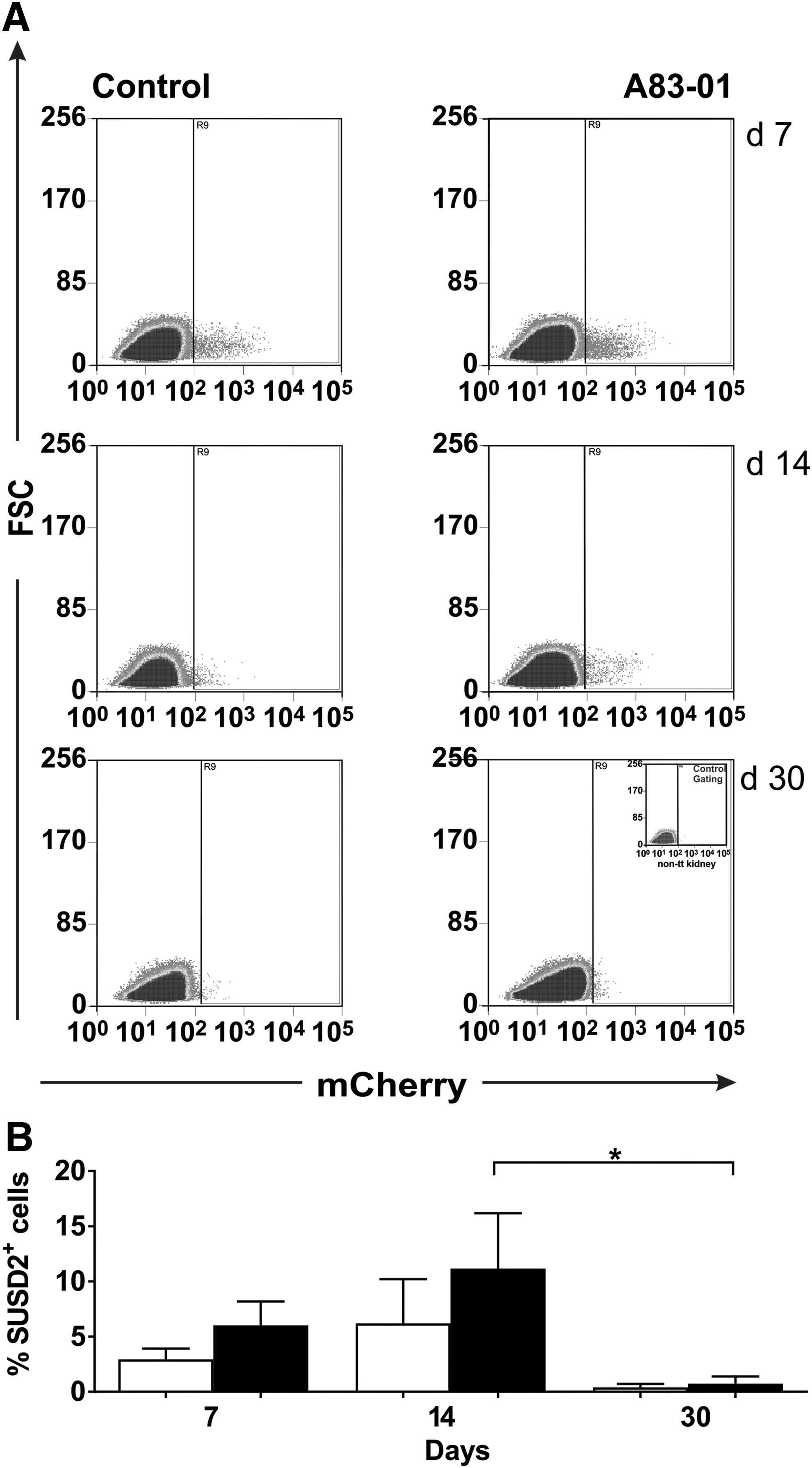
Flow cytometry traces of kidney derived single cells from mice engrafted with human mCherry-labeled SUSD2+ eMSCs analyzed at different times posttransplantation.
mCherry positive eMSCs were present in tissue grafts
The presence of mCherry-labeled cells was further examined in sections of kidneys by fluorescence microscopy. eMSCs were detected by mCherry fluorescence indicating that expression was not lost following in vivo transplantation. As expected from the PCR and flow cytometric results, clusters of mCherry/red cells were prominent at 7 days under the kidney capsule but diminished at 14 days in both groups and were undetectable at 30 days (Fig. 4). In both control and treated groups, mCherry positive cells did not invade the kidney cortex.
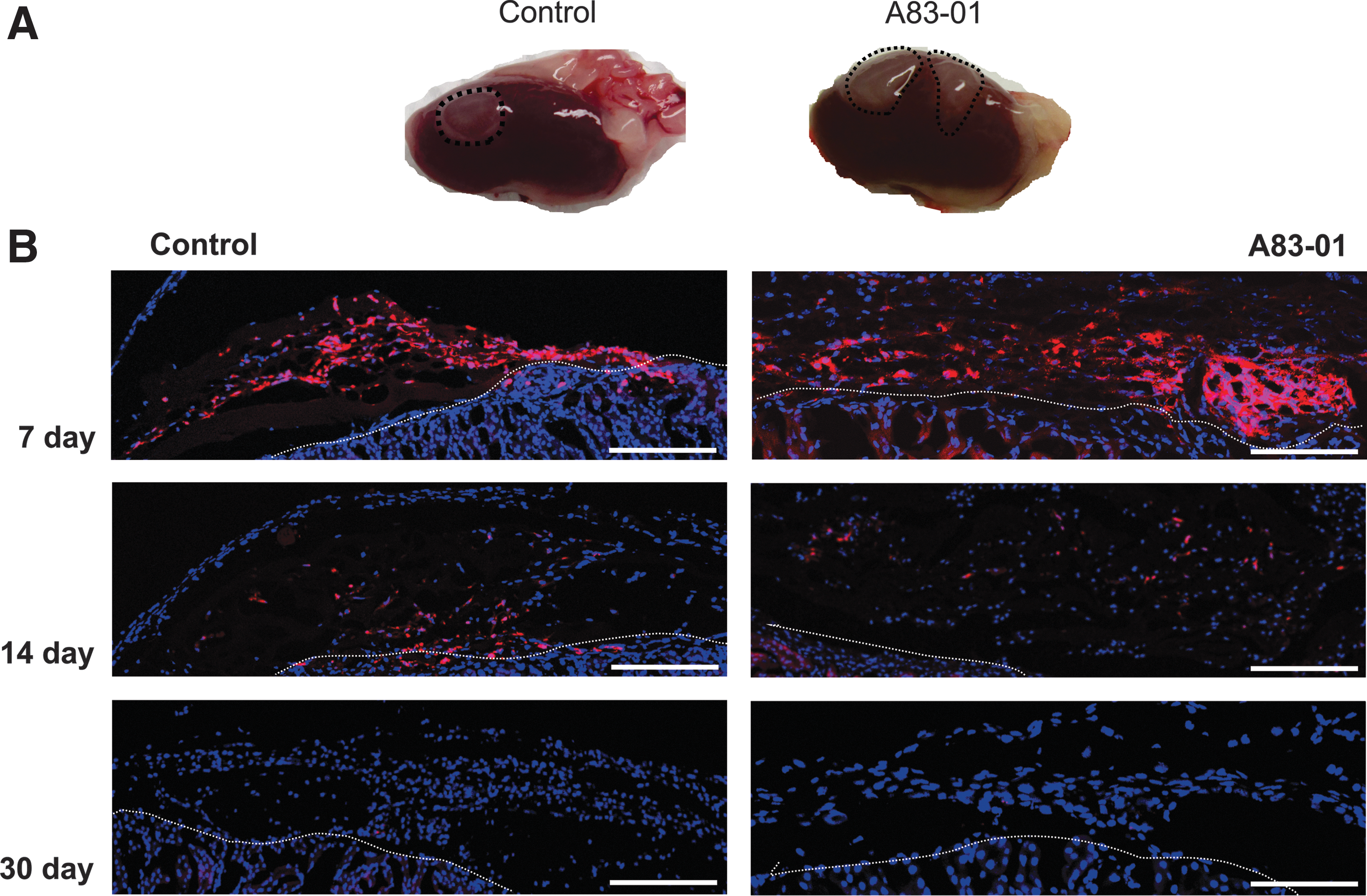
Human endometrial SUSD2+ mCherry positive cells in kidney tissue explants.
Discussion
This study demonstrated that human eMSCs can be efficiently transduced with mCherry using a lentiviral vector and that treatment with A83-01 enhances their survival in vivo compared to our previous study in immunocompromised rats [5]. Specifically, mCherry reporter gene transduction results in durable reporter expression in long-term eMSC culture (up to 4 weeks in vitro). We demonstrated, using three independent ex vivo methods, that eMSCs cultured in 5% O2 in SFM survive for at least 30 days in immunocompromised mice compared to our previous study where very few Dio+ cells were detected at only 14 days, although the human nature of those cells was not confirmed [5]. Our ex vivo data also show that treatment of eMSC with A83-01 in vitro has no detrimental effect on their survival in vivo.
eMSCs cultured in serum medium in normoxic conditions spontaneously differentiated and by P6 only 10% were still SUSD2+ [5]. Culturing eMSCs in 5% of O2 in SFM partially rescued this spontaneous differentiation [13]. In the present study, further improvements were made by continuous culture in 5% O2 and treatment with A83-01 in late passage cultures, resulting in a homogeneous population where >95% expressed the SUSD2 surface marker for each patient sample [11,13]. This has important implications for autologous use of eMSCs [6,17]. Culture-expanded MSCs used in research and clinical trials are often a heterogeneous population of cells with reduced functional properties, which may be one of the reasons for the variable outcomes observed in clinical trials [6,18]. Clinical outcomes can be improved by generating cells by culturing them in conditions that maintain their potency. However, it is important to demonstrate that A83-01 culture expanded cells have capacity to differentiate in vivo and do not generate tumors.
MSC therapies are proving to be beneficial [18]; however, there is still no clear evidence on how long they survive in vivo, primarily due to the lack of appropriate labeling of cells. Various cell labeling methods have been used to track human cells by genetically labeling them with reporter genes such as luciferase gene, GFP [19], mCherry [20], fluorescent proteins Katushka2S and IRFP [21], or directly labeling the cells with nanoparticles such as superparamagnetic iron oxide nanocomposites and NIR815 dye [22,23]. Genetic methods label cells and their progeny permanently, providing a distinct advantage over nonpermanent labeling, which is diluted as cells undergo proliferation or leak into the surroundings upon cell death and are taken up by host cells [5,20,22]. Lentiviruses are advantageous for permanent cell labeling with transduction approaches because of their ability to infect both dividing and nondividing cells, low immunogenicity, and high transduction efficiency [20,22,24]. Furthermore, the lentiviral mCherry transduction process does not affect the biological characteristics of human MSCs [4,20,25]. In our study, we showed that mCherry protein expression and intensity were maintained throughout a prolonged (4 weeks) cell culture period following transduction. Although there was a lag in proliferation following sorting of mCherry positive cells, eMSCs recovered rapidly and overall propagation was not affected. The lag phase could be due to the polybrene used to enhance the transduction process, which inhibits human MSC proliferation [26]. The concentration of polybrene used in this study (3 μg/mL) was optimized to balance between high transduction efficiency and maintaining cell proliferation. Permanent labeling of eMSCs using a lentiviral vector encoding the mCherry gene for this fluorescent protein provides an ex vivo way to investigate their survival, as well as transformation, differentiation, migration, and cellular interaction, in vivo in preclinical animal models of disease.
The human genome comprises ∼20% repetitive DNA sequences, one of which is Alu sequence [27,28]. It is the most abundant of the transposable elements interspersed throughout the human genome and makes up 13% of the human gDNA (∼1 million copies) [27,29]. By taking advantage of Alu sequence abundance, we could detect small numbers of human cells in tissues following transplantation for up to 30 days in mice transplanted with eMSCs pretreated with and without A83-01. They were detected predominantly in kidneys but also in skin and spleen. Since the fibrin gel is not completely sealed, some eMSCs may have escaped to nearby sites such as the incision and spleen during the procedure. This shows that the cells do not migrate systemically when fibrin gel is used. Although flow cytometry analysis did not show statistical significance, eMSCs could be detected in these local sites longer in the A83-01 treated eMSC group than the control, implying the possibility of increased survival following this in vitro pretreatment.
Human cells were not detected in tissues distant from the kidney, such as liver and uterus and the control kidney (by flow cytometry), indicating that for future clinical application, local delivery is possible and potentially safe in the clinical setting. The identity of transplanted cells was further confirmed by the detection of the mCherry transgene bands by qualitative PCR. However, we were not able to detect human mCherry eMSCs at later time points at the same detection level as Alu sequences. This may be because of the decreasing number of total surviving human mCherry positive eMSCs and also due to the low mCherry copy number per cell which fell below the limit of detection by PCR [20]. Delivering MSCs to a local site and confining them in a fibrin gel may keep them to the site of intended use compared to intravenous administration of MSCs. Cells delivered through intravenous route are also trapped in the lungs on the first pass and/or are lost in the circulation by an instant blood-mediated inflammatory reaction [3].
To test the survival of control or A83-01 treated eMSCs in vivo, further complementary ex vivo methods were utilized. mCherry positive cells were detected in both A83-01 treated and nontreated cell groups up until 30-day posttransplantation by flow cytometry. Interestingly, there was a significant decrease in the percentage of SUSD2+/mCherry+ eMSCs in both groups in the second fortnight following transplantation. By 30 days, <1% of human cells were detectable. This indicates that some of the implanted SUSD2 positive cells may have differentiated into SUSD2 negative fibroblasts because not all of the mCherry cells were SUSD2 positive. Even though mCherry cells were detectable at earlier time points using immunofluorescence staining, it was difficult to identify them at the 30-day time point, possibly due to their rarity. No mCherry+ migratory human cells were observed in our study, unlike those observed when primary SUSD2+ eMSCs were transplanted in the subrenal capsule of NSG mice [8]. This could be because Masuda et al. used primary SUSD2+ eMSCs, which were delivered in DMEM, as opposed to the current study using P6-cultured eMSCs in a fibrin gel. In addition, mice in the Masuda et al. study were hormonally regulated by ovariectomizing and treating with exogenous estrogen and progesterone, which could have promoted their differentiation into endometrial cell types.
A limitation of this study is the choice of immunocompromised animal model in assessing the survival of the A83-01 treated and nontreated eMSCs as it does not mimic the pathophysiological immune environment in patients who need cell therapy or regenerative medicine. However, to understand the role of MSCs alone and their interaction with innate immune cells, NSG mice are good model as their lack of natural killer (NK) cells enables longer persistence compared to other immunocompromised mouse models. Further studies using an immunocompetent animal model or disease models will provide further insight into whether pretreating eMSCs with A83-01 provides any added benefits for healing and repairing processes imperative for a cell therapy and clinical translation. In addition, our previous study is the first to target TGF beta signaling to prevent culture induced spontaneous differentiation of eMSCs [11]. Therefore, our first attempt was to target the pathway after the eMSCs had been in culture for a prolonged period and when there was evidence of substantial differentiation to fibroblasts.
In conclusion, this study demonstrates the ability to permanently and safely genetically tag eMSCs and shows their ability to survive longer than our previous study with our improved eMSC culturing protocol using a small molecule inhibitor. In fact, the persistence of transplanted eMSCs detected in this study is the longest they have been demonstrated to survive in vivo. Likewise, the decreased percentage of the SUSD2 positive population detected ex vivo indicates in vivo differentiation. The use of small molecule to maintain eMSC stemness had no long-term detrimental effects on cell persistence and may in fact be beneficial. Using A83-01 treated eMSCs in animal models may identify approaches that are suitable for clinical translation.
Footnotes
Acknowledgments
This work was supported by National Health and Medical Research Council (NHMRC) of Australia project grant 1081944 (C.E.G., J.A.W.), an NHMRC Senior Research Fellowship (1042298) (C.E.G.), a Monash Graduate Scholarship (S.G.), and the Victorian government's Operational Infrastructure Support Program.
Author Disclosure Statement
No competing financial interests exist.