
Abstract
Neural stem cells (NSCs) are characterized as self-renewing cell populations with the ability to differentiate into the multiple tissue types of the central nervous system. These cells can differentiate into mature neurons, astrocytes, and oligodendrocytes. This category of stem cells has been shown to be a promisingly effective treatment for neurodegenerative diseases and neuronal injury. Most treatment studies with NSCs in animal models use embryonic brain-derived NSCs. This approach presents both ethical and feasibility issues for translation to human patients. Adult tissue is a more practical source of stem cells for transplantation therapies in humans. Some adult tissues such as adipose tissue and bone marrow contain a wide variety of stem cell populations, some of which have been shown to be similar to embryonic stem cells, possessing many pluripotent properties. Of these stem cell populations, some are able to respond to neuronal growth factors and can be expanded in vitro, forming neurospheres analogous to cells harvested from embryonic brain tissue. In this study, we describe a method for the collection and culture of cells from adipose tissue that directly, without going through intermediates such as mesenchymal stem cells, results in a population of NSCs that are able to be expanded in vitro and be differentiated into functional neuronal cells. These adipose-derived NSCs display a similar phenotype to those directly derived from embryonic brain. When differentiated into neurons, cells derived from adipose tissue have spontaneous spiking activity with network characteristics similar to that of neuronal cultures.
Introduction
N
NSCs have also been shown to be an effective method of treatment in animal models of neurodegenerative diseases and injuries such as Parkinson's disease, Huntington's disease, stroke, and spinal cord injury where neuronal populations are depleted [3]. In some cases, the treatment effect from NSCs is thought to be provided by trophic support to the injured tissue [4]. Other studies have shown transplanted NSCs to differentiate into functional neurons that successfully integrate into the host tissue [5]. Currently, there are several hurdles for translating the many successful studies, in which NSCs have been used to improve neurological conditions in animal models, to successful treatments in human patients. These are largely based on the methods used for deriving NSCs.
NSCs can be derived from a variety of sources, but the most common source for research purposes is embryonic brain tissue due to ready availability from animal sources and ease of use with a short time from tissue harvest to the generation of neurospheres [3]. Another commonly used source of NSCs is embryonic stem cells, where pluripotent stem cells are derived from a blastocyst, expanded, and differentiated into multipotent NSCs [6]. Ethical considerations regarding the use of embryonic tissue on a large scale makes these options less attractive for therapeutic use. Another consideration in using NSCs from embryonic sources is rejection by the host as the NSCs are not derived from the patient.
One solution to tissue rejection and the ethical concerns with embryonic tissue sources is to generate NSCs from differentiated tissue of the patient who will receive the cells [7 –9]. This can be achieved by using “induced pluripotent stem cells” (iPSCs) as the starting material. iPSCs are derived from postmitotic cells such as fibroblasts from a patient's skin, and induced, through exposure to dedifferentiation factors, to become pluripotent cells; these can then be differentiated into NSCs just as embryonic stem cells. An extended period (∼6 months) of time is required from the initial tissue harvest to deriving the first generation of NSCs when using the iPSC route [10].
NSCs can also be generated from adult sources by direct transdifferentiation of cells cultured from adult tissue into NSCs, without going through a pluripotent stem cell stage, as is done with iPSCs. Some of these sources include adult adipose tissue, bone marrow, and skin cells. Previously, these tissues have been used to transdifferentiate stromal-derived stem cells, mesenchymal stem cells, and fibroblasts into NSCs with the use of a wide variety of growth factors and other organic compounds [9,11 –15]. Utilizing these strategies has proven effective to generate functional neurons in vitro. Methods such as these, however, also require a lengthy period of time from the initial tissue harvest to generation of a uniform population of NSCs.
An ideal solution would be a method that can quickly, reliably, and consistently produce NSCs from adult tissue, making autologous NSC treatments a more viable option for patients. Adult adipose tissue is known to contain a mixed population of stem cells consisting mainly of multipotent stem cells restricted to the mesodermal lineages [16]. A small subset of stem cells found in adipose tissue has previously been shown to be pluripotent-like stem cells that have the ability to differentiate into all three germ layers [17]. In this case, cells positive for SSEA-3, a marker for pluripotent cells, are isolated from adipose tissue or from mesenchymal stem cells that were derived from adipose tissue with the use of flow cytometry cell sorting and then cultured under the same conditions as embryonic stem cells; these cells then are able to be differentiated into NSCs.
Embryonic-like stem cells are also found in other tissues such as bone marrow where a similar method involving cell sorting can be used to isolate stem cells with pluripotent properties from bone marrow [18]. The purpose of this study was to determine if a subpopulation of stem cells is present in adult adipose tissue that can be directly expanded into NSCs by applying the widely used neurosphere culture methods [19], thereby reducing the expense and time necessary to derive and expand NSCs. Establishing a method for generating NSCs without the need for embryonic tissues or adult tissue intermediates will ultimately increase the utility of NSCs for both clinical and research applications.
Materials and Methods
Cell culture media
NSC growth media consisted of Neurobasal media containing 1× Glutamax, 1× B-27 (without vitamin A), 1× N2, 1× Pen/Strep, heparin 5 μg/mL (ThermoFisher/Life Technologies), 20 ng/mL basic fibroblast growth factor (bFGF) (Stemgent), and 20 ng/mL epidermal growth factor (EGF) (Sigma). Neuronal differentiation media used for spontaneous differentiation consisted of Neurobasal media, 1× B-27, 1× Glutamax, and 1× Pen/Strep [20]. All materials were purchased from Invitrogen, except when indicated otherwise.
Neurosphere culture to derive embryonic neural stem cells
All procedures involving animals conformed to the NIH guidelines for vertebrate animal use and were approved by the Central Michigan University Institutional Animal Care and Use Committee. Embryonic-derived NSCs were harvested from the cortex of E18 Sprague–Dawley rats. The dam was euthanized with CO2 inhalation before embryo dissection. For dissection, the cortex was removed from five embryos and placed in growth media. The tissue was gently triturated (15–30 times) with a P1000 pipette until tissue was completely dissociated. The cell suspension was then centrifuged at 1,200 RPM for 5 min, the supernatant was removed, cell pellet resuspended in 1 mL of growth media, and a viable cell count performed. Cells were plated in growth media at a density of 6 × 104/mL and 1.2 × 105/mL in a T-25 adherent tissue culture flask (MidSci). Cells were incubated at 37°C, 5% CO2, with saturated humidity. A full media change was performed 3 days after the initial plating, and then, cells were incubated in growth media until neurospheres formed (∼1.5 weeks).
Neurosphere culture to derive adipose neural stem cells
Adipose tissue was harvested from adult Sprague–Dawley rats (see Fig. 1 for timeline of procedures). The animal was euthanized with CO2 inhalation before tissue harvest. The inguinal and subcutaneous adipose tissue was removed and ∼10 mL of adipose tissue was transferred to a 50 mL tube. Next, the tissue was washed extensively with phosphate-buffered saline (PBS) to remove blood cells. The tissue was minced on a sterilized glass Petri dish with a razor blade. The tissue was then digested with 40 U/mL type I collagenase (Sigma) at 37°C. The tissue was gently triturated with a 10 mL pipette every 15 min until the tissue was completely dissociated. Any remaining intact tissue was removed and the cell suspension was then centrifuged at 1,200 RPM for 5 min. The top layer, consisting of mature adipocytes, and supernatant were removed, leaving the stromal vascular fraction (SVF) in a dense pellet at the bottom of the centrifuge tube. Pelleted cells were resuspended in 1 mL of growth media, and a viable cell count performed. Cells were plated in growth media at a density of 2.0 × 107/mL and 4.0 × 107/mL in a T-25 tissue culture flask. For optimization of cell seeding density, cells were seeded in a 24-well plate at densities from 6.25 × 105cells/mL to 1.6 × 108cells/mL. Cells were incubated at 37°C, 5% CO2 with saturated humidity.
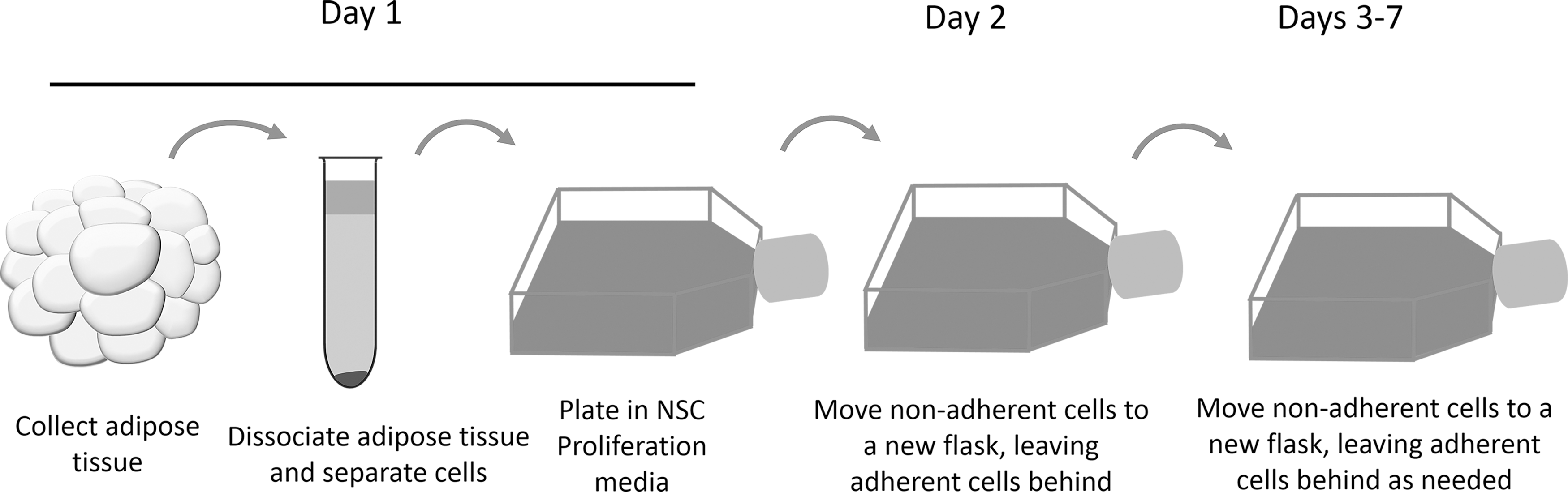
Timeline for culturing neurospheres from adult adipose tissue.
Collection of nonadherent cells and a full media change were performed 1 day after the initial plating. The media containing the nonadherent cells was collected and placed in a 15 mL tube. Next, the flask was washed with 5 mL of PBS to remove any remaining nonadherent cells. The adherent stromal cells were discarded. The cell suspension was then centrifuged at 1,200 RPM for 5 min, supernatant removed, and all cells resuspended in growth media, plated in a new T-25 cell culture flask, and incubated in growth media until neurospheres formed (∼1.5 weeks). If a large number of adherent cells formed in the flask, the nonadherent cells were collected and replated again as described above.
Cell culture maintenance/expansion
Once spheres formed and grew to a substantial size (1–2 weeks after plating), they were passaged and replated. To passage the spheres, the media containing the spheres was removed and placed in a 15 mL tube. The cell culture flask was washed with 5 mL of PBS, which was added to the 15 mL tube. Spheres were centrifuged at 500 RPM to pellet the spheres, supernatant removed, cells resuspended in 3 mL of Accutase (ThermoFisher/Life Technologies), and incubated at 37°C for 5 min. Next, the spheres were gently triturated with a P1000 pipette until no visible spheres remained. These NSCs were then replated in growth media at a density of 2.5–5 × 105cells/mL.
Neurite outgrowth
NSCs were plated as single cells in six-well plates that were coated with poly-D-lysine (10 μg/mL) and laminin (10 μg/mL), at a density of 625 cells/cm2 in 2 mL of differentiation media. Five random pictures of each cell type were captured at 20 × 1 and 2 weeks after plating. The NeuronJ plug-in for ImageJ was used for measuring neurite length and counting neurites per cell [21]. Only cells that had traceable neurites were included and the total neurite length from the sampled image was normalized to the number of cells which had been traced to achieve the average process length per cell.
Electrophysiology
For Multielectrode array (MEA) experiments, six-well MEA plates (Multichannel Systems) were used, with each of the six wells containing nine electrodes. Before plating, MEA plates were sterilized by autoclaving. MEA plates were treated with FCS for 30 min at 37°C, washed four times with sterile dH2O and allowed to dry completely. Once dry, a 5 μL drop of 0.1% polyethyleneimine was placed directly over the electrodes of each well and incubated at 37°C for 30 min. Following incubation, the plates were washed four times with sterile dH2O, and then allowed to dry completely. Once dry, a 5 μL drop of 50 μg/mL laminin was placed directly over the electrodes and the MEA plates were incubated at 37°C for 30 min. While the laminin was incubating, cells were prepared for plating. NSCs were passaged and viable cells counted. Media was added to the cell suspension such that 2.5 × 104 cells were contained in 5 μL of media. Once the laminin incubation was complete, the laminin drop was aspirated and 5 μL of the cell suspension was immediately plated directly over the electrodes and incubated at 37°C, 5% CO2 for 30 min to allow the cells to adhere. Once the cells had adhered to the surface, the wells were slowly flooded with NSC differentiation media, and plates were returned to the incubator; half media changes were performed every 4 days. Recordings were taken 24 days after cells were plated with an MEA 2100 Lite head stage and amplifier with a sample rate of 10,000 Hz. To assess the contribution of glutamatergic signaling and sodium channels to spiking, recordings were repeated in the presence of glutamate antagonists NBQX (10 μm), AP-5 (50 μm), and tetrodotoxin (1 μm).
All MEA analyses were done offline with MC_Rack software (Multichannel Systems) and NeuroExplorer. Spike counting was done with MC_Rack with spikes counted when the extracellular-recorded signal exceeded five standard deviations of the baseline noise [22]. Bursting data were analyzed using the burst analysis tool in NeuroExplorer with the Interval Algorithm Parameters set at max interval to start burst at 10 ms, Max interval to end burst at 10 ms, minimal interval between burst 10 ms, minimal duration of burst at 20 ms, and minimal number of spikes in a burst at four spikes.
Quantitative real-time polymerase chain reaction
To compare gene expression between adipose neural stem cells (aNSCs) and embryonic neural stem cells (eNSCs) as neurospheres, quantitative real-time polymerase chain reaction (qRT-PCR) was used to compare expression of neuronal lineage markers. Total RNA was harvested from neurospheres using the RNeasy Kit (Qiagen). Complementary DNA synthesis was performed using the High Capacity RNA-cDNA Kit (Applied Biosystems) following the manufacturers' instructions on a BioRad C1000 thermal cycler. Quantitative PCR was performed in triplicate on a StepOnePlus Real-Time PCR machine (Applied Biosystems) using SYBR Green PCR Master Mix (Applied Biosystems) in a total volume of 20 μL. Gene expression was normalized to glyceraldehyde 3-phosphate dehydrogenase. Results were analyzed using the double delta cycle threshold (CT) method and are graphically presented as fold expression of eNSCs [23]. Primers used for gene targets and the type of cells which express the target are listed in Table 1.
GAPDH, glyceraldehyde 3-phosphate dehydrogenase; GFAP, glial fibrillary acidic protein.
Immunocytochemistry
For neurosphere staining, fully grown spheres were plated on coverslips coated with poly-D-Lysine (Neuvitro ptg-12-pdl) and coated with laminin (10 μg/mL) for 1 h at 37°C. Spheres were incubated overnight to adhere to the coverslips before staining. For differentiation, 1 × 105 cells were plated on poly-D-lysine- and laminin-coated coverslips. Cells were allowed to spontaneously differentiate under the conditions described above for 4 weeks with media changes every 3–4 days. For immunostaining, cells were fixed in 4% paraformaldehyde for 10 min at room temperature and then washed twice with ice-cold PBS. Cells were permeabilized in 0.1% PBS-Triton X-100 (PBST) for 10 min and washed twice with PBS. Cells were blocked for 1 h at room temperature in 10% donkey or goat serum in PBST. The following primary antibodies were used in overnight incubations at 4°C at specified dilutions: Nestin (Novus NBP1-02419) 1:300, Sox2 (Abcam ab97959) 1:500, glial fibrillary acidic protein (GFAP, Abcam ab53554) 1:500, TujI (Abcam ab7751) 1:200, myelin basic protein (Abcam ab40390) 1:500, and oligodendrocyte-specific protein (Abcam ab7474) 1:300 [15]. Cells were then washed twice with PBS and incubated in appropriate secondary antibody (donkey ∝-goat or ∝-rabbit conjugated to Alexa Fluor 568, Thermo A11057 and A10042 or goat ∝-mouse conjugated to Alexa Fluor 488, Thermo A11001) at 1:500 in 1% donkey or goat serum in PBS in the dark. Cells were washed twice with PBS, mounted with Sigma Fluoroshield with DAPI, and imaged using a Nikon A1R confocal microscope using lasers with wavelengths of 403, 488, and 561 nm with a 60× objective and a step size of 0.25 μm.
Results
For this study, our first goal was to develop a method that allows for neurospheres to be cultured directly from the SVF of adult adipose tissue. This was accomplished by using a defined serum-free neuronal growth media and implementing a process to select for nonadherent cells which could generate neurospheres, followed by optimizing plating densities to determine ideal plating conditions for healthy neurosphere formation. The second goal was to characterize the NSCs grown as neurospheres by morphology, expression of neuronal markers, and electrophysiology in comparison to neurospheres derived from embryonic cortical tissue as a positive control.
Optimization of cell plating densities for neurosphere formation
To determine the optimal cell plating densities single cells in the SVF of adult adipose tissue were plated at densities ranging from 6.25 × 105 to 1.6 × 108 (Table 2). The vast majority of cells plated did not form spheres and remained as single cells in suspension, as adherent cells with fibrous morphology, or died after cell plating. The optimal plating densities range was determined to be from 1.0 × 107 to 8.0 × 107 with lower densities not yielding any spheres and higher densities not yielding any healthy spheres. We concluded that the cells found in adipose tissue are capable of generating primary neurospheres using our cell culture methods and that those cells capable of generating neurospheres are only present in very small quantities.
Morphology
Neurospheres derived from adult adipose tissue are morphologically indistinguishable from neurospheres derived from embryonic cortical tissue (Fig. 2A, B). When cells were plated as single cells on an adherent surface and allowed to differentiate spontaneously by removal of growth factors, a high degree of similarity between cells from the two tissue sources was observed at multiple time points (Fig. 2C–F). There was no significant difference in total process length per cell at either 1 or 2 weeks after differentiation (Fig. 2G). At 1 week after plating, eNSCs had an average process length of 110 μm per cell and aNSCs had an average process length of 117 μm per cell. At 2 weeks after plating, eNSCs had an average process length of 226 μm per cell and aNSCs had an average process length of 241 μm per cell. At 1 week, eNSCs had an average of 6.6 neurites per cell, and aNSCs had an average of 6.1 processes per cell. At 2 weeks after plating, eNSCs had an average of 7.6 neurites per cell, and aNSCs had an average of 7.7 neurites per cell. There was no significant difference in the number of neurites per cell at either time point between NSCs derived from the two different sources.
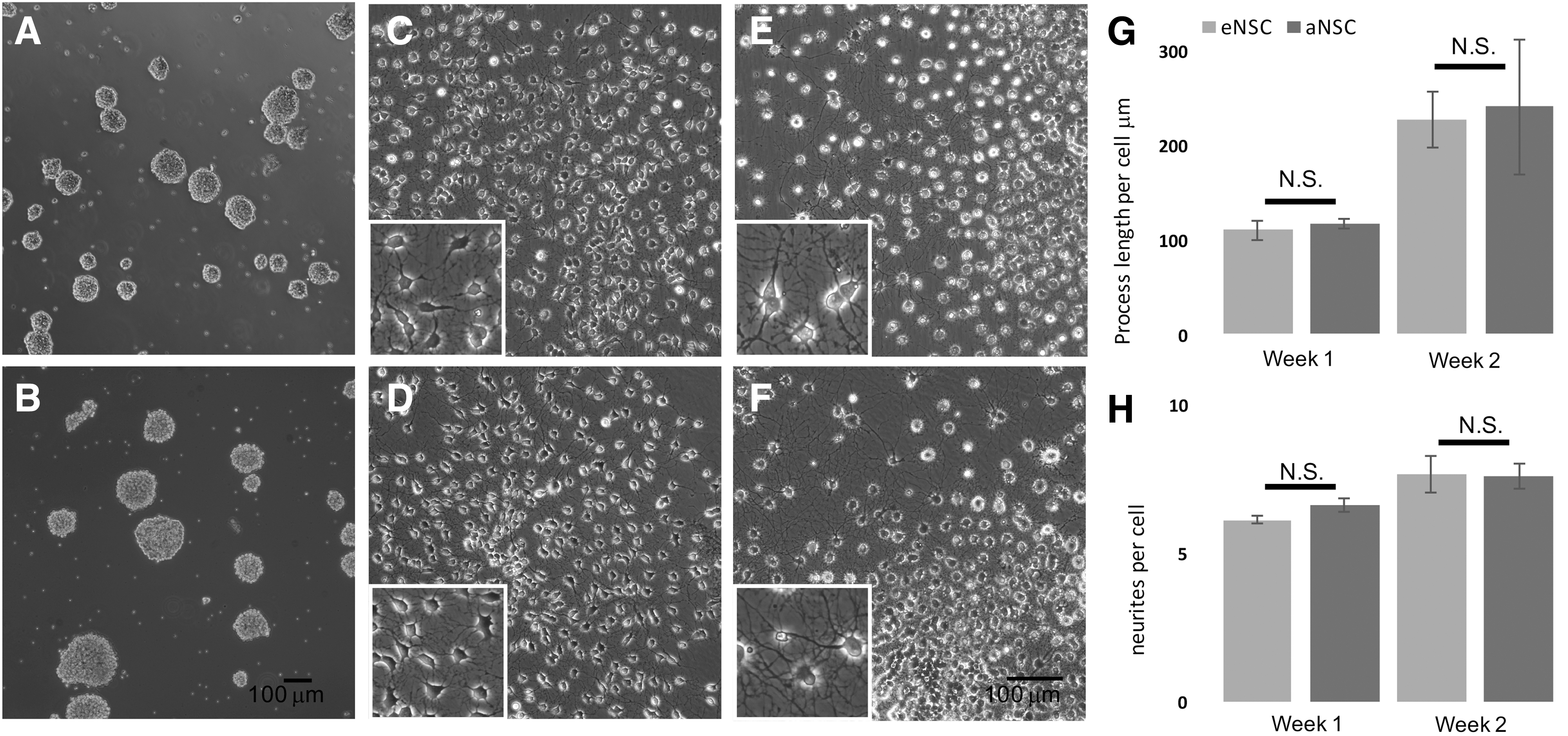
Morphological characteristics of differentiated aNSCs compared with differentiated eNSCs.
Electrophysiological characteristics
When NSCs were cultured on MEA plates in differentiation media, spiking was observed as little as 2 weeks after plating and cultures showed robust activity at 3–4 weeks after plating. Comparisons of electrophysiological activities at three and a half weeks, when the cultures had matured, revealed differences between aNSC and eNSC spiking frequency and burst characteristics, but not bursting frequency (Fig. 3). Cultures derived from aNSCs had a significantly greater spiking frequency, which was more than three times the rate of eNSC cultures (Fig. 3C). The frequency of bursting was slightly higher in the aNSC cultures but did not differ significantly from the eNSC cultures (Fig. 3D). Furthermore, cultures from eNSCs had significantly greater spikes per burst, with twice as many spikes per burst compared with aNSC cultures (Fig. 3E). The mean burst duration was also significantly longer for eNSC cultures, being twice as long as for aNSC cultures (Fig. 3F). The eNSC cultures had a higher percentage of spikes within bursts compared with aNSC cultures with an average of 86.7% of spikes occurring within a burst compared to an average of 50.9% of spikes occurring within bursts (Fig. 3G). For the aNSC cultures, 38% of electrodes detected spikes, and for eNSC cultures, 18% of electrodes detected spikes.

Electrophysiological characteristics of differentiated aNSCs compared to differentiated eNSCs.
Previous studies with eNSC-derived neurons demonstrated spiking activity to be dependent on both sodium channels and glutamate receptors [22]. When NBQX and AP-5 (which are antagonists to AMPA/kinate and NMDA receptors, respectively) were added to the media of spiking aNSC-derived neuronal cultures, spiking was completely ablated and returned once the antagonists were washed away (Fig. 4A). Following the addition of tetrodotoxin (which blocks sodium channels) to the same cultures the following day, activity was nearly ablated (Fig. 4B). Some cultures of differentiated aNSCs which were spiking did not have a strong response to the glutamate antagonists and also did not respond to the addition of tetrodotoxin the following day (data not shown).

Neuronal characteristics of differentiated adipose-derived neural stem cells.
Marker expression
Various neuronal lineage markers were compared between neurospheres derived from adult adipose tissue and embryonic cortex utilizing comparative qRT-PCR. As determined by mRNA expression, the neurospheres derived from both tissue sources show a high level of similarity based on the neuronal lineage markers analyzed (Fig. 5). NSCs derived from both tissue sources contain heterogeneous populations, composed of NSCs, lineage-specific precursors, and postmitotic neuronal and glial cells. There were no significant differences between cell sources for synaptophysin, Glt1, VMAT2, Olig1, or GFAP. Sox2, which is expressed by multipotent NSCs that are not lineage restricted, was slightly decreased in aNSCs, being 0.84-fold of the eNSCs. The difference in Sox2 expression was not considered to be statistically significant with a P-value of 0.057, but was approaching the level of significance.
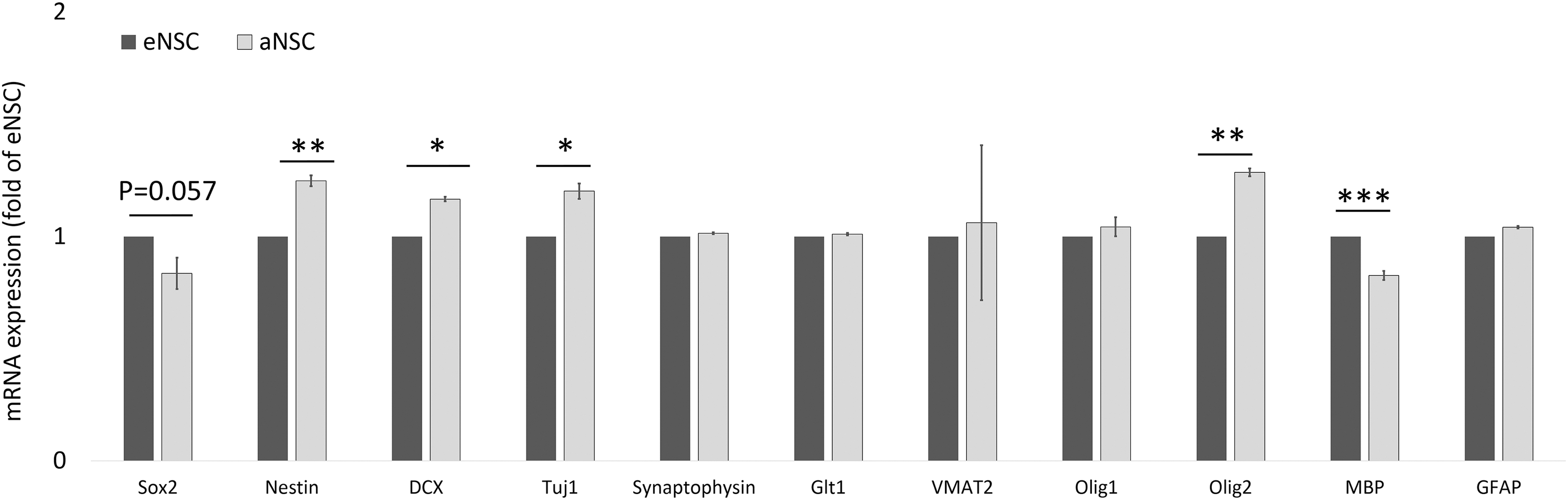
mRNA expression in neurospheres derived from adult adipose tissue compared to embryonic neurospheres. Based on levels of mRNA detected, neurospheres derived from either tissue source show a high degree of similarity. Significance was determined with Student's t-tests. *P < 0.05, **P < 0.01, ***P < 0.001.
Slightly higher levels of Nestin (a marker of NSCs that are not lineage restricted) were observed in aNSCs at 1.25-fold of eNSCs. Doublecortin (DCX) and Tuj1 (both markers of neuron restricted precursors) were also slightly increased with aNSCs expressing at 1.17- and 1.2-fold, respectively, compared with eNSCs. Olig2, which is a marker of oligodendrocyte precursors or motor neurons, was also slightly upregulated in aNSCs at 1.29-fold of eNSCs. Slightly decreased levels of mRNA for myelin basic protein, a marker of Schwann cells, was observed in aNSCs at 0.83-fold the level of eNSCs. Immunostaining for nestin and Sox2 was positive in undifferentiated neurospheres, while spontaneously differentiated aNCSs stained positive for Tuj1 and GFAP (Fig. 6). Immunostaining of myelin basic protein and oligodendrocyte-specific protein was negative in the differentiated aNSCs (data not shown).
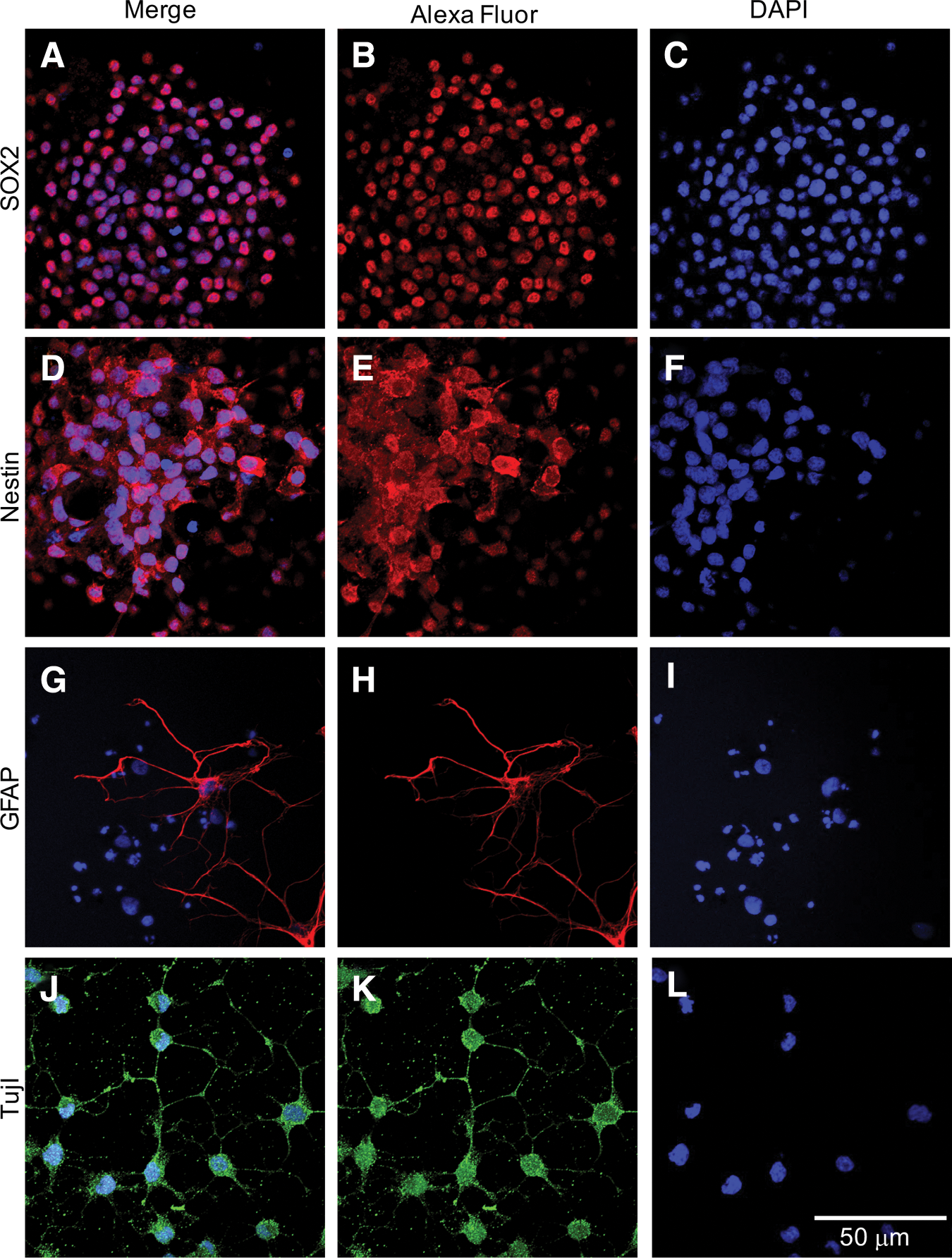
Expression of markers for immature neural stem cells and mature neuronal and glial cells.
Discussion
In this study, we determined that through implementation of a simple cell culture protocol, a highly pure population of neurospheres can successfully be cultured directly from adult adipose tissue from rats. These cells show a high degree of similarity to neurospheres cultured from embryonic brain tissue with respect to their morphology. Furthermore, they were able to successfully be differentiated into electrophysiologically active neuronal cells. Finally, gene expression of various neuronal lineage markers was similar between neurospheres derived from embryonic versus adipose tissue sources.
The high number of seeded cells that was found to be necessary for the initial formation of neurospheres is likely reflective of the low quantities of stem cells found in peripheral adult tissues that are capable of responding to the growth factors used to selectively expand NSCs. For example, a previous study found a very small population of stem cells in bone marrow to possess the pluripotent properties of embryonic stem cells, and this population only represented 0.02% of all bone marrow-derived mononuclear cells [24]. The same group also reported that once sorted and cultured in a similar manner to embryonic stem cells, neurospheres can be generated from this discrete population of cells [25]. A similar population of stem cells with pluripotent properties represents a very small percentage of the cells found in human adipose tissue [17].
The neurospheres derived from adipose tissue have a similar morphology to neurospheres derived from embryonic cortical tissue and are similar to previous reports of neurospheres derived from embryonic and other adult sources [13,19]. The NSCs derived from adipose tissue are self-renewing and can be expanded and passaged repeatedly. We have so far been able to maintain the neurospheres in vitro beyond passage 25, with the cells retaining their self-renewing capabilities and requiring passage every 6–9 days. Based on the observed similarity of aNSCs to eNSCs and previously described neurospheres, we proceeded to compare the adipose derived neurospheres to embryonic neurospheres to see if the aNSCs closely recapitulate the phenotype of eNSCs. When NSCs are grown on an adherent surface and growth factors (EGF and bFGF) are removed (differentiation media), the NSCs can spontaneously differentiate and extend neurites to make connections with other cells in culture. There are various molecular regulatory mechanisms related to the formation and growth of neurites, and measuring the length of neurites in culture can be a sensitive measure to detect differences between populations due to changes in these molecular regulatory mechanisms as well as to detect differences in cellular health [26 –28]. Our results showed that after 1 or 2 weeks, neurite length was similar between groups, meaning that NSCs derived from either tissue are both capable of extending neurites to make connections with neighboring cells with similar levels of success and suggesting that aNSCs have the molecular mechanisms for neurite outgrowth similar to eNSCs. In addition, NSCs from both sources showed comparable numbers of processes per cell. Based on these similarities, it would be expected that NSCs from both tissue sources are capable of forming active neuronal networks and would show a high degree of similarity in gene expression.
To test the ability of NSCs derived from adipose versus embryonic tissue to form electrophysiologically active populations, we cultured them under the same differentiation conditions on MEAs. Spontaneous activity was recorded three and a half weeks after plating when the differentiated NSCs were expected to be active. The differentiated aNSCs were able to spike, with spiking observed throughout the culture dish. This spiking was, in some cases, dependent on glutamatergic stimulation that was blocked with the addition of glutamate antagonists. As expected, the same cultures which responded to the glutamate antagonists also responded to the addition of the sodium channel blocker tetrodotoxin. Cultures that did not have a response to the glutamate antagonist also did not respond to the sodium channel blocker as neurons would be expected to. Based on this observation, we believe the adipose-derived NSCs to be able to differentiate into functional neurons that can form functional networks under the differentiation conditions used. When differentiated, aNSCs were directly compared to differentiated eNSCs for spontaneous activity, some unexpected differences were found in spiking frequency and burst characteristics, with the differentiated aNSCs primarily spiking in a tonic manner, while the differentiated eNSCs primarily spiked in a bursting manner. In primary neuronal cultures and in cultures of NSCs which are differentiated into cortical neurons on MEAs, the neuronal networks mature from spontaneously spiking occasionally in the first week to spiking tonically with occasional bursts and then transitioning to robust bursting activity with a low level of tonic spiking after 2–4 weeks [22,29]. To our knowledge, there has not yet been any report on the in vitro developmental timeline of spontaneously differentiated NSCs to compare our results to.
However, based on what is known regarding the stereotypical development of primary neuronal cultures and directed differentiation of stem cells into cortical neurons in vitro, the most likely explanation for the differences in spiking and bursting observed in this study is that the spontaneously differentiated aNSCs could be developing networks slower than the eNSCs or the aNSCs are not making fully mature networks that would allow for the robust bursting that was observed in the differentiated eNSC cultures. Another explanation could be that NSCs from the two sources differentiated into different proportions of neuronal subsets such as differences in the ratio of inhibitory to excitatory neurons. Based on these findings, further experiments, which utilize directed differentiation or patch clamp techniques, are necessary to further explore the neuronal differentiation of NSCs derived from adipose tissue.
To assess the expression of neuronal lineage markers, comparative qRT-PCR and immunostaining were used. No major differences were found in mRNA expression between NSCs of either tissue source for the markers used which include genes for undifferentiated NSCs, lineage-restricted NSCs, and mature markers for neuron and glial lineages. Gene expression of SOX2, a marker of pluripotent and uncommitted NSCs [30,31], was slightly decreased in the aNSC population. Nestin mRNA expression was slightly higher in the aNSCs which, taken together with the decrease in SOX2 mRNA expression, may indicate that aNSCs as a population are more actively differentiating into lineage-restricted precursors. SOX2 and nestin protein expression was prevalent in undifferentiated aNCSs, indicating the capacity of these cells for self-renewal and to maintain a neuronal lineage. The aNSCs also expressed higher levels of DCX and Tuj1, both expressed at different stages of progenitors that are restricted to the neuronal lineage, suggesting that aNSCs could more likely differentiate into neurons than eNSCs.
Both populations expressed similar mRNA levels of the mature neuronal marker synaptophysin and of VMAT2, a marker expressed by neurons that use amine neurotransmitters. Both populations of NSCs also expressed similar mRNA levels of Glt1, which is found in mature astrocytes and glutamatergic neurons. Together with the comparable expression levels of GFAP, which is found primarily in astrocytes and glial-restricted progenitors, both populations are very similar with respect to astrocytes and mature neuronal cells. Differentiated aNSCs were immunoreactive for GFAP staining, as well as for the mature neuronal marker Tuj1, confirming at the protein level the potential of aNSCs to develop along a glial and a neuronal lineage. Olig1 and Olig2 are both expressed by oligodendrocyte-restricted progenitors and mature oligodendrocytes, while Olig2 is also expressed by motor neurons and motor neuron-restricted progenitors. Given that Olig1 is very similar between groups and Olig2 is expressed at a higher level in the aNSC population, aNSCs may also have a higher propensity to differentiate into motor neurons. Myelin basic protein, which is expressed in Schwann cells, was slightly decreased in aNSCs, likely indicating that the aNSCs may be less prone to differentiate into Schwann cells than the eNSCs. Overall, the mRNA expression data did not show differences at a high order of magnitude, which indicates that aNSCs and eNSCs are highly similar. Immunostaining confirmed that undifferentiated neurospheres derived from adipose tissue are positive for markers of NSCs with the ability to self-renew. When allowed to differentiate spontaneously, matured cells were positive for markers of both mature neurons and astrocytes. Although cells were not positive for oligodendrocyte-specific markers, they were able to differentiate into one of the glial lineages. It is likely that the aNSCs could differentiate into mature oligodendrocytes if a differentiation media was used, which specifically promoted oligodendrocyte differentiation.
Our comparison of aNSCs to eNSCs suggests that adult adipose tissue is a suitable source to derive NSCs that can be differentiated into neurons efficiently. The cell culture method utilized in this study for the growth of NSCs directly from adipose tissue is, to our knowledge, the quickest and simplest method of deriving primary neurospheres from adult tissue, taking only 1–2 weeks for the formation of primary neurospheres. These primary neurospheres were then able to be passaged and expanded in vitro. A method such as this could potentially be useful for autologous NSC transplantation procedures. If implemented in human patients, this method would require minimally invasive techniques for the harvest of adipose tissue and have a quicker turnaround time, compared to other methods, to generate a sufficient number of NSCs for cell transplantation.
The method described in this study also requires less resources than some of the other methods used to grow NSCs from adult tissues. For example, iPSC induction followed by neuronal differentiation and the culture and differentiation of pluripotent-like cells found in other peripheral tissues both require the use of feeder cells. Moreover, expansion of pluripotent-like cells harvested from other peripheral tissues requires multiple rounds of cell sorting, which translates to a timeline spanning several months and a large amount of resources being utilized before the generation of NSCs [10,17,18]. The method described in this study also results in the formation of neurospheres in less time than is required for transdifferentiation methods, which use adherent cells from bone marrow or adipose stem cells, usually after multiple passages [11].
NSCs generated from adult tissues by methods such as iPSC differentiation and transdifferentiation have been shown to retain many aspects of the epigenetic regulations, the epigenetic memory, of the tissue from which they were sourced [32]. While longer in vitro culture might allow cells to shed their epigenetic memory, it would likely be present in NSCs that are maintained in vitro for only a short period. This is likely the case for neurospheres transdifferentiated from adherent adipose stem cells (mesenchymal stem cells) in as little as 2 weeks [9]. We do not anticipate epigenetic memory to be prominent in NSCs derived directly from adipose tissue as described in this study. As we are targeting and expanding those cells that are the most capable of directly responding to the NSC growth factors, the expectation is that they would likely not have as strong of an epigenetic memory as that associated with multipotent stem cells or postmitotic cells of the tissue from which they were sourced. However, further study is necessary to test this prediction; it would be especially worthwhile to carry out these experiments with aNSCs derived from human adipose tissue by the method described here.
There are some ways in which the efficiency of generating neurospheres from adipose tissue could potentially be improved. For the purpose of this study, the culture conditions were kept as close as possible to those used for the eNSCs, to not bias the comparison between the cell types. However, to improve the efficiency for future experiments, the media formulation could be altered in multiple ways, such as the addition of different growth factors to the media could potentially increase the efficiency in deriving NSCs from adipose tissue. For example, BDNF and GDNF have been used to differentiate pluripotent stem cells isolated from adipose tissue into NSCs [11]. There are also other basal media formulations and NSC supplements which have recently been developed that could be used instead of the Neurobasal, B27, and N2 combination used here.
The next important step will be to find out if neurospheres can be expanded directly from human adipose tissue, using the protocol applied in this study in proof-of-principle experiments with rat tissue.
Footnotes
Acknowledgments
This study was supported by the CMU Office of Research and Graduate Studies (Graduate Student Research and Creative Endeavors Grant to E.D.P.). We thank Akash Pal for assistance with experiments and William Medendorp for assistance with statistical analysis.
Author Disclosure Statement
No competing financial interests exist.