
Abstract
Mobilization of mesenchymal stem cells (MSCs) is an attractive strategy for cell therapy. Our previous study demonstrated that MSCs can be mobilized in circulating blood by short-term hypoxia, and hypoxia-inducible factor-1α is essential for MSC mobilization. In the present study, the effect of the hypoxia-mimicking agent CoCl2 was examined on MSC mobilization. The results indicated that the frequency of circulating MSCs increased slightly by administration of CoCl2. However, the mobilization efficiency was low. Considering the critical role of stromal cell-derived factor-1α (SDF-1)/CXCR4 axis in the regulation of MSC migration, the effects of granulocyte colony-stimulating factor (G-CSF) and the CXCR4 antagonist AMD3100 were investigated on MSC mobilization. The experiments were notably demonstrated in animals preconditioned with CoCl2. The frequency of colony-forming unit fibroblast and the proportion of CD45−CD90+ cells did not significantly increase in the peripheral blood of rats treated with G-CSF and/or AMD3100 alone. The concomitant administration of G-CSF with CoCl2 could not stimulate the release of MSCs. However, AMD3100 dramatically increased MSC mobilization efficiency in rats pretreated with CoCl2. Furthermore, we identified and compared the multilineage differentiation capacities of MSCs derived from bone marrow (BM-MSCs) and mobilized peripheral blood (PB-MSCs). The results indicated that PB-MSCs exhibited higher osteogenic potential and lower adipogenic differentiation as compared with BM-MSCs. The findings may inform studies investigating mechanisms of the regulation of MSC mobilization and can aid in the development of clinically useful therapeutic agents.
Introduction
M
The mobilization of endogenous stem and progenitor cells is considered an appealing strategy for cell therapy [5 –7]. This approach has been established clinically using granulocyte colony-stimulating factor (G-CSF) to mobilize hematopoietic stem cells (HSCs) for bone marrow (BM) transplantation [8]. G-CSF is the most frequently used agent for HSC mobilization. Stromal cell-derived factor-1 (SDF-1)/CXCR4 axis is essential for the mobilization of HSCs induced by G-CSF [9] AMD3100, a neutralizing CXCR4 antagonist, which can induce the rapid release of HSCs into the circulation and acts synergistically with G-CSF in the induction of cell mobilization [10]. The mobilization of MSCs is also of considerable therapeutic interest. A number of studies have attempted to induce MSC mobilization using conventional agents, such as G-CSF and AMD3100. However, the results were largely disappointing [11 –14].
The previous study conducted by our group demonstrated that MSCs can be mobilized to the circulating blood by hypoxia, whereas the transcription factor hypoxia-inducible factor-1α (HIF-1α) was shown to be essential for hypoxia-induced MSC mobilization [15]. HIF-1 is a heterodimer composed of a constitutively expressed HIF-1β subunit, and a HIF-1α subunit, whose expression and transcriptional activity are precisely regulated by the cellular O2 concentration. Under normoxia, HIF-1α is hydroxylated by proline hydroxylase domain proteins (PHD) that are required for the binding of von Hippel–Lindau protein (VHL) and the recognition subunit of an E3 ubiquitin-protein ligase that targets HIF-1α for proteasomal degradation. Under hypoxia, PHD is inactivated, which in turn, inhibits the degradation and leads to the accumulation and stabilization of HIF-1α [16]. The identification of HIF-1α suggested that this target molecule might be used to explore mobilizing agents, such as cobalt chloride (CoCl2) [17]. Previous studies indicated that CoCl2 abolishes the HIF-1α-VHL interaction through the blockade of PHD activity through the metal iron-binding domain. The exposure of cells to CoCl2 inhibits HIF-1α ubiquitination and the stabilization of HIF-1α expression [18 –20]. The hypoxia preconditioning can be mimicked by CoCl2 treatment that stabilizes HIF-1α [21].
In the present study, the effects of the hypoxia-mimicking agent, CoCl2, were investigated on MSC mobilization. The number of MSCs in the blood circulation increased slightly following treatment with CoCl2. However, the mobilization efficiency was low. Considering the critical role of SDF-1/CXCR4 axis in the regulation of MSC migration, we speculated that the combination of G-CSF and/or AMD3100 (CXCR4 antagonist) might improve the mobilization efficiency of MSCs. Thus, the effects of G-CSF and AMD3100 were examined on MSC mobilization, especially in animals preconditioned with CoCl2.
Materials and Methods
Animals
Adult male Sprague–Dawley rats (6 weeks, 200–220 g) were provided by the Experimental Animal Center of the Zhejiang University. They were housed under controlled conditions of light and temperature with free access to water and food. All animal investigations were in accordance with the Guide for the Care and Use of Laboratory Animals published by NIH and approved by the Institutional Animal Care Committee of the Zhejiang University.
Grouping
(1) To investigate the CoCl2-induced mobilization effect on MSCs, rats were divided into four groups (five rats in each) namely, normal saline 3 days treatment group (NS 3 days), CoCl2 3 days treatment group (CoCl2 3 days), normal saline 7 days treatment group (NS 7 days), and CoCl2 7 days treatment group (CoCl2 7 days) group. Rats were administered CoCl2 (Sinopharm Chemical Reagent Co., Shanghai, China) every day, at a concentration of 10 mg/kg by intraperitoneal injection, whereas the rats in the control group were treated intraperitoneally with an equal volume of normal saline.
(2) To determine the combined effect of CoCl2-induced MSCs' mobilization in the presence of G-CSF or AMD3100, the rats were divided into six groups (five rats in each group) namely, (a) normal saline 7 days treatment group (NS 7 days group, rats received the same treatment as mentioned above), (b) CoCl2 7 days treatment group (CoCl2 7 days group, rats received the same treatment as mentioned above), (c) G-CSF group, (d) CoCl2 with G-CSF group, (e) AMD3100 group, and (f) CoCl2 with AMD3100 group. In group c, rats were administered an equal amount of NS on the first and second days, and G-CSF (100 μg/kg; Jiuyuan gene Co., Hangzhou, China) for 5 days starting from the third day, whereas in group d, rats were administered CoCl2 10 mg/kg/d for 2 days, and subsequently G-CSF 100 μg/kg at 2 h following intraperitoneal injection of CoCl2 10 mg/kg for 5 days. In group e during 1–7 days, rats were administered equal amounts of normal saline for the period of 1–7 days, and were treated with AMD3100 (5 mg/kg, Sigma-Aldrich, St. Louis, MO) by intraperitoneal injection on the eighth day. Peripheral blood (PB) was collected following 1 h of the injection. In group f, the rats initially received CoCl2 (10 mg/kg/d) for 7 days and were administered AMD3100 (5 mg/kg) on the eighth day by intraperitoneal injection. Peripheral blood was collected following 1 h postinjection. Subsequently, mononuclear cells (MNCs) were isolated from the peripheral blood and BM for the determination of the number of MSCs using a colony-forming unit fibroblastic (CFU-F) assay. The proportion of CD45−CD90+ cell population was estimated using flow cytometry.
BM and peripheral blood cell preparation
The animals of each group were anesthetized with 4% chloral hydrate (500 mg/kg; Sigma-Aldrich). Peripheral blood (PB) (8–10 mL) was collected from the postcaval vein in heparinized tubes. BM was obtained from rat femoral and tibialis tissues as described previously [15]. Briefly, the muscles with the entire connective tissue were detached and the epiphyses were removed. The marrow was harvested by inserting an 18-gauge syringe needle into one end of the bone shaft and flushing the contents into a 60-mm culture dish that contained Dulbecco's modified Eagle's medium–low glucose (LG-DMEM) (Invitrogen, Carlsbad, CA) supplemented with 20% (vol/vol) fetal bovine serum (FBS) (Invitrogen). A single cell suspension was obtained by passing the cell culture samples through needles of decreasing size. The MNCs derived from the PB and the BM were separated by Lymphoprep (Haoyang Biological Manufacture Co. Ltd, Tianjin, China).
CFU-F assay
For CFU-F assays, MNCs derived from 6 mL PB of each rat were placed in a 25-cm culture flask in the presence of a proliferation culture medium that consisted of LG-DMEM supplemented with 20% (vol/vol) FBS. The MNCs that were derived from the BM were plated at a density of 3 × 106 cells per 12.5-cm culture flask using the same proliferation culture medium. Adherent colonies (>50 cells) that were derived from CFU-Fs were counted on day 10 for the presence of PB cells and on day 14 for the presence of BM cells.
Flow cytometry assay
Following cell counting, the concentration of MNCs in the peripheral blood and the BM was adjusted to 107 cells/mL by the additions of PBS. A total of 100 μL was placed into a 1.5-mL EP tube. Concomitantly, monocolor and bicolor isotype control groups were prepared. The antibody staining for the flow cytometry assay was conducted as follows: CD90-PE (Santa Cruz Biotechnology, Santa Cruz, CA, 5 μL/tube) and CD45-FITC (Invitrogen, 2 μL/tube) were incubated in the dark and at room temperature for 30 min. The samples were rinsed twice with PBS, resuspended in 400 μL PBS, and finally added to the flow tubes for flow cytometric detection.
MSCs derived from the peripheral blood of rats, treated with CoCl2 and AMD3100 (PB-MSCs), were cultured in parallel with MSCs derived from BM of the untreated rats (BM-MSCs). Membrane antigen expression on PB-MSCs and BM-MSCs was determined at Passage 4 by flow cytometry assay. A total of 1 × 105 cells from single cell suspensions were incubated for 30 min at room temperature with monoclonal antibodies against rat antigens, including direct phycoerythrin- or fluorescein isothiocyanate-conjugated anti-rat monoclonal antibodies recognizing CD44, CD90, CD73, and CD45. Irrelevant isotype-identical antibodies served as a negative control. After washing, the samples were analyzed using CellQuest software.
Enzyme-linked immunosorbent assay
The BM that was derived from the left femur of each group was flushed 20 times with 1 mL of ice-cold PBS, following centrifugation at 1,000 g for 5 min. The flushing supernatant was collected and the concentration of vascular endothelial growth factor (VEGF) in the BM was analyzed by enzyme-linked immunosorbent assay (ELISA), using commercially available kits (Boster, Wuhan, China), according to the instructions provided by the manufacturer.
Western blotting
BM cells were flushed from two femurs using 1 mL ice-cold PBS containing 10 μM phenylmethanesulfonyl fluoride and 10 μL of 100 × protease inhibitor cocktail (Biovision, Mountain View, CA). Following centrifugation at 4°C, the nuclear protein from BM cells was isolated using the Nuclear Protein Extraction Kit (Biovision) according to the instructions provided by the manufacturer. The protein contents of the cell lysates were determined using the Micro BCA Kit (Thermo, Rockford, IL). The protein from the cell lysates was mixed with 4 × loading buffer containing 10 mM dithiothreitol and boiled for 10 min before electrophoresis on a 10% sodium dodecyl sulfate–polyacrylamide gel. Following transfer to polyvinylidene fluoride membranes and blocking, the membranes were incubated overnight at 4°C with 1:500 diluted rabbit anti-rat HIF-1α mAb (Thermo) and/or 1:1,000 rabbit anti-rat VEGF mAb (Santa Cruz) and/or 1:2,000 mouse anti-β-actin mAb (Sigma-Aldrich). Following several washes in TBST, the membranes were subsequently incubated for 1 h with horseradish peroxidase-conjugated goat anti-rabbit IgG antibody (Sigma-Aldrich) diluted as 1:3,000 in blocking buffer. The membranes were washed three times with TBST and analyzed by chemiluminescence (Thermo).
Generation and culture of MSCs
MSCs derived from BM and/or PB were trypsinized (0.25% trypsin-EDTA, Invitrogen) and resuspended in culture medium, consisting of LG-DMEM and supplemented with 20% (v/v) FBS. They were seeded at a density of 10,000 cells/cm2 and assigned as passage 1. The culture medium was subsequently replaced every 3 days. The cells used in subsequent experiments were of passage 4 (P4).
Differentiation assay
The differentiation potential capacities of the MSCs that were derived from mobilized PB and BM (P4) were evaluated as follows:
Osteogenesis
The cells were seeded into six-well plates in the proliferation medium at a density of 10,000 cells/cm2. Following 2 days of culture, the medium was replaced with osteogenic induction medium consisting of LG-DMEM with 10% FBS, 0.1 μM dexamethasone (Sigma-Aldrich), 10 mM β-glycerophosphate (Sigma-Aldrich), and 50 μM ascorbic acid (Sigma-Aldrich). The medium was replaced with fresh medium every 3 days. The expression levels of the osteogenesis-specific genes namely, Runx2 and Bglap2, were analyzed on 0, 3, and 7 days following induction by real-time RT-PCR. The mineralized areas were revealed by von Kossa stain.
Adipogenesis
The cells were seeded in six-well plates at a density of 20,000 cells/cm2 and cultured in the proliferation medium consisting of LG-DMEM and supplemented with 20% FBS. Confluent cultures were treated with adipogenic induction medium consisting of LG-DMED with 1× adipogenic stimulatory supplement (Stem Cell Technologies, Hangzhou, China) and further cultured for 21 days. The medium was exchanged every 3 days. On day 21, the cells were stained with Oil Red O (Sigma-Aldrich) stain. For quantitative analysis, the absorbance was detected at 570 nm after destaining with isopropanol for 30 min. The expression levels of the adipogenesis- related genes, PPARγ and LPL, were analyzed by real-time RT-PCR at 0, 3, and 7 days time periods following induction of adipogenesis.
Chondrogenesis
Cells at 80% confluency were trypsinized with 0.25% (vol/vol) trypsin-EDTA, washed in PBS, and resuspended in high-glucose DMEM containing 0.1 μM dexamethasone (Sigma-Aldrich), 1 mM sodium pyruvate (Invitrogen), 1× insulin–transferrin–selenium (Invitrogen), 200 μM ascorbic acid (Sigma-Aldrich), 10 ng/mL transforming growth factor-1 (PeproTech, London, United Kingdom), and 10 ng/mL transforming growth factor-3 (PeproTech). Viable cells were counted and seeded at a density of 5 × 105 cells per pellet in 15-mL conical tubes. Cells were centrifuged at 500 g to the bottom of the tubes and allowed to form compact cell pellets, then incubated in a humidified atmosphere at 37°C with 5% CO2, with medium changes every 3 days. Pellets were embedded in paraffin after 21 days in culture. Cartilage glycosaminoglycans were detected by staining with Toluidine Blue (Sigma-Aldrich). Sulfated glycosaminoglycan (sGAG) content was quantified using the Blyscan sGAG assay (Biocolor). The expression levels of the chondrogenesis-related genes, SOX9 and COL2A, were analyzed by real-time RT-PCR on 0, 7, and 14 days following induction.
Real-time PCR assay
Total cellular RNA was extracted using TRIzol reagent (Invitrogen) according to the protocol provided by the manufacturer. Total RNA (1 μg) was used for cDNA synthesis using the PrimeScript™ RT-PCR Kit (TaKaRa). Quantitative polymerase chain reaction (qPCR) was conducted in a Light Cycler system (ABI 7500) using SYBR Premix Ex Taq (TaKaRa). Each sample was tested in triplicate. The primer sequences are listed in Table 1.
Statistical analysis
All data are presented as mean ± SEM. Data were analyzed using routine homogeneity test of variance. The differences between the mean values of each group were assessed by analysis of variance for multiple comparisons using SPSS 16.0. Oil Red O quantification and sGAG assay was analyzed using Student's t-test. A P-value of less than 0.05 (P < 0.05) was considered significant.
Results
Administration of CoCl2 for 7 days increases the number of MSCs in peripheral blood
Initially, the CFU-F and flow cytometry assays were employed to detect the changes in the number of MSCs in the peripheral blood and the BM in rats that were successively administered CoCl2 at a dose of 10 mg/kg/d for 3 days and/or 7 days. The results indicated that the number of CFU-Fs in the peripheral blood and the BM in rats that received CoCl2 (10 mg/kg/d) for 3 days did not show significant differences compared with the control group (NS group). However, the number of CFU-Fs in the peripheral blood increased markedly in rats that received CoCl2 at a dose of 10 mg/kg/d for 7 days compared with the control group (2.37 ± 0.19 vs.1.27 ± 0.08 CFU-Fs/mL, P < 0.05) (Fig. 1A). Furthermore, the number of MSCs was increased in the BM of the CoCl2 7-day group (18.40 ± 1.50 vs. 12.80 ± 1.39 CFU-Fs/1 × 106, P < 0.05) (Fig. 1B). The results of the flow cytometry further revealed that the proportion of the CD45−CD90+cell population in the peripheral blood and BM were elevated in the CoCl2 7-day group compared with those of the control group (Fig. 1C, D).
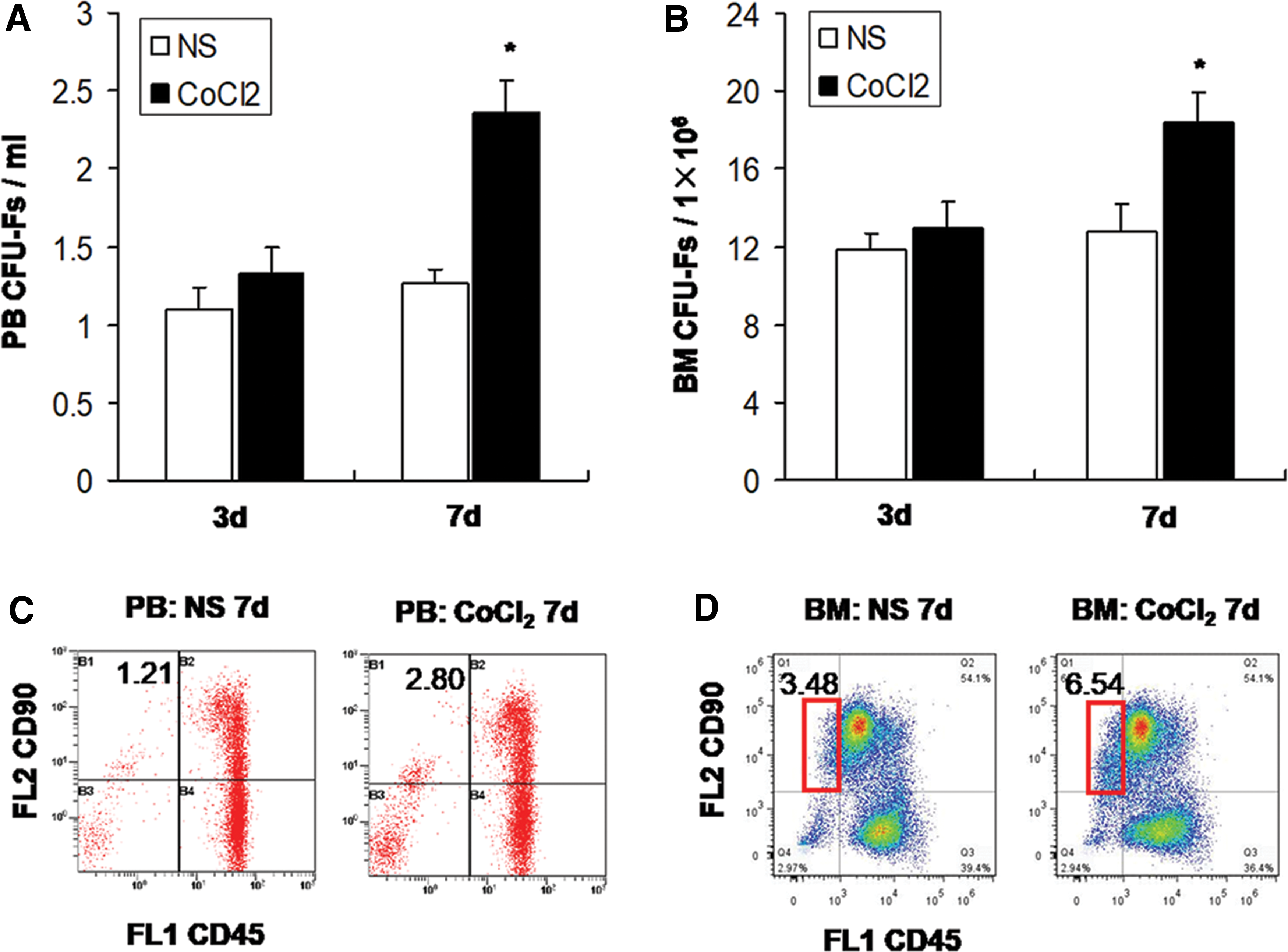
Mobilization effect of CoCl2 on MSCs in rats.
Administration of CoCl2 upregulates HIF-1α expression and increases VEGF concentration in the BM of rats
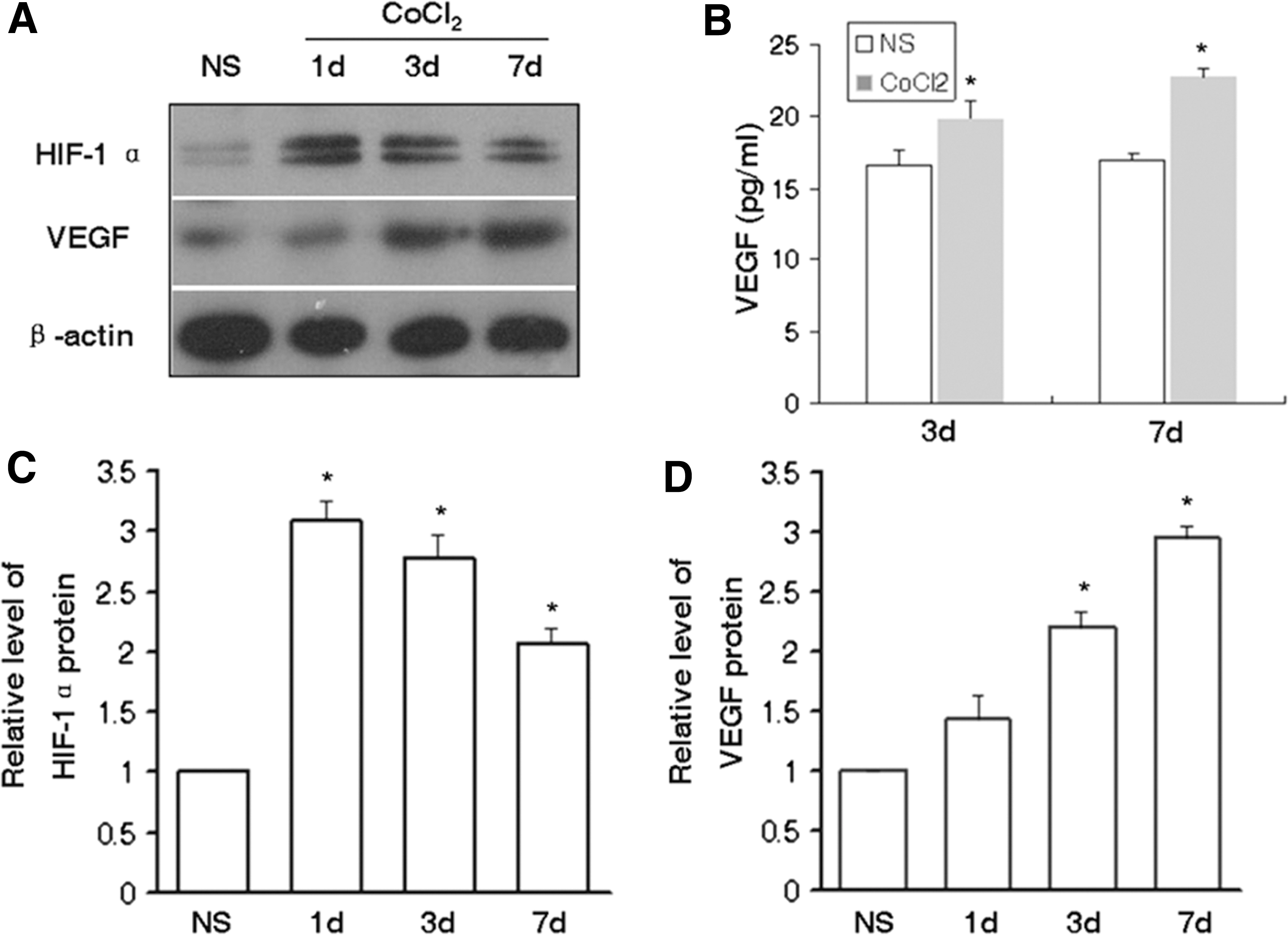
The expression of HIF-1α in rats that were treated with CoCl2 was investigated following the initial observations regarding the increase in the number of MSCs. As shown in Fig. 2A, C, HIF-1α was expressed at a low level in the control group, whereas the levels increased significantly in rats that received CoCl2 for 1, 3, and 7 days, especially in the CoCl2 1-day group, reaching a level of three-fold higher than the normal control. VEGF is a well-known HIF-1α downstream gene, and one of the most significant growth factors involved in the mobilization and migration of MSCs. Consequently, the concentrations of VEGF in the BM were examined. The results of the western blotting assays indicated that the expression of VEGF was significantly increased following administration of CoCl2 for 3 days, whereas VEGF levels remained at a high level during day 7 (Fig. 2A, D). Data derived by the ELISA assay further confirmed that the concentration levels of VEGF were significantly increased in rats that received CoCl2 for 3 and 7 days, respectively (Fig. 2B).
CXCR4 antagonist AMD3100 promotes MSCs mobilization in the peripheral blood of rats preconditioned with CoCl2
The aforementioned results indicated that the number of BM-MSCs increased in rats that were treated with CoCl2. Considering the critical role of the SDF-1/CXCR4 axis in MSC retention, we speculated that the combination of the agents G-CSF and/or the CXCR4 antagonist AMD3100 might promote the release of MSCs and enhance their CoCl2-induced mobilization efficiency. Therefore, the mobilization effects of G-CSF and AMD3100 combined with CoCl2 were tested on MSCs. Data demonstrated that the number of CFU-Fs in the peripheral blood did not significantly increase in rats treated with G-CSF and/or AMD3100 alone (P > 0.05, Fig. 3A). In addition, CoCl2 used in combination with G-CSF could not promote the release of MSCs. It is interesting to note that AMD3100 dramatically increased MSCs mobilization efficiency in rats that were pretreated with CoCl2. The number of CFU-Fs was increased significantly in combination with the CoCl2 and AMD3100 groups, compared with the CoCl2 group alone (3.83 ± 0.32 vs. 2.37 ± 0.19 CFU-Fs/mL, P < 0.05) (Fig. 3A).

Mobilization effect of CoCl2 combined with G-CSF and AMD3100 on MSCs.
In addition, the changes of CFU-Fs in the BM were detected. The number of CFU-Fs in the G-CSF group was elevated compared with the NS control group (16.4 ± 1.33 vs.12.8 ± 1.39 CFU-Fs/1 × 106, P = 0.075), whereas the corresponding number in the AMD3100 group was decreased compared with the normal control group (9.00 ± 0.71 vs.12.8 ± 1.39 CFU-Fs/1 × 106, P = 0.096). The differences, however, did not reach statistical significance. Furthermore, the number of the CFU-Fs in the BM of the CoCl2 with AMD3100 (Co+A) group was significantly decreased compared with that noted in the CoCl2 group (13.8 ± 1.07 vs.18.4 ± 1.50, < 0.05) (Fig. 3B).
The percentage of the CD45−CD90+ cell population was examined in each group using flow cytometry. Data revealed that treatment of rats with G-CSF or AMD3100 did not increase the proportion and number of the CD45−CD90+ cell population in the peripheral blood. However, the administration of CoCl2 for 7 days could elevate the proportion of the CD45−CD90+ cell population in the peripheral blood (2.96 ± 0.08 vs. 1.45% ± 0.28%, P < 0.05). The percentage of the CD45−CD90+ cell population in the peripheral blood was further increased in rats receiving CoCl2 with AMD3100 compared with the CoCl2 group (3.53 ± 0.17 vs. 2.96% ± 0.08%, P < 0.05).
The aforementioned results demonstrate that the CXCR4 antagonist AMD3100 promotes MSC mobilization in the peripheral blood of rats preconditioned with CoCl2.
The immunophenotype and differentiation potential of MSCs derived from mobilized PB of rats treated with CoCl2 and AMD3100
Immunophenotype of mobilized PB-MSCs is similar to those of BM-MSCs
The cell surface antigen expression of adherent cells derived from the peripheral blood of rats treated with CoCl2 and AMD3100 (PB-MSCs) was detected by flow cytometry. PB-MSCs were positive for CD90 (Thy-1) and CD44 (homing-associated cell adhesion molecule) and moderately expressed CD73 (cluster of differentiation 73), but were negative for CD45 (leukocyte common antigen) (Fig. 4). Therefore, the cell surface immunophenotype of PB-MSCs was therefore comparable to that of MSCs derived from BM of the untreated rats (BM-MSCs).
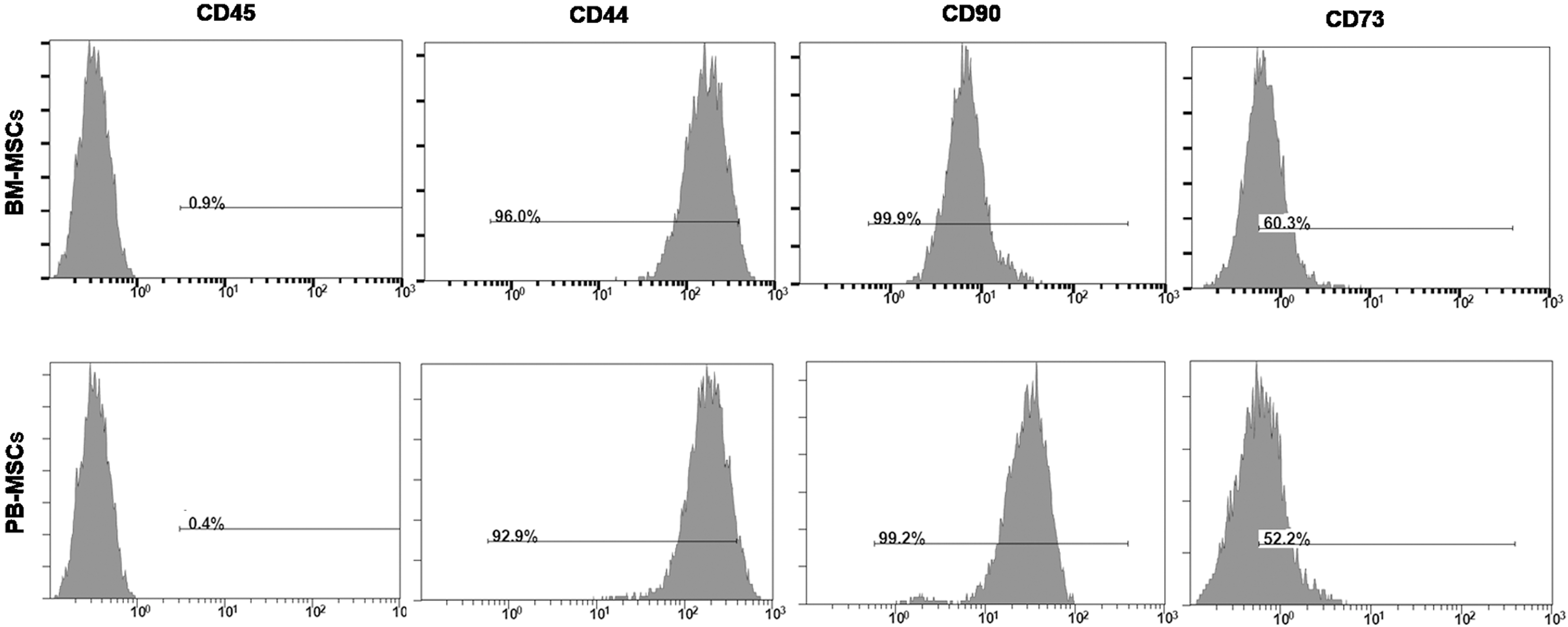
Immunophenotype of PB-MSCs and BM-MSCs. Membrane antigen expression on PB-MSCs and BM-MSCs was determined at passage 4 by flow cytometry. Gray areas represent the fluorescence intensity with specific antibodies (Abs) for membrane antigen. BM-MSC, bone marrow–mesenchymal stem cell; PB-MSC, peripheral blood–mesenchymal stem cell.
PB-MSCs exhibit a potent osteogenic differentiation capacity compared with BM-MSCs
PB-MSCs were cultured in parallel with BM-MSCs and the differentiation capacity was examined. The homogeneous layer of the mobilized fibroblast-like cells that were derived from PB (Fig. 5Aa) was similar to that noted in the BM (Fig. 5Ab). The morphological observation indicated that PB-MSCs contained traces of calcium salt during the 7 days of osteogenic induction (Fig. 5Ad), whereas BM-MSCs exhibited less calcium salt secretions (Fig. 5Ac). On day 14 of the differentiation, additional traces of calcium salt were noted in the BM-MSC group (Fig. 5Ae), whereas the differentiation efficiency was higher in the PB-MSC group (Fig. 5Af). Furthermore, von Kossa staining revealed a greater extent of mineralization with more detectable bone nodules in osteogenic culture of PB-MSCs than in that of BM-MSCs (Fig. 5Ag, h).
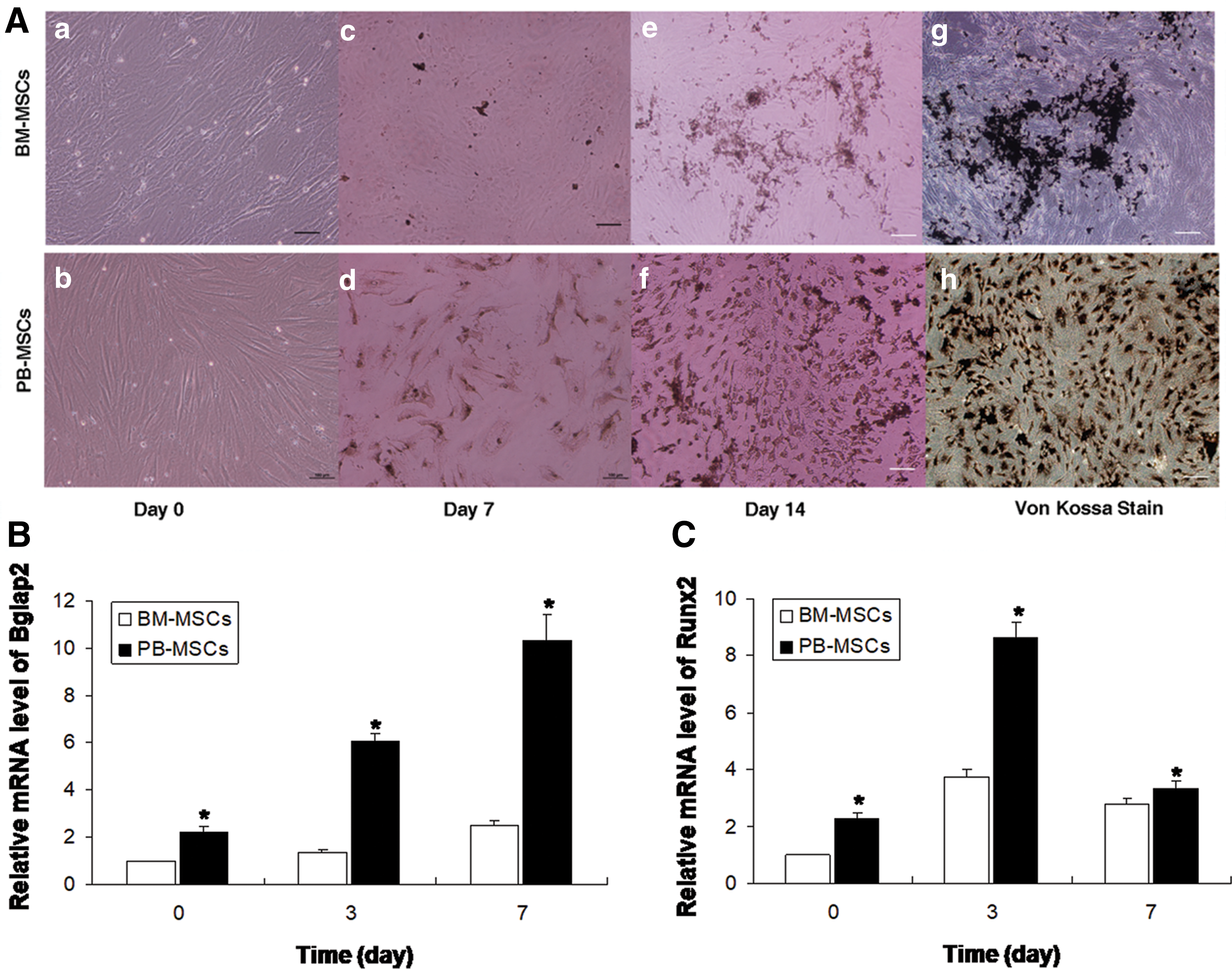
In vitro osteogenic potential of the PB-MSCs and BM-MSCs.
On 0 day following induction of differentiation, the expression levels of the genes Bglap2 and Runx2 in the PB-MSC group were significantly higher than those in the BM-MSC (Fig. 5B, P < 0.05) group. This finding confirmed that the PB-MSC group had a higher baseline expression level of the osteogenic differentiation-related genes. On 3 days following induction of differentiation, Bglap2 mRNA expression levels increased significantly in the PB-MSC group. This increase was noted until day 7. In addition, the Bglap2 expression at each differentiation time point was significantly increased in the PB-MSC compared with the BM-MSC groups (P < 0.05). In contrast to Bglap2, Runx2 expression presented a consistent trend of expression at each time point in both groups, although it was significantly increased at day 3 following the induction of differentiation in the PB-MSC compared with the BM-MSC groups (P < 0.05). The results revealed that PB-MSCs exhibited more potent osteogenic differentiation potential compared with the BM-MSCs.
PB-MSCs exhibit a weaker adipogenic differentiation capacity compared with BM-MSCs
Following induction of adipogenic differentiation for 7 days, the cells of the PB-MSC and BM-MSC groups indicated round or polygonal shapes and irregular arrangement. Following induction of adipogenic differentiation for 21 days, the clustered high-refractive lipid droplets were evident in the cytoplasm of both groups of cells. The droplets appeared orange by Oil Red O staining. Interestingly, the PB-MSCs group displayed a lower percentage of fat-containing cells compared with the BM-MSCs group (Fig. 6A). Quantitation of dye content in the adipogenic cultures confirmed the histochemical observations (Fig. 6B).
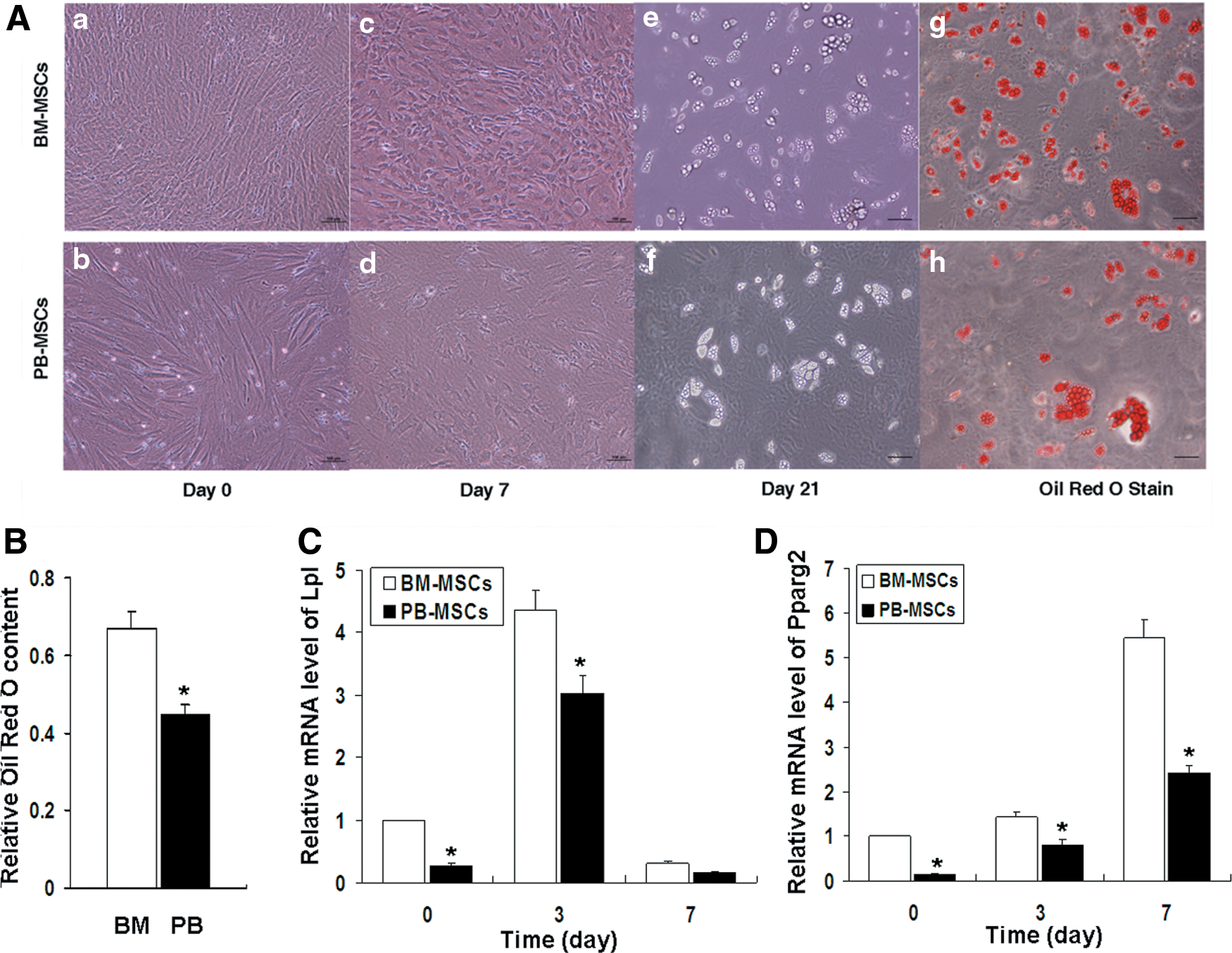
In vitro adipogenic differentiation potential of the PB-MSCs and BM-MSCs groups.
The mRNA analysis (Fig. 6C, D) revealed that on 0 day following induction of differentiation, the expression levels of the Lpl and Pparg2 genes in the PB-MSC group were lower than those in the BM-MSC group (P < 0.05). This indicated that the PB-MSC group exhibited a lower baseline expression level of adipogenic differentiation-related genes. On day 3, Lp1 expression in the PB-MSC group was significantly increased compared with that on day 0, although it was still lower than that in the BM-MSC group (P < 0.05). In addition, Pparg2 mRNA expression levels at different time points were significantly lower in the PB-MSC compared with the BM-MSC groups (P < 0.05). The results revealed that PB-MSCs exhibited weaker adipogenic differentiation potential, compared with the BM-MSCs.
PB-MSCs and BM-MSCs have comparable chondrogenic differentiation potential
As a pellet culture system, in vitro chondrogenesis was performed to evaluate the chondrogenesis potential of PB-MSCs and BM-MSCs. During chondrogenesis, the pellets increased in size due to the production of extracellular matrix. On day 14 of differentiation, the pellet sizes of PB-MSCs and BM-MSCs were similar (data not shown). Histologically, each cell pellet displayed a cartilage-specific metachromasia with Toluidine Blue staining. Moreover, the staining intensities of Toluidine Blue were similar between PB-MSCs and BM-MSCs (Fig. 7A). The biochemical analysis also showed similar sGAG levels (P > 0.05, Fig. 7B).
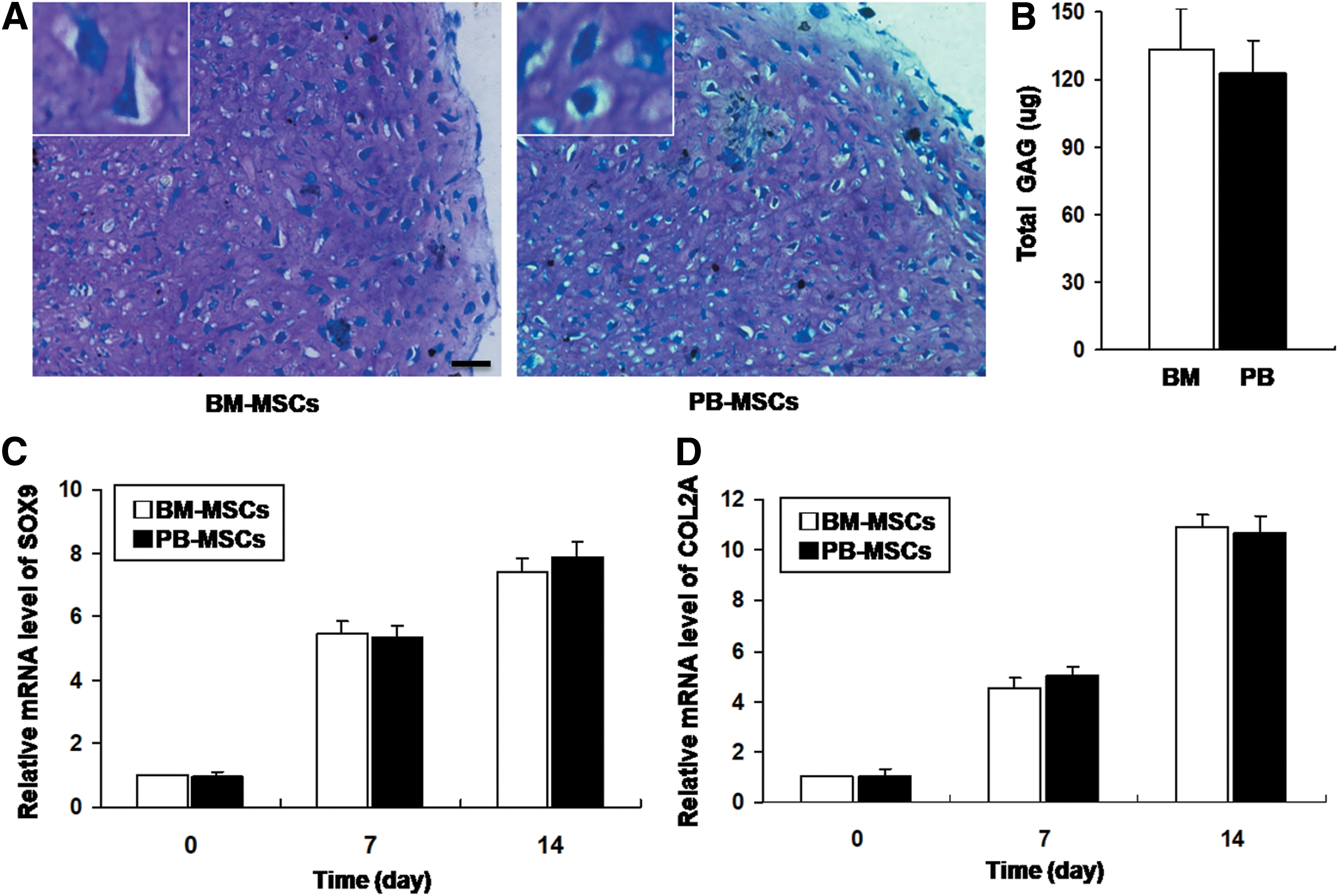
In vitro chondrogenic differentiation potential of the PB-MSCs and BM-MSCs groups.
The chondrogenic differentiation markers, SOX9 and COL2A, were analyzed by quantitative RT-PCR. The results indicated that the mRNA levels of SOX9 and COL2A accumulatively increased by the induction for 0, 7, and 14 days (Fig. 7C, D), which also suggested the effective induction. However, the mRNA expression of SOX9 and COL2A was comparable between the two groups (P > 0.05). The above results indicated that the BM-MSCs and PB-MSCs had a comparable chondrogenic differentiation potential.
Discussion
Hypoxia is a significant environmental factor that affects the biological processes associated with MSCs namely, proliferation, differentiation, and migration [22]. The previous studies conducted by our group demonstrated that MSCs can be mobilized in circulating blood by hypoxia through the induction of the transcription factor HIF-1α [15]. CoCl2 is a hypoxia-mimicking agent that has been used to activate the HIF-1α signaling pathway in a variety of cells. CoCl2 chelates Fe2+ in cells, and suppresses the activity of the prolyl hydroxylase enzyme, thereby decreasing the degradation of HIF-1α under normoxic condition [23,24]. Kudo et al. reported that preconditioning with CoCl2 increased the expression of HIF-1α and improved the migration of transplanted MSCs in an acute kidney injury model [19]. Moreover, recent studies have demonstrated that CoCl2 promoted the proliferation and migration of MSCs in vitro [25,26]. In the present study, MSCs were mobilized in the peripheral blood of rats by continuous administration of CoCl2 at a dose of 10 mg/kg for 7 days.
Whether MSCs can be isolated from peripheral blood remains controversial [13]. The discrepancies noted in different studies may be attributed to the relatively low frequency of MSCs in the circulation. In the present study, we report that low levels of MSCs were detected in the PB of normal rats at a steady state. Moreover, the circulating blood MSC pool was dramatically increased in animals treated with CoCl2 for 7 days. Data further demonstrated that the number of BM MSCs increased slightly in the CoCl2-treated group, suggesting that CoCl2 also influences the BM pool while inducing MSC mobilization. A large number of studies have shown that hypoxia (1–2% O2) promotes the proliferation of MSCs [22]. Thus, CoCl2 might induce a similar hypoxic environment, which results in an increase in MSCs in BM.
The mechanism of CoCl2-induced MSC mobilization into the bloodstream remains to be determined. Our previous study demonstrated that MSCs can be mobilized in circulating blood by short-term hypoxia and HIF-1α was upregulated during hypoxic exposure. Inhibition of HIF-1α expression by YC-1, a widely used HIF-1α inhibitor, remarkably reduced the number of mobilized MSCs, suggesting that HIF-1α is essential for hypoxia-induced MSC mobilization [15]. The results of the present study further revealed that the administration of CoCl2 could stably upregulate HIF-1α protein in the BM cells of rats. The mobilization of MSCs by CoCl2 was accompanied by an increase in HIF-1α, implying that HIF-1α may play a role in this process.
The results indicated that CoCl2 increased the number of MSCs in the circulation. However, the mobilization efficiency was low. Considering the critical role of the SDF-1/CXCR4 axis in the regulation of MSC migration and homing [27 –29], it was hypothesized that the combination of the agents G-CSF and/or AMD3100 might improve the mobilization efficiency of MSCs. G-CSF has been clinically applied to mobilize HSCs for the efficient HSC collection from PB. A number of studies have attempted to induce MSC mobilization using G-CSF. However, limited successes have been reported on the isolation of G-CSF-mobilized MSCs from the PB [13,14,30]. The results of the present study indicated that G-CSF alone could not induce MSC mobilization into circulation. This finding is consistent with the majority of previous studies.
AMD3100 is a blocker of the SDF-1 receptor CXCR4, which can competitively bind to CXCR4 and effectively block the SDF-1/CXCR4 axis [10]. Increasing data have indicated that AMD3100 can rapidly induce the mobilization of HSCs both in patients and in healthy donors [31,32]. Interestingly, results of clinical trials have shown that AM3100 plus G-CSF allows the collection of a high yield of HSCs with fewer apheresis sessions in patients with non-Hodgkin's lymphoma (NHL) and multiple myeloma (MM) [33,34]. AMD3100 (plerixafor/Mozobil®) was recently approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for use in combination with G-CSF to mobilize HSCs in patients with NHL and MM. Some studies have further revealed that AMD3100 can induce mobilization of other CXCR4+ cells, such as endothelial progenitor cells [35,36]. Recent findings indicated that combination of growth factors with AMD3100 could modulate the stem cell peripheral proliferation and mobilization [37,38]. In the present study, the single administration of AMD3100 could not significantly increase the number of MSCs in the PB of rats. However, the administration of AMD3100 in rats that received CoCl2 for 7 days could significantly increase MSC mobilization efficiency. Concomitantly, the number of MSCs in the BM was decreased in AMD3100-treated groups. The aforementioned results indicated that AMD3100 may promote the release of MSCs to circulation.
The results of present study indicated that MSCs cannot be mobilized by G-CSF or AMD3100 alone, implying that the mechanisms of MSC mobilization are probably different from those involved in HSC mobilization. Mobilization of stem and progenitor cells is a multistage process and related to a number of direct or indirect mechanisms [39]. Local factors involved in the retention of stem cells at their specific niches are proven importantly responsible for mobilization. HSCs mainly reside in a perivascular niche in the BM, which is often localized near the trabecular bone [40]. L molecules, such as VCAM-1, SDF-1, SCF, and their receptors are essential for HSC retention. Among these factors, SDF-1 and its sole receptor CXCR4 are speculated to be critical [41]. Although the disruption of the SDF-1/CXCR4 chemotactic interaction within the BM is the primary mechanism underlying HSC mobilization induced by G-CSF and AMD3100, the effects on HSC niches are distinct. G-CSF induces gradual proteolytic degradation of BM SDF-1 through neutrophil elastase, accompanied by an increase in its receptor CXCR4 in the BM. In addition, the levels of this chemokine and its receptor in the circulation were not affected. The decrease in BM SDF-1 that formed a gradient between BM and peripheral blood was correlated with stem cell mobilization [42]. However, AMD3100 increased the release of the SDF-1 in the circulation. Hence, HSCs are capable of being mobilized in response to the elevated SDF-1 levels in the circulation following AMD3100 treatment [43]. In this study, we found that AMD3100, but not G-CSF, increased the MSC mobilization in rats that were pretreated with CoCl2, suggesting diverse mechanisms.
The present results indicated that AMD3100 dramatically increased the MSC mobilization in rats pretreated with CoCl2, implying that SDF-1α/CXCR4 axis also plays a role in this process. Thus, CXCR4 might be speculated to be expressed on the cell surface of MSCs at a low level. A recent study indicated that the expression level was significantly improved and the CXCR4+ MSC population increased in the CoCl2-induced hypoxia treatment [44]. BM is the richest and most reliable reservoir for MSCs. In the hypoxic niche, AMD3100 might block the binding of SDF-1 and CXCR4, consequently promoting the egress of MSCs from BM to circulation. In addition, we found that AMD3100 dramatically increased the mobilization of MSCs in the presence of CoCl2, whereas the number of BM CFU-Fs in the same group was decreased significantly. Consistent with our results, Wise et al. [45] also revealed that AMD3100 administration leads to a significant decrease in the number of BM CFU-Fs during hypoxia (5% O2). AMD3100 rapidly induced the mobilization of MSCs into circulation under hypoxia, which might lead to a decrease in BM MSCs. However, the mechanisms underlying the synergistic effect of CoCl2 and AMD3100 are complex and require further investigation.
Despite the increased knowledge of MSCs during the past years, the exact location of MSCs remains elusive. New insights and evidences suggest that perivascular zone is the MSC niche in vivo [46,47]. The results from the present study together with those from previous studies indicated that hypoxia is a sensitive stimulus inducing MSC mobilization [15,39]. VEGF, a cytokine known to induce vasodilation and vascular permeability, is regulated by hypoxia [48]. Thus, the concentrations of VEGF in the BM were examined in this study. The results showed that the VEGF concentration in the BM increased following administration with CoCl2 in the rats for 3 days. The increase was higher at 7 days. Previous studies have suggested that VEGF is one of the major regulating factors required for the mobilization of MSCs [37,38]. We have previously demonstrated that VEGF may increase the angiogenesis in BM niche [15]. These evidences imply that VEGF might be an important factor regulating the mobilization of MSCs.
In the present study, the multilineage differentiation potential of MSCs from the BM and the PB were identified and compared. PB-MSCs exhibited potent osteogenic potential, whereas they demonstrated weaker adipogenic differentiation compared with BM-MSCs. The mobilized PB-derived MSCs possessed superior osteogenic potential suggesting their use for bone regeneration therapies in clinical practice. However, the differences of MSCs from mobilized PB and BM are yet unclear. The different characteristics might be attributed to the following: (1) The effect of CoCl2 and/or AMD3100 exposure. Although we used MSCs of passage 4 to examine the differentiation potential, the influence of chemical agents in vivo could not be excluded. Yoo et al. [49] recently demonstrated that the preconditioning of MSCs with CoCl2 enhanced the osteogenic differentiation and inhibited the adipogenesis in vitro. (2) The heterogeneity of MSCs. PB-MSCs and BM-MSCs might be derived from different subclones, resulting in diverse differentiation potentials [50]. (3) All the circulating MSCs may not originate from BM, but partially from other non-BM tissues, although the phenotype of PB-MSCs is comparable to that of BM-MSCs. (4) The physiological characteristics of MSCs may change spontaneously when detached from the BM niche. Nevertheless, our knowledge on PB MSCs is yet very limited, and thus, additional studies are essential to clarify the biological behaviors.
Strategies to enhance the mobilization of MSCs into the bloodstream are of great therapeutic interest as they would facilitate the collection of MSCs from the PB and their subsequent homing to injured tissues. In this study, we identified an effective protocol that selectively mobilizes the MSCs. However, it should be emphasized that there are questions that remain to be answered before the introduction of new mobilization regimes into the clinic.
In conclusion, the CXCR4 antagonist AMD3100 promoted MSCs mobilization in the PB of rats preconditioned with the hypoxia-mimicking agent CoCl2. The findings may add an insight in the mechanism underlying the regulation of MSC mobilization and aid the development of clinically useful therapeutic agents.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81500114, 31570994, and 81520108002), National key Basic Research Program (2015CB964900), and Zhejiang provincial grants (2012ZDA015, LY15C100001).
Author Disclosure Statement
The authors declare no competing financial interests.