
Abstract
Haploid cells facilitate genetic screening of recessive mutations for a single set of chromosomes. Haploid embryonic stem cells (haESCs) have been achieved in several species and widely utilized in genetic screens. The fact that haESCs undergo substantial diploidization during differentiation has limited the screening to other haploid cell types. In this study, we report a method to establish haploid neural stem cells (haNSCs) by selection for a Pax6 reporter. We inserted a green fluorescence protein (GFP) marker gene by homologous recombination into the Pax6 locus of an haESC line. GFP-positive haploid cells could be sorted and further cultured in the NSC medium for more than 30 passages. The established haNSCs expressed neural lineage markers and could differentiate into neurons, oligodendroglia, and astrocytes. Our study shows the feasibility of deriving haploid proliferative somatic cell lines using a genetically encoded reporter that suggest a system for genetic screening of neural and retinal development.
Introduction
H
haESCs have been established in several mammalian species, including rodents and primates [4 –6] facilitating forward and reverse genetic screening of gene function [7,8]. Notably, human haESCs have been reported last year [9,10], which provide a valuable platform to study the genetics of human diseases [3]. However, haESCs undergo diploidization during self-renewal and differentiation, and periodic sorting for haploid cells is essential, although the exact mechanism of diploidization remains unknown.
Although haESCs possess a pluripotent differentiation potential, diploidization has been a major impediment for the generation of haploid differentiated cell types. Proliferative somatic haploid cell types are highly desirable for genetic screening of lineage specification and target gene discovery for drug action and resistance. Previously, haploid differentiated cells have been detected transiently both in vivo and in vitro [11], but terminal differentiated haploid cells have not been achieved until very recently [12]. In addition, the properties of differentiated haploid cells were not determined.
Neural stem cells (NSCs) are multipotent and can differentiate into a variety of neurons and glia cells. NSCs can be obtained from ESCs and self-renew in culture in response to epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF) [13]. NSCs are also of interest for screening of pathways of clinical relevance.
In this study, we use a genetically encoded reporter for obtaining haploid neural stem cell (haNSC) cultures. Pax6 is a transcription factor that is expressed during neurogenesis specifically of the developing central nervous system [14]. Neural-specific green fluorescent protein (GFP) expression can be applied to trace neural progenitor cells (NPCs) during differentiation of haESCs [15]. We introduced a Pax6-GFP reporter into an haESC line by CRISPR/Cas9-mediated gene targeting [16,17]. haNSCs were derived from differentiated Pax6-GFP haESCs by fluorescence-activated cell sorting (FACS) purification and further cultured in NSCs medium. These haNSC cultures maintained haploidy and neuronal differentiation potential over extensive passages suggesting a system for lineage-specific genetic screening.
Materials and Methods
Mice
All the animals were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. and housed in Nankai University Animal Center. All experiments are guided by the Institutional Animal Care and Use Committee of Nankai University Animal Center. Cell lines in this work were derived from the housed mice and followed the guidance of animal care and use.
Derivation of haESCs
Oocytes were collected from superovulated 129Sv/Jae female mice and activated in CZB medium [18] containing SrCl2 for 6 h. Activated parthenogenetic haploid embryos were cultured in KOSM/AA medium (Millipore) [19] at 37°C, 5% CO2 until morula stage. To derive haESCs, the embryos that reached morulae were transferred to modified serum medium in feeder-cell precoated four-well plates.
The modified serum medium consists of Dulbecco's modified Eagle medium (DMEM)/F12 (Gibco) supplemented with 10% knockout serum replacement (KOSR; Gibco) and 7% fetal bovine serum (FBS; BI), 1 mM sodium pyruvate (Sigma), 100 μM β-mercaptoethanol (Sigma), 100 mg/mL Strep-Pen (Gibco), 1,500 U/mL leukemia inhibitory factor (LIF), 3 μM CHIR99021 (Gene-operation), and 1 μM PD0325901 (Gene-operation). For purifying haESCs, haESCs were incubated with 7.5 μg/mL Hoechst 33342 (Invitrogen) for 20 min at 37°C. Subsequently, the haploid (1n) peak was gated to sort cells with a diploid control on BD FACS Aria II (BD Biosciences).
Plasmid construction, transfection, and PCR verification
The reporter cassette was designed to fuse the GFP reading frame with the coding region sequence (CDS) of Pax6 through a T2A sequence that encodes a self-cleaving linker peptide [20]. The T2A-eGFP-bGHpA and SV40-neoR-SV40pA fragments were PCR amplified from pcNDA3.1 (+) (Addgene), and Pax6-HAL and Pax6-HAR were amplified from the genomic DNA of 129Sv/Jae mice. All the fragments were inserted into the pEASY-Blunt Simple plasmid using restriction enzyme ligation method. The vector pSpCas9n (BB) −2A-GFP (PX461; Addgene) was used to express Cas9n and sgRNA. The sequence of sgRNAs were designed referring to Feng Zhang's Laboratory website (
Transfections were performed using a NEON (Invitrogen) instrument set to 1300 V, three pulses, and 10 ms. When performing PCR verification, we used three pairs of primer*-P1 Forward, P1 Reverse (for amplifying the wild-type Pax6 arm); P2 Forward, P2 Reverse (for amplifying the 5′ arm); and P3 Forward, P3 Reverse (for amplifying the 3′ arm). Then we purified the PCR product of P1 and sequenced it. All the primers and sgRNA-targeting sequences used in vector construction were listed in Supplementary Table S1 (Supplementary Data are available online at
Neural differentiation of haESCs
To generate haploid neural progenitor cells (haNPCs), haESCs with high percentage of haploid cells were selected for the differentiation. To form embryonic bodies (EBs), haESCs were cultured with EB medium in noncoated Petri dishes (Falcon) for 4 days. Briefly the EB medium contains DMEM/F12 (Gibco) supplemented with 20% KOSR, 1 mM sodium pyruvate (Sigma), 100 μM β-mercaptoethanol (Sigma), and 100 mg/mL Strep-Pen (Gibco). The EB aggregates were plated on fibronectin-precoated Petri dish on fourth day with medium changing to N2B27 medium (TaKaRa). After 10 days, GFP-positive haploid cells were sorted by FACS machine according to the cell size and green fluorescence. Harvested haNPCs were further cultured in N2B27 medium supplemented with 10 ng/mL EGF (PeproTech) and 10 ng/mL bFGF (PeproTech) for proliferation.
Neuronal specification of haNSCs
To address whether haNSCs could differentiate to neural subtypes with a haploidy genome, neuronal and oligodendrocyte differentiation were introduced in haNSCs for 14 days according to previous report [21] with slight modification. For characterizing the NSC properties, haNSCs were suspended in noncoated dishes to form floating aggregates in the differentiation medium.
For neuronal differentiation, haNSCs were plated into Poly-D-lysine (PDL) (Sigma) and Laminin (Sigma)-precoated dishes and cultured in N2B27 medium supplemented with 10 ng/mL brain derived neurotrophic factor (BDNF) and 10 ng/mL neurotrophins-3 (NT-3) to generate neurons. At 2 weeks, the differentiated NSCs were examined for expression of neuronal (Tuj1, MAP2, and NeuN) and oligodendroglial (O4) markers. For astrocyte differentiation, haNSCs were exposed to 1% FBS and bone morphogenetic protein 4 (10 ng/mL) in N2B27 medium for 5 days followed by glial fibrillary acidic protein (GFAP) staining.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde (Sigma) overnight at 4°C and then permeabilized with 0.3% Triton X-100 (Sigma) for 1 h at room temperature (RT). Nonspecific sites were blocked with 3% bovine serum albumin for 1 h at RT. Primary antibodies, including anti Nestin (Abcam), Sox1 (Abcam), Pax6 (Abcam), MAP2 (Abcam), Tuj1 (Abcam), NeuN (Abcam), O4 (RD), and GFAP (Abcam), were incubated with the samples overnight at 4°C. The cells were treated with secondary antibody with fluorescence for 1 h at RT, respectively.
Karyotype analysis
haESCs or haNPCs were incubated with 0.2 μg/mL nocodazole (Sigma) overnight. After trypsinization, the haNSCs were suspended in KCl (0.075 M) at 37°C for 30 min. The samples treated with hypotonic solution were fixed in methanol:acetic acid (3:1) for 20 min twice and dropped onto precooled slides. Cells were stained with Giemsa for 15 min before observation.
Quantitative PCR
Total RNA was extracted from cells using TRIzol Reagent (Invitrogen), and cDNA was synthesized using the Prime Script™ RT Reagent Kit with gDNA Eraser (TaKaRa). Quantitative PCR was performed on ABI QuantStudio™ 6 Flex machine with FS Universal SYBR Green Master (Roche). Relative expression levels were normalized to Gapdh. Average and standard deviation are of three independent experiments. All primers are shown in Supplementary Table S1.
Results
Derivation of Pax6-GFP haESCs by combining CRISPR and homologous recombination
To derive haESCs with robust pluripotency, we used a modified serum medium [22] supplemented with chemical inhibitors of GSK3 and MEK kinases (2i) and LIF. Haploid embryos were prepared from superovulated 129Sv/Jae female mice. Parthenogenetic haploid embryos were generated by activation of the collected oocytes using SrCl2. Embryos were subsequently cultured to the morula stage in KSOM/AA medium. Four morulae developed from which one haESC line (129PH-1) was obtained (Supplementary Fig. S1A) and expressed pluripotent markers, including OCT4, NANOG, and SSEA-1 (Supplementary Fig. S1B).
Alkaline phosphatase staining further confirmed a pluripotent cell identity (Supplementary Fig. S1C). The 129PH-1 cells showed a typical domed colony morphology (Fig. 1A). The haESCs could be expanded and maintained haploidy with occasional sorting of the 1n population using Hoechst33342 staining (Fig. 1B, C). For introducing a GFP marker into the Pax6 gene, we constructed a targeting vector containing left and right homology arms, the enhanced green fluorescent protein (eGFP) reading frame, and an SV40-driven neomycin resistance gene (neoR) (Fig. 1D and Supplementary Fig. S1D).
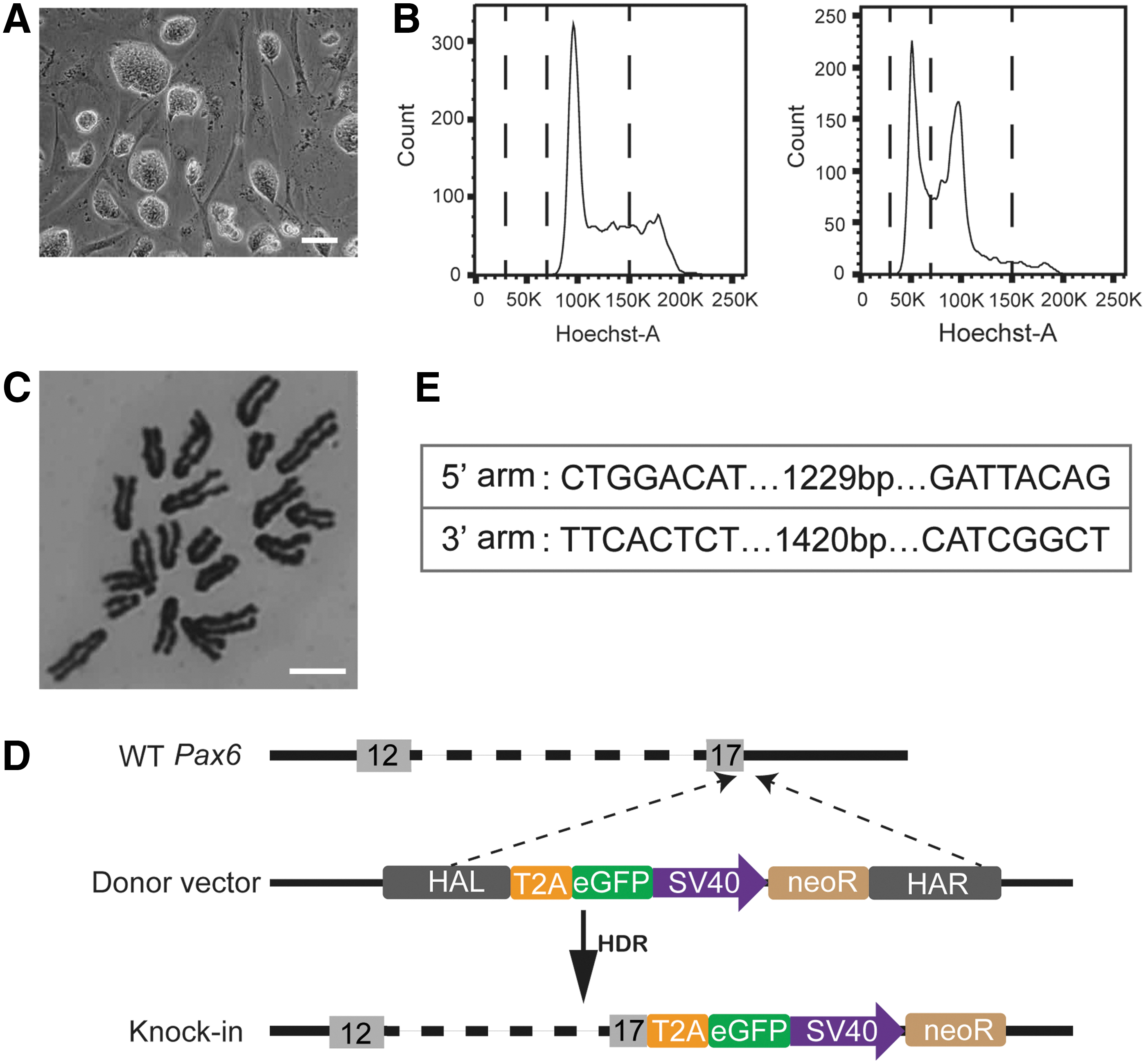
Strategy of CRISPR/Cas9-mediated reporter knockin at the Pax6 locus in mouse haESCs.
In the electronic transfection process, 2 μg Cas9-sgRNA1, 2 μg Cas9-sgRNA2, and 4 μg donor vector were cotransfected into around 106 haESCs. The Cas9-sgRNA coexpression plasmid carried an eGFP gene to indicate the transfection efficiency. Two days after transfection, GFP-positive haploid cells that indicated Cas9-positive cells were purified by FACS. Approximately, 14% of the 1n population were GFP positive (Fig. 2A). Purified GFP-positive haESCs were subsequently cultured with G418 to select for the neoR resistance cassette of the targeting vector. After 7 days, several haESC colonies appeared, whereas no colonies were observed in the nontransfected control group (Fig. 2B).
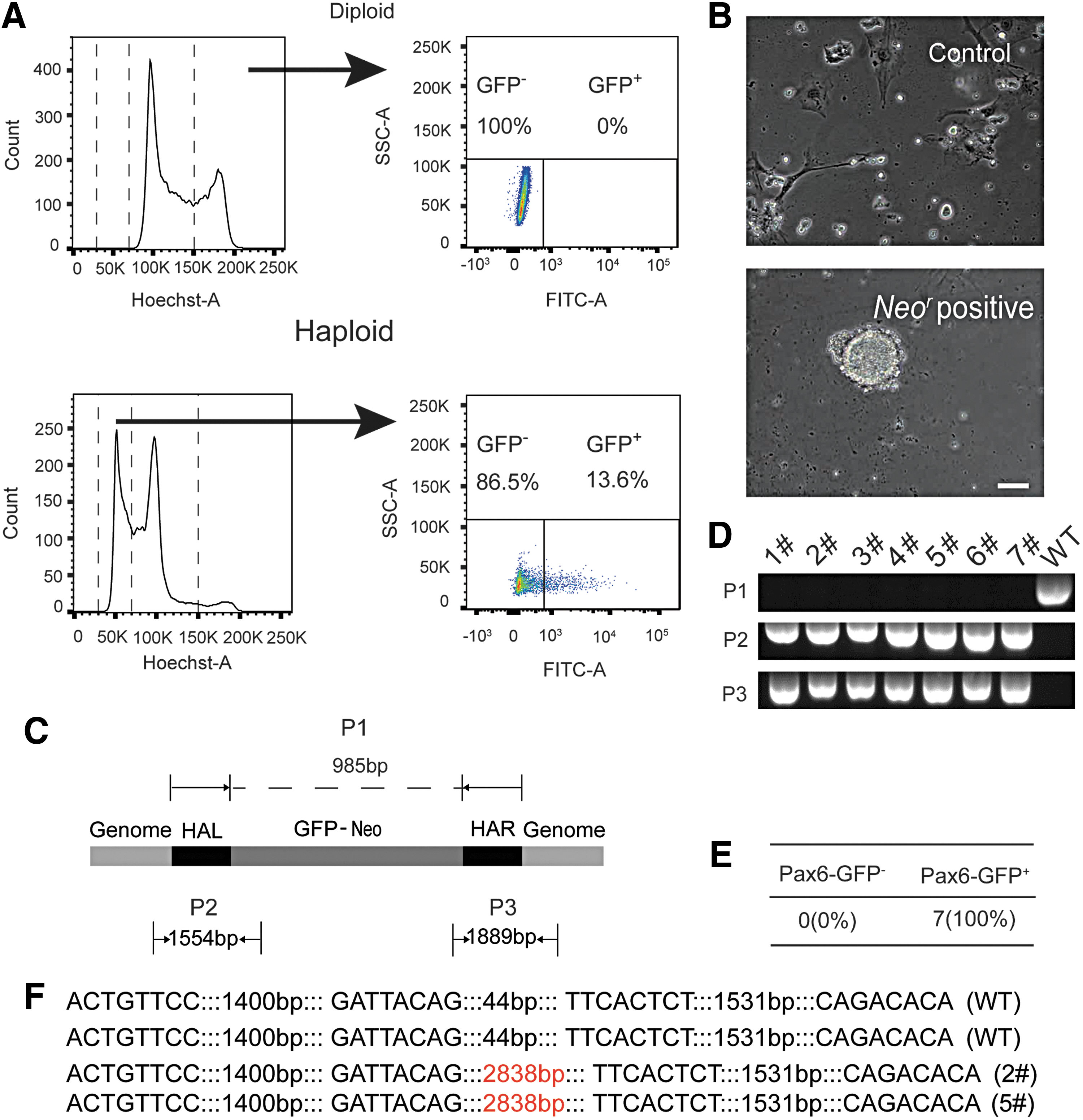
Derivation of Pax6-GFP haESCs through CRISPR/Cas9.
We used three primer pairs to identify the wild-type Pax6 locus (P1), the insertion of the left homology region (P2), and the right homologous region (P3) (Fig. 2C) in seven randomly picked subclones. In all clones, the P2 and P3 fragments were observed indicating the insertion of the GFP marker gene, whereas the wild-type fragment (P1) was absent (Fig. 2D). This result indicated that in all seven subclones the GFP sequence has been correctly inserted and was present in a homozygous or hemizygous state (Fig. 2E). Three of the seven clones retained a substantial fraction of haploid cells, whereas the other four clones had already diploidized (Supplementary Fig. S3A, B and Supplementary Table S2).
Sequencing of the reporter allele in two haploid subclones confirmed the predicted structure of the GFP sequence inserted into the Pax6 locus (Fig. 2F and Supplementary Fig. S2). We chose one Pax6-GFP haESC line for subsequent attempts of neural differentiation.
Derivation of haNSCs using the Pax6-GFP reporter
Differentiation of ESCs to NPCs has been achieved using cell aggregation in suspension [23] or adherent monolayer culture protocols [24]. Although, haESCs have the potential for neuroectodermal development, differentiated neural cells have been found to diploidize rapidly indicating a need for optimized methods to enrich the haploid neural precursors [7,11]. Therefore, we developed a simplified strategy of neural differentiation using the Pax6-GFP reporter (Fig. 3A). Newly sorted haESCs were aggregated to form EBs and cultured in nontreated dishes for 4 days in medium without 2i-inhibitors and LIF. On day 4, the EBs began to express Pax6 as observed by GFP fluorescence signals under the microscope (Fig. 3B).
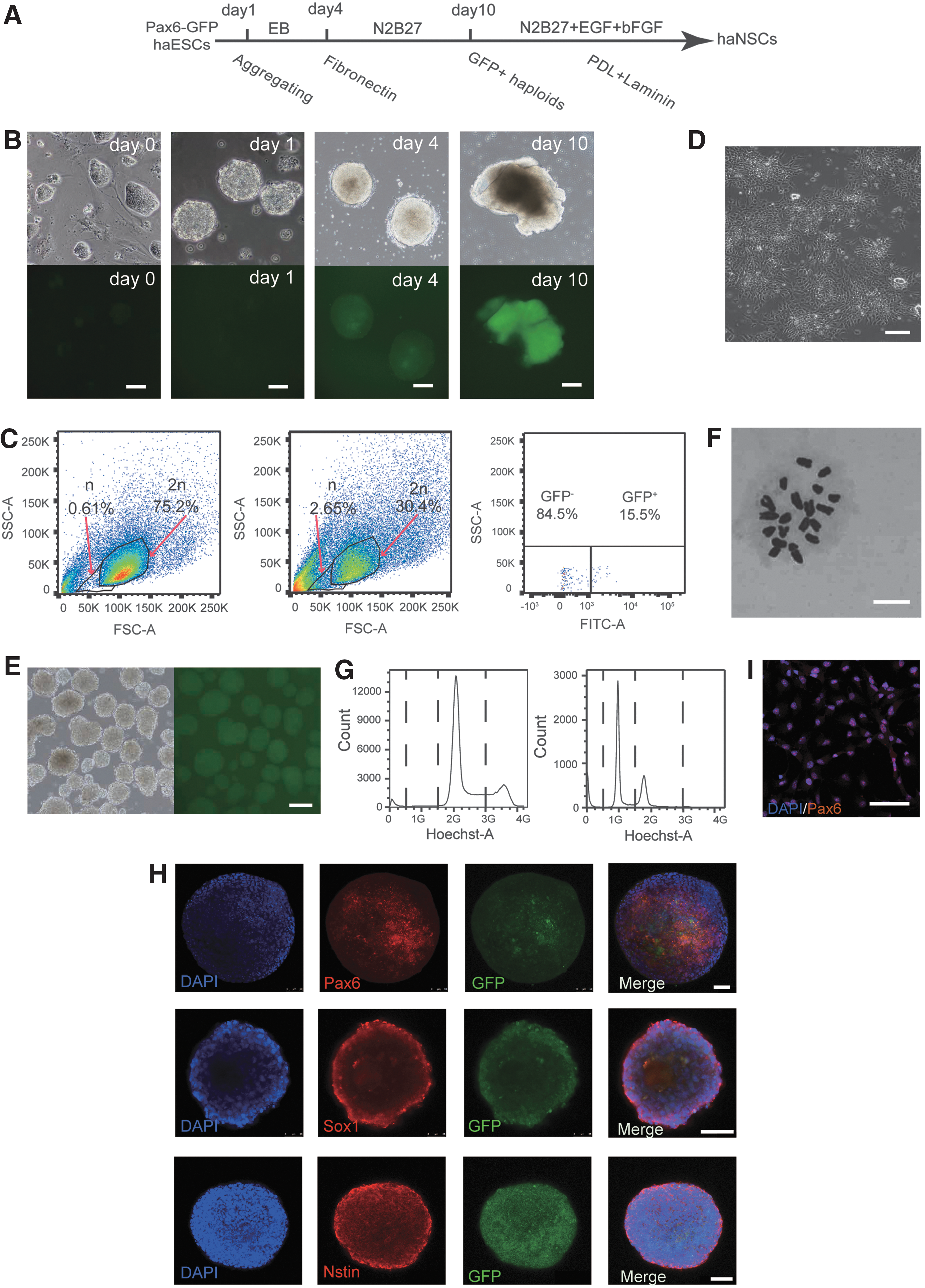
Derivation of a haNSC line using the Pax6-GFP reporter.
Subsequently, EBs were plated on fibronectin-precoated dishes and further cultured in N2B27 medium [24], which is an efficient medium for culturing neural cells. On day 10, a considerable percentage of cells expressed GFP and a changing culture morphology could be observed (Fig. 3B). For isolation of the GFP-positive presumptive NPCs we performed FACS. To avoid staining with DNA dyes, such as Hoechst33342 that can be toxic to cells and affect their survival [25], we enriched haploid cells using forward and side scatter to select small cell size. Diploid NPCs were used to define a suitable sorting gate that is specific for a haploid population (Fig. 3C). Of these, 15.5% Pax6-GFP-positive cells in the haploid populations could be sorted and were subsequently plated into PDL- and Laminin-precoated four-well plates.
Around 0.5 million sorted cells were cultured in one well in N2B27 medium with EGF and bFGF (NSC medium). The haNPCs showed standard NSC morphology (tiny and bipolar), and could be aggregated to form neural spheres in a nontreated dish (Fig. 3D, E). The early passage (p2) spheres expressed Pax6, Sox1, and Nestin (Fig. 3H), and could proliferate many passages in NSC medium as spheres. We disassociated the spheres after 30 passages and replated the single cells, staining them with PAX6 antibody. As expected, the neural spheres could express Pax6 after expansion (Fig. 3I). Additionally, the late-passage NSCs also could form neural spheres, expressing Pax6, Sox1, and Nestin (Supplementary Fig. S4A). Furthermore, we checked the genome of haNSCs by chromosome spread and FACS analysis.
An intact haploid karyotype was maintained after more than 30 passages with the NSC medium (Fig. 3F, G), which suggests a novel haploid cell system that is suitable for neuronal genetic screening.
haNSCs possess a neural differentiation potential
Previous studies have suggested that haploid genomes might not be compatible with forming a neuroectodermal cell fate [11]. Therefore, it was important to establish whether our haNSCs possess an intact neural differentiation potential after expansion. To assess whether our Pax6-GFP reporter system works well, the GFP-positive and GFP-negative cells were sorted and replated separately in the NSC medium, thereafter stained with Pax6 antibody. The results showed that only GFP-positive cells can recognize the Pax6 antibody (Fig. 4A and Supplementary Fig. S4B).
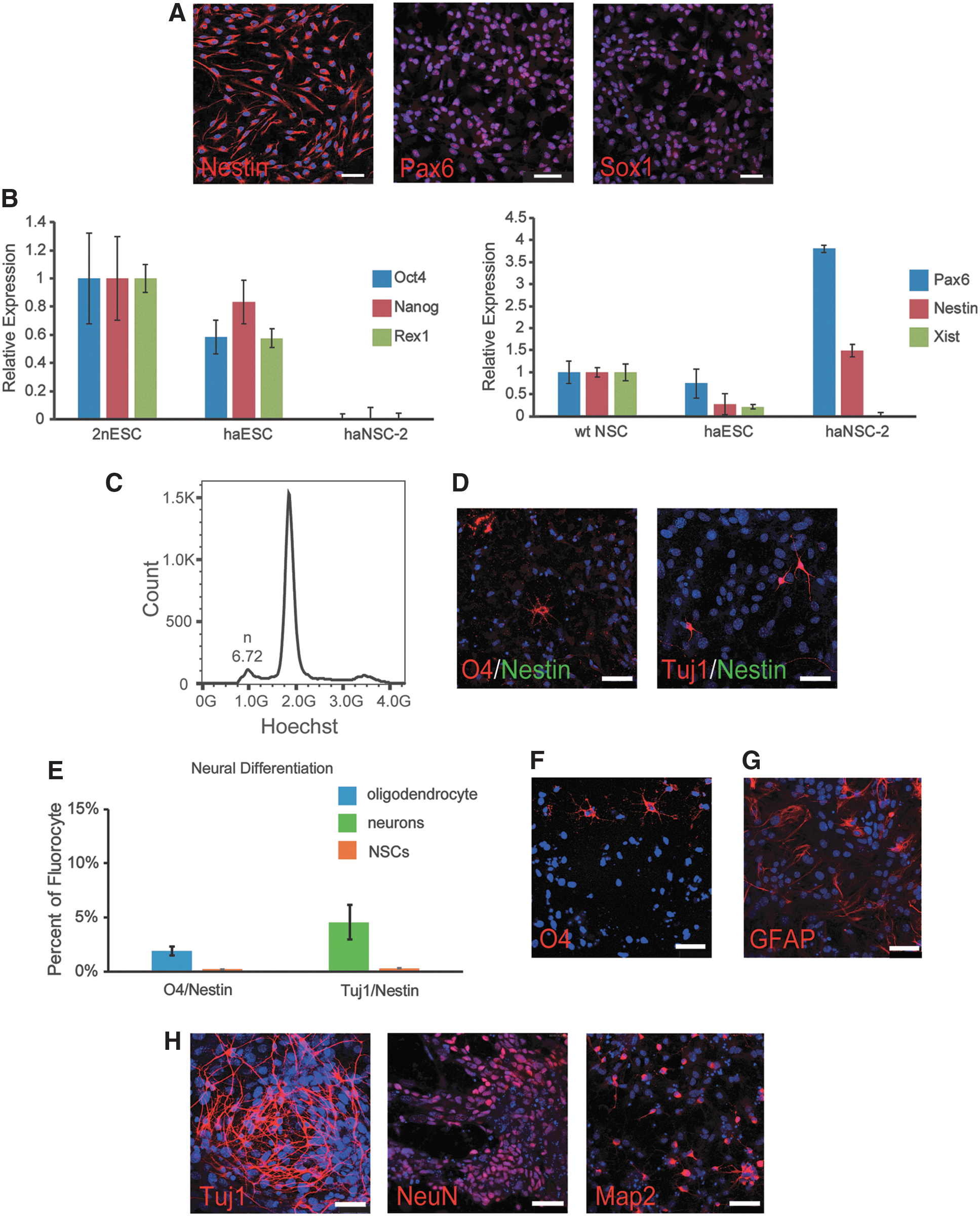
Identification of derived haNSCs.
As expected, haNSCs expressed the neural markers Nestin, Sox1, and Pax6 as observed by immunostaining (Fig. 4A) and reverse transcription polymerase chain reaction (RT-PCR) (Fig. 4B). In contrast, haNSCs did not express pluripotent markers, including Oct4, Nanog, and Rex1 consistent with the absence of haESCs from these cultures (Fig. 4B). Xist is a lncRNA that regulates X chromosome inactivation in female cells [26,27]. To investigate if X inactivation was initiated, we checked Xist expression in haNSCs. According to RT-PCR, haNSCs expressed drastically reduced amounts of Xist compared with diploid NSCs suggesting that the single X chromosome in haNSCs was in an active configuration (Fig. 4B) and X inactivation had not been initiated in the differentiated state.
We further tested the ability of haNSCs for differentiation into neurons, oligodendroglia, and astrocytes using specific growth factors [13,28]. First, we performed neuronal and oligodendrocyte differentiation with specific medium [21] in the presence of ROCK inhibitor (Y-27632), and analyzed the DNA content of the differentiated cells by Hoechst33342 staining. We found there was a small population of haploid cells during sorting (Fig. 4C), and the sorted haploid cells were O4 and Tuj1 positive by immunostaining confirmation (Fig. 4D). Furthermore, the positive cell quantitative statistics of Nestin, O4, and Tuj1 showed that haploid cells were barely NSCs after differentiation (Fig. 4E), which demonstrated haNSCs had further differentiation potentials in haploidy genome.
Next, we assessed multiple differentiation potentials of haNSCs to diverse neurons and glia by specific protocols. Immunostaining demonstrated that markers for oligodendroglia (O4) and astrocytes (GFAP) were positive in differentiated cells from haNSCs (Fig. 4F, G) and several markers for neurons (Tuj1, NeuN, and Map2) were observed by immunostaining after differentiation, too (Fig. 4H). We performed the same differentiation experiments and found that the late-passage (p32) haNSCs could also differentiate to neurons, oligodendroglia, and astrocytes (Supplementary Fig. S4C). Taken together, our results suggested that the established haNSCs possess a multipotent differentiation potential similar to diploid NSC cultures.
Discussion
Our data demonstrate a method for deriving haNSCs from haESCs, which are neural lineage-specific haploid adult stem cells. Up to date, haESCs have shown great advantages in genetic screening, including the identification of target genes of ricin toxicity [7], and exit from self-renewal [8]. However, the fact that haESCs tend to diploidize during self-renewal or differentiation has compromised the development of other haploid cell types. The exact mechanism of diploidization remains unclear, although a few hypotheses were dressed out. One possibility was the result of mistakes in mitosis other than cell fusion of two individual haploid cells [11]. Another hypothesis was mistaken cell cycle check point triggering the diploidization. To overcome this issue, shortening the time of G2/M has improved maintaining haploidy in haESCs [29].
Recently, analysis of cell cycle dynamics in haESCs by single-cell imaging has suggested that some haESCs failed to complete mitosis [30]. Since it was difficult to avoid diploidization of haESCs, we optimized the differentiation and FACS processes to generate haNSCs. First, we introduced an NPC-specific Pax6-GFP reporter into haESCs genome for facilitating the isolation of haNPCs that emerged in differentiating cultures by green fluorescence. After selection in seven out of seven subclones, homozygous or hemizygous insertion of the reporter was observed illustrating the advantage of haESCs in generating homozygous genotypes. Second, as haploid cells show a smaller cell size than diploid ones [31], we introduced a different way to enrich haploid cells avoiding Hoechst33342 staining.
Cell sorting based on cell size was easy to handle and less harmful to the cells, resulting in improved survival with similar accuracy than the DNA staining method. Our derivation of haNSCs also raised another fascinating issue, which was, how can haESCs differentiate with only one X chromosome? Whereas Xist was clearly upregulated in differentiated female cells, haNSCs appeared to express very little if any Xist suggesting that dosage compensation was not established through X inactivation in haNSCs.
Previous studies claimed that haESCs could differentiate into haNSC-like cells through haEpiSCs [32], but functional haNPCs to neurons have only recently been demonstrated [12]. Xu et al. [12] used a haESC line carrying Oct4-GFP reporter to perform neural differentiation, whereby a GFP-negative state indicated that the haploid differentiated cells had abandoned the pluripotent state. Although neural-specific markers showed that haploidy could be maintained in NSCs and neurons in this study, a stable haploid somatic cell line had not been obtained, which would have been suitable for application in lineage-specific screening [12].
In this study, we generate multipotent haNSCs by Pax6-GFP reporter system (sensitive indicator for NSCs) and cell size (low cytotoxicity for sorting haploid cells). Importantly, our haNSC line maintains robust proliferation ability and multipotency to form neurons and glia. Unfortunately, we did not succeed in generating haploid terminally differentiated astrocytes, and the mechanism behind it requires more investigations. Nevertheless, our haNSCs could be expanded for more than 30 passages with EGF and bFGF in vitro, which could provide abundant haploid functional adult stem cells with great values as cell resource for screening. Recently, human haESCs have been derived and it has been reported that haploidy can be maintained in all three germ layers in vivo and in vitro [9].
Our findings reveal that haESCs can differentiate into haploid adult stem cells (NSCs) and maintained a haploid karyotype over many passages. This novel cell system suggests a promising tool for studying genes that regulate neurogenesis and neural disorders.
In summary, we have generated functional haNSCs from haESCs in vitro, which show great value in neural lineage genetic screening. Our Pax6-GFP haESCs also supply a platform to understand mechanisms regulating ESCs to NSCs.
Footnotes
Acknowledgments
This study was funded by the National Natural Science Foundation of China (31501186 and 31671538 to L.S.), and the Natural Science Foundation of Tianjin City (15JCZDJC65300 to L.S.).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.