
Abstract
Glucocorticoids are steroid hormones used as anti-inflammatory treatments. However, this strong immunomodulation causes undesirable side effects that impair bones, such as osteoporosis. Glucocorticoid therapy is a major risk factor for developing steroid-induced osteonecrosis of the femur head (ONFH). Since ONFH is incurable, therapy with mesenchymal stem cells (MSCs) that can differentiate into osteoblasts are a first-line choice. Bone marrow-derived MSCs (BM-MSCs) are often used as a source of stem cell therapy for ONFH, but their proliferative activity is impaired after steroid treatment. Adipose tissue-derived MSCs (AT-MSCs) may be an attractive alternative source; however, it is unknown whether AT-MSCs from steroid-induced ONFH (sAT-MSCs) have the same differentiation ability as BM-MSCs or normal AT-MSCs (nAT-MSCs). In this study, we demonstrate that nAT-MSCs chronically exposed to glucocorticoids show lower alkaline phosphatase activity leading to reduced osteogenic differentiation ability. This impaired osteogenesis is mediated by high expression of Dickkopf1 (Dkk-1) that inhibits wnt/β-catenin signaling. Increased Dkk-1 also causes impaired osteogenesis along with reductions in bone regenerative capacity in sAT-MSCs. Of note, plasma Dkk-1 levels are elevated in steroid-induced ONFH patients. Collectively, our findings suggest that glucocorticoid-induced expression of Dkk-1 could be a key factor in modulating the differentiation ability of MSCs used for ONFH and other stem cell therapies.
Introduction
M
Glucocorticoids, a drug class analogous to endogenous steroid hormones, are an important anti-inflammatory drug, which has been used for their strong immunomodulatory effects [8]. Therefore, they are widely used to treat a broad range of indications [8]. However, despite their benefits, glucocorticoids cause several complications, such as diabetes, hypertension, and osteoporosis [9,10]. Both beneficial and side effects are known to be mediated through the glucocorticoid receptor, which is expressed in most cell types [9,10].
Among these side effects, impairment of bone tissues, such as in osteoporosis, is considered as the most common complication [11]. Osteoporosis is a catabolic bone disease, which manifests as a decrease of both bone mass and density, as well as degradation of bone microstructure which makes the bone fragile [12]. Glucocorticoids induce osteoporosis by impairment of calcium absorption and increased calcium excretion in kidneys [12]. In bone tissue, osteoblasts are directly affected and their proliferation and maturation is impaired by glucocorticoids [12,13]. In addition, suppression of osteoblast function is reported to be associated with changes in wnt/β-catenin signaling, an important signal for bone remodeling and development [14]. Previous studies demonstrated that glucocorticoids upregulated antagonists for wnt/β-catenin signaling, including Dickkopf1 (Dkk-1) and secreted frizzled-related protein-1, in osteoblasts [14,15]. Importantly, in addition to osteoporosis, long-term and high-dose glucocorticoid therapy is also known as the major risk factor for steroid-induced osteonecrosis of the femoral head (ONFH) [16,17], a bone tissue ischemia involving death of osteocytes [18,19].
Glucocorticoids are also known to support osteogenic differentiation of MSCs in vitro [20 –22]. On the other hand, it has been reported that glucocorticoids suppress the osteogenic differentiation of a murine osteoblast cell line, MC3T3-E1 [15]. Previous studies reported that excessive amounts of glucocorticoids cause MSCs to favor to the adipocyte lineage rather than bone or muscle lineages [11,23]. However, very few studies have focused on the influences of long-term and high-dose glucocorticoid treatments on the characteristics and differentiation ability of patient-derived MSCs. This raises questions as to how excessive glucocorticoids affect MSCs differentiation ability; however, the influences of high-dose glucocorticoid treatment on endogenous MSCs have not been elucidated. Moreover, studies on the characteristics and the therapeutic function of MSCs derived from steroid-induced ONFH patients have not been well focused.
In this study, we aimed to clarify the mechanism of how glucocorticoids impair the differentiation of MSCs into osteoblasts to determine the possibility of using glucocorticoid-treated MSCs for clinical use of bone fractures or bone-related diseases. We investigated the general characteristics of bone marrow- (BM-) and adipose tissue-derived (AT-) MSCs derived from ONFH patients undergoing high-dose treatment with glucocorticoids. Our study shows an elevated expression of Dkk-1 in AT-MSCs derived from steroid-induced ONFH (sAT-MSC) patients and its inhibition successfully restores apparent bone regenerative capacity. Hence, further studies related to the molecular mechanisms of steroid-treated AT-MSCs would be necessary to evaluate them for use in autologous stem cell transplantation.
Materials and Methods
Isolation and culture of BM-MSCs and AT-MSCs
All human cell experiments were approved by the Ethics Committee of the University of Tsukuba. Human BM and ATs from steroid-induced ONFH patients (BM: n = 13, maximum dose of glucocorticoid = 46.5 ± 12.1 mg, current dose of glucocorticoid = 7.96 ± 6.42 mg, age = 40.5 ± 10.0; AT: n = 6, maximum dose of glucocorticoid = 70.0 ± 29.5 mg, current dose of glucocorticoid = 8.58 ± 2.25 mg, male = 2, female = 4, age = 41.5 ± 11.2) and traumatic ONFH patients (BM: n = 5, no systemic glucocorticoids administration, age = 47.5 ± 9.9; AT: n = 6, no systemic glucocorticoids administration, male = 2, female = 4, age = 60.5 ± 20.4) were obtained from the Department of Orthopedic Surgery, University of Tsukuba Hospital (Tsukuba, Japan).
AT-MSCs were isolated as previously described [24]. Briefly, human AT was minced with scissors and treated with 0.1% collagenase (Nitta Gelatin, Osaka, Japan) in phosphate-buffered saline (PBS), 20% fetal bovine serum (FBS; Hyclon, UT) for 45 min at 37°C. After incubation, samples were centrifuged and cultured in a maintenance medium [Iscove's modified Dulbecco's medium (IMDM; Invitrogen, CA)/10% FBS/5 ng/mL basic fibroblast growth factor (PeproTech, London, United Kingdom), and 0.1% (v/v) penicillin–streptomycin (100 U/mL penicillin, 0.1 mg/mL streptomycin; Invitrogen)].
BM-MSCs were isolated as previously described [25]. Briefly, BM-derived cells were put on a density gradient buffer (Histopaque 1.083 g/cm3; Sigma-Aldrich, MO) and centrifuged at 2,000 rpm for 20 min at room temperature. Cells were harvested from middle layer and then cultured by the same method as used for AT-MSCs described above.
Colony-forming unit of fibroblast assay
For primary BM aspirate, 2 mL BM with acid citrate dextrose solution was diluted five times and seeded at 500 μL/well in six-well plates (SUMILON, Tokyo, Japan) in maintenance medium. For cultured BM-MSCs, BM-MSCs were seeded at 100 cells/well in six-well plates (SUMILON) in maintenance medium at P1. After 2 weeks, both primary BM and BM-MSCs were fixed and stained with 0.5% Crystal Violet. Visible colonies were scored by macroscopic observation.
Long-term glucocorticoid exposure in vitro
For long-term glucocorticoid exposure assay, isolated cells from AT were directly cultured in maintenance medium with 100, 10, or 1 nM of dexamethasone (Sigma-Aldrich). AT-MSCs were seeded at 4 × 103 cells/cm2 in a T25 flask (SUMILON). After five passages, dexamethasone-exposed cells were used for further experiments.
In vitro differentiation assay
AT-MSCs were evaluated for their differentiation ability into osteocytes, adipocyte, and chondrocyte. AT-MSCs were seeded in four-well in vitro fertilization plates (BD Falcon, MA) at passages 5–8. Adipogenic differentiation was initiated by incubating in adipogenic differentiation medium [IMDM (Invitrogen) +10% FBS, 0.5 mM 3-isobutyl 1-methylxantine (Sigma-Aldrich), 10 μg/mL insulin (Wako), 0.1 mM dexamethasone (Sigma-Aldrich), and 200 μM indomethacin (Sigma-Aldrich)] as described previously [20], and the adipogenic ability was examined by Oil Red O staining (Muto Pure Chemicals, Tokyo, Japan). For the quantitation of adipogenic differentiation, Oil Red O was dissolved in isopropanol containing 2% IGEPAL® (Sigma-Aldrich) solution and quantified by spectrometry at 492 nm.
Osteogenic differentiation was induced by incubation of cells in osteogenic differentiation medium [IMDM with 1% FBS, 10 mM β-glycerol (Sigma-Aldrich), 200 μM ascorbic acid (Sigma-Aldrich), 0.1 mM dexamethasone (Sigma-Aldrich), and 20 ng/mL epidermal growth factor (PeproTech)] as described previously [24]. Osteogenic differentiation was examined using 1% Alizarin Red S (Wako, Osaka, Japan) solution and alkaline phosphatase (ALP) staining solution (Sigma-Aldrich). For the quantification of osteogenic differentiation, Alizarin Red S was dissolved in Plank-Rychlo (Kanto-Kagaku, Tokyo, Japan) and analyzed by spectrometry at 480 nm. ALP activity was examined histologically with the Leukocyte Alkaline Phosphatase Kit (Sigma-Aldrich) according to the manufacturer's instructions.
Chondrogenic differentiation was performed by aggregate culture methods. 2.5 × 105 cells were seeded in low-cell adhesion 96-well plates (SUMILON) using chondrocyte differentiation medium [IMDM +1% FBS, 1% Insulin-Transferrin-Selenium-premix (BD Bioscience), 4 mM proline (Sigma-Aldrich), 50 μg/mL ascorbic acid, 0.1 mM dexamethasone, 1 mM sodium pyruvate (Sigma-Aldrich), 10 ng/mL transforming growth factor-β1 (PeproTech), and 20 ng/mL bone morphogenetic protein-2 (BMP-2; PeproTech)] as described previously [26]. The spheroids were fixed and embedded in optimum cutting temperature compounds (Sakura Finetek, Tokyo, Japan). Frozen sections were cut to 7 μm thickness and stained with Toluidine Blue and Hematoxylin and Eosin (H&E; Muto Pure Chemicals). To quantify differentiation, proteoglycans were measured by use of the Mucopolysaccharide Assay Kit (Cosmo Bio, Tokyo, Japan). The assay was performed according to the manufacturer's instructions.
Analysis of immune phenotype
AT-MSCs were harvested and suspended in PBS containing 2% FBS and incubated with anti-CD13, anti-CD14, anti-CD31, anti-CD34, anti-CD45, anti-CD73, anti-CD90, anti-CD105, anti-CD166, anti-HLA-ABC, and anti-HLA-DR antibodies (BD Pharmingen, CA) at 4°C for 30 min. After incubation, the stained cells were washed and 15,000 cells at a time were analyzed using a Gallios flow cytometer (Beckman Coulter, CA). Data were processed with the help of FlowJo software (Tree Star, Ashland, OR). A positive expression profile was defined as a level of fluorescence greater than 99% of the corresponding isotype control-stained sample.
Gene expression analysis
Total RNA was isolated from cells using the extraction reagent (Sepasol-RNA I Super G; Nacalai Tesque, Kyoto, Japan) and reverse transcription (RT) was performed with 1 μg of the total RNA using the RT Polymerase Chain Reaction Kit (RT-PCR; Toyobo, Osaka, Japan), following the manufacturer's instructions. The expression levels of target genes were analyzed using a 7500 Fast Real-Time PCR machine (Applied Biosystems, CA) with Thunderbird SYBR qPCR Mix (Toyobo). Experiments were carried out in triplicate and data were calculated using the 2−ΔΔCT method. Data were normalized to β-actin mRNA. The sequences of the primers used for quantitative RT-PCR (qRT-PCR) are shown in Table 1.
ALP, alkaline phosphatase; Dkk-1, Dickkopf1; PPARγ, peroxisome proliferator-activated receptor gamma.
ALP overexpression with retroviral constructs
Prepared AT-MSCs were infected using cell-free retroviral supernatant from PT67 cells producing MSCV–hALP–IRES–EGFP or MSCV–IRES–EGFP for 8 h with 4 μg/mL polybrene (Sigma-Aldrich). After 8 h, the medium was changed to fresh medium and cells were expanded. Subsequently, green fluorescent protein-positive cells were sorted by a Moflo XDP (Beckman Coulter) and expanded for further experiments. The expression of human ALP mRNA was confirmed by qRT-PCR.
Establishment of shDkk-1 AT-MSCs
OmicsLink shRNA Expression clone targeted Dkk-1 and scrambled control gene (GeneCopoeia, MD) were transfected into HEK293T cells using the Lenti-Pac HIV Expression Packaging Kit (GeneCopoeia) and pseudoviral particles were harvested. These particles were then transduced into target cells and selected with 4 μg/mL puromycin. The effect of shRNA of Dkk-1 was assessed by qRT-PCR analysis.
Western blot
Cells were harvested and homogenized with RIPA buffer at 4°C. Twenty micrograms of whole cell extracts in each well were electrophoretically separated on sodium dodecyl sulfate–polyacrylamide gel and then electrotransferred to polyvinylidene difluoride membranes (Merck Millipore, MA). The membranes were immunoblotted with primary antibody (anti-β-catenin antibody: 1:2,000, sc-7199; Santa Cruz Biotechnology, TX; anti-actin antibody: 1:2,000, sc-1616; Santa Cruz Biotechnology; anti-Dkk-1: 1:1,000, AF1096; R&D, MN). After extensive washing, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (Vector Laboratories, CA), and positive signals were analyzed by a luminescence imager (ImageQuant LAS4000; GE Healthcare, NY) using chemiluminescence reagents (Merck Millipore). Data were normalized to β-actin protein level.
Enzyme-linked immunosorbent assay
Human BM was aspirated from anterior iliac crests for concentrated autologous bone marrow aspirate transplantation therapy for ONFH patients [27]. The Dkk-1 (the DuoSet ELISA Development Kit; R&D) level in aliquots of serum was measured using respective kits according to the manufacturer's instructions. Results were calculated by interpolation from a standard curve obtained using a series of recombinant human Dkk-1 concentrations.
Critical-sized calvarial defect model
All animal experiments were carried out under accepted principles of laboratory animal care (Approval number: 17-085, Guide for the Care and Use of Laboratory Animals, University of Tsukuba) and were approved by the Institutional Animal Care and Use Committee of the University of Tsukuba. BALB/c nu/nu mice (8-week-old) (Oriental Yeast, Tokyo, Japan) were used as recipients. The critical-sized calvarial defect model was generated as described previously [28]. Briefly, mice were anesthetized using tribromoethanol (125 mg/kg; Wako) and nonhealing critical-sized (4 mm) calvarial defects were created in the parietal bone of adult BALB/c nu/nu mice using a biopsy punch (Kai Industries, Tokyo, Japan). Importantly, the dura mater was left undisturbed. Then, 1 × 106 AT-MSCs were transplanted with a collagen scaffold (Atelocollagen sponge MIGHTY®; KOKEN, Tokyo, Japan) into defects. The mice were divided into six groups: PBS (scaffold with PBS, N = 5), normal AT-MSC (nAT-MSC) (N = 5), sAT-MSC (N = 5), nAT-MSC transfected with mock (N = 5), sAT-MSC transfected with mock (N = 5), and sAT-MSC transfected with shDkk-1 (N = 5). The calcified defect area was assessed by microcomputed tomography using the Aloka LaTheta LCT-100 system (Hitachi, Tokyo, Japan).
Histological analysis
Mice were sacrificed 3 months after transplantation of MSCs and skulls were dissected. For H&E staining, the skulls were fixed and decalcified. The fixed tissues were embedded in paraffin, and 5 μm serial sections were prepared. The sections were stained with H&E solution (Muto Pure Chemicals). To assess whether transplanted MSCs contribute to the bone regeneration, immunohistological staining with anti-human osteopontin (hOPN) antibody (1:200, AF1433; R&D) was performed. All staining was observed under a BZ-X700 microscope (Keyence, Osaka, Japan).
Statistical analysis
Data were presented as mean ± standard deviation. The comparison between two groups was analyzed by student's t-test. Tukey–Kramer test was used to analyze the differences among more than three groups after one-way analysis of variance. Statistical analysis was performed using GraphPad Prism 5 software (GraphPad Software, CA). Statistical significance was taken as P ≤ 0.05.
Results
Chronic glucocorticoid treatment impairs the number of MSCs in BM and their proliferation potential in vitro
To analyze how long-term glucocorticoid treatment affects BM-MSC characteristics, colony-formation assays using primary BM aspirates from both traumatic (a cell source for normal BM-MSCs) or steroid-induced ONFH patients were performed. It has been reported that the MSC pool in the proximal femur of steroid-induced ONFH patients decreased, whereas the function of their BM-MSCs were impaired [29,30]. Consistent with the results, we found that steroid-induced ONFH patients had less colony-forming unit of fibroblast than traumatic ONFH patients (Steroid: 48.1 ± 48.6 vs. Traumatic: 91.3 ± 54.9, Steroid: n = 36, Traumatic: n = 12, P < 0.05) (Supplementary Fig. S1A; Supplementary Data are available online at
Steroid-induced ONFH patient derived AT-MSCs show less osteogenic potential
To investigate the differences in characteristics between MSCs derived from BM and AT, we next isolated and analyzed AT-MSCs derived from steroid-induced ONFH patients. We found that these AT-MSCs possess a higher and more stable proliferative activity than BM-MSCs (Supplementary Fig. S1), suggesting that AT-MSCs would be an alternative useful source for future clinical applications. however, the effect of steroids on AT-MSCs is still unknown [31]. To determine this, AT-MSCs derived from steroid-induced ONFH patients who received high dose glucocorticoid therapy (current dose: > 5 mg/day, period: > 2 years) (sAT-MSCs) were characterized and compared with the AT-MSCs derived from traumatic ONFH patients (nAT-MSCs). Both sAT-MSCs and nAT-MSCs showed a fibroblastic morphology (Fig. 1A) and there was no significant difference in the proliferative activity between nAT-MSCs (doubling time: 43.3 ± 7.14 h) and sAT-MSCs (doubling time: 40.7 ± 8.07 h) (Fig. 1B, C). Furthermore, we analyzed the immune phenotype of nAT-MSCs and sAT-MSCs and found that both AT-MSCs showed MSC-like immune phenotypes (CD13+, CD73

Steroid-induced ONFH patient-derived AT-MSCs showed less osteogenic potential compared with AT-MSCs derived from traumatic ONFH patients.
We next examined the osteogenic differentiation ability of nAT-MSCs and sAT-MSCs at day 14 and 21 and found that both nAT-MSCs and sAT-MSCs could differentiate into osteoblasts. However, sAT-MSCs showed a lower differentiation ability than nAT-MSCs (Day 14: 0.089-fold; Day 21: 0.43-fold vs. nAT-MSCs) (Fig. 1E, F). In addition, the chondrogenic differentiation ability of sAT-MSCs was also reduced compared with nAT-MSCs. Adipogenic differentiation ability was also examined and no significant differences in adipogenesis were observed in both nAT-MSCs and sAT-MSCs (Fig. 1G–J and Supplementary Fig. S2A).
To clarify the molecular mechanism of how osteogenic differentiation is impaired in sAT-MSCs, we analyzed the expression of osteogenic-related genes at day 0, 7, and 14 after differentiation. Consistent with the results of Alizarin Red S staining, gene expression of bone ALP was decreased in sAT-MSCs (Day 0: 0.049-fold, Day 7: 0.088-fold, Day 14: 0.23-fold decrease vs. nAT-MSC) (Fig. 1K). On the contrary, no significant changes in the expression of other osteogenic-related genes, such as Runx2, osteocalcin, and osteopontin, were observed.
We next evaluated the ALP enzymatic activity in differentiated cells by ALP staining and found that sAT-MSCs showed reduced ALP activity on day 14 postdifferentiation (Fig. 1L). To confirm whether ALP is responsible for the impairment of osteogenesis in sAT-MSCs, ALP-overexpressing sAT-MSCs were established by a retroviral infection and infected cells were confirmed by RT-PCR (Fig. 1M). After 14 days of differentiation, the calcium component was detected in the ALP-overexpressing sAT-MSCs as similar level as nAT-MSCs, whereas no improvement of calcification was observed in mock-transfected sAT-MSCs (Fig. 1N). These data show that overexpression of ALP may rescue the osteogenic differentiation ability of sAT-MSCs.
Several studies have reported that MSCs contribute to tissue repair due to their potential to produce many cytokines and chemokines instead of their differentiated cells [21]. From this perspective, we assessed the expression of secretion factors promoting bone regeneration, such as BMP-2, in sAT-MSCs, and found that BMP-2 was downregulated in sAT-MSCs (Supplementary Fig. S2B).
sAT-MSCs had no differences in cell morphology, proliferation ability, immune phenotype, and adipogenic capacities compared with the nAT-MSCs. On the other hand, the osteogenic and chondrogenic abilities of sAT-MSCs were significantly downregulated compared with nAT-MSCs. Of note, overexpression of ALP in sAT-MSCs rescued osteogenic differentiation ability, suggesting that sAT-MSCs possess osteogenic differentiation ability that centers around the expression of ALP.
Chronic dexamethasone exposure promotes the in vitro proliferation of MSCs in a concentration-dependent manner
We next analyzed how different concentrations of glucocorticoids (1, 10, or 100 nM dexamethasone) affect AT-MSCs differentiation. In all dosage conditions, AT-MSCs showed fibroblastic morphology and no morphological change was observed between glucocorticoid-treated and untreated AT-MSCs (Fig. 2A). However, contrary to adipose-derived rat stromal cells reported previously [32], dexamethasone promoted AT-MSCs proliferative activity in a concentration-dependent manner (Doubling time: no Dex, 50.4 ± 7.17 h; 1 nM, 35.7 ± 5.37 h; 10 nM, 28.2 ± 4.76 h, and 100 nM, 29.1 ± 7.14 h) (Fig. 2B, C). In addition, there was no difference in the MSC-like immune phenotype (in all conditions, AT-MSCs were positive for CD13, CD73, CD90, CD105, CD166, HLA-ABC, and negative for CD14, CD31, CD34, CD45, and HLA-DR) (Supplementary Fig. S3A). Thus, dexamethasone treatment impaired only proliferative capacity, but not the immune phenotype of AT-MSCs.
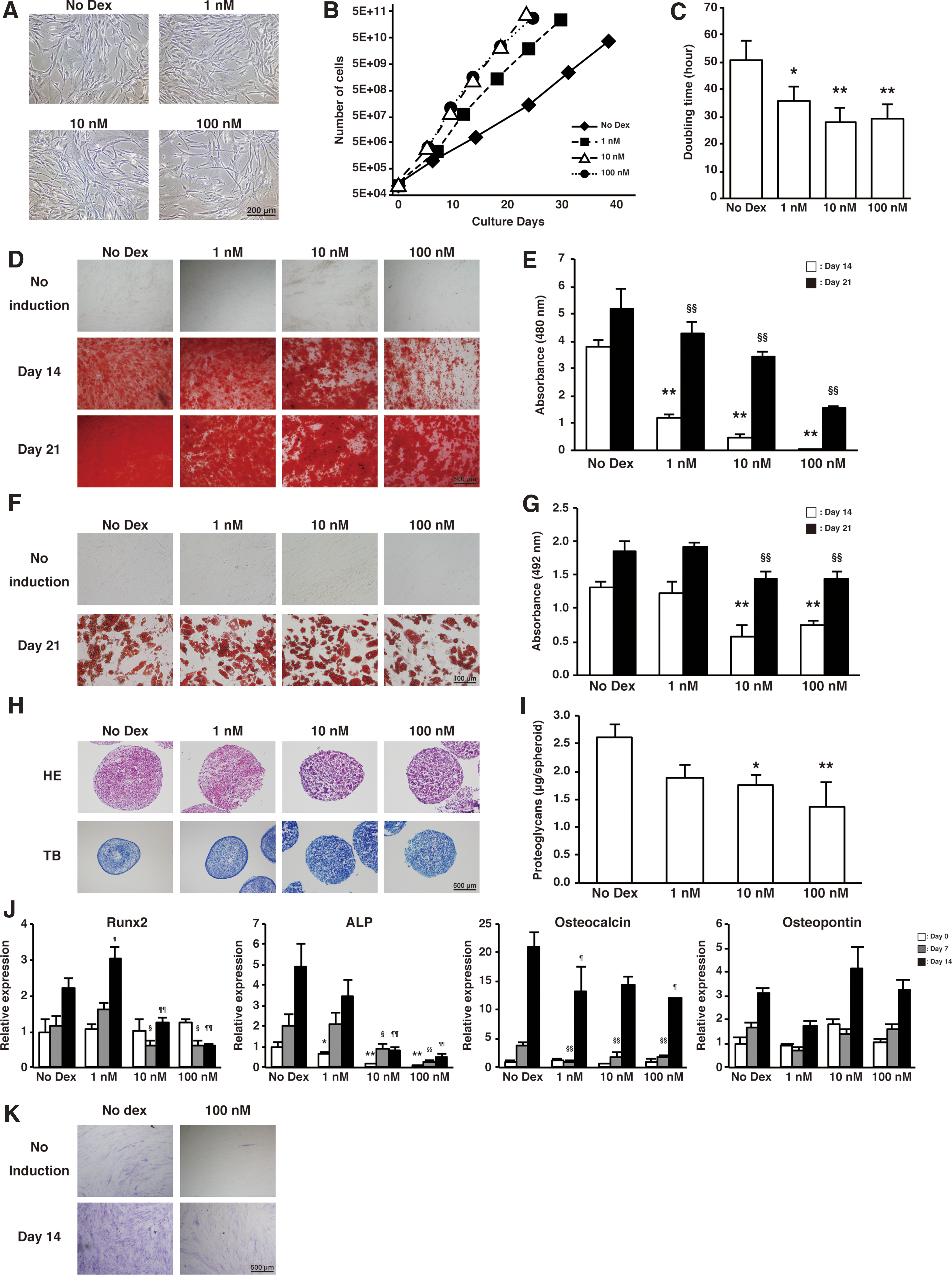
Characterization of AT-MSCs exposed to dexamethasone.
Chronic dexamethasone treatment impairs the in vitro osteogenic differentiation ability of MSCs
We next analyzed how glucocorticoids influence AT-MSCs differentiation ability toward osteogenic, adipogenic, and chondrogenic lineages. Significant reduction of osteogenic differentiation ability was observed in dexamethasone-treated nAT-MSCs compared with untreated nAT-MSCs in a concentration-dependent manner (Day 14: 1 nM, 0.82-fold; 10 nM, 0.67-fold; 100 nM, 0.3-fold, Day 21: 1 nM, 0.92-fold, 10 nM, 0.70-fold; 100 nM, 0.44-fold vs. No Dex), whereas slight reduction of adipogenic and chondrogenic differentiation abilities was observed in dexamethasone-treated nAT-MSCs (Fig. 2D-I).
To clarify how the osteogenic differentiation mechanism was suppressed by the treatment of dexamethasone in nAT-MSCs, osteogenic-related genes were analyzed 7 and 14 days postdifferentiation. As shown in Fig. 2J, ALP gene expression was decreased concomitant to concentration increases of dexamethasone (Fig. 2J). In addition, expression of Runx2 and osteocalcin were also downregulated in the presence of dexamethasone. We next examined the enzymatic activity of ALP by ALP staining and found that 100 nM dexamethasone-treated nAT-MSCs showed impaired ALP activity at 14 days postdifferentiation (Fig. 2K). In addition, BMP-2, which promotes bone regeneration, was also downregulated in a concentration-dependent manner to increases of dexamethasone (Supplementary Fig. S3B).
Taken together, these data indicated that continuous dexamethasone treatment of nAT-MSCs in vitro impaired their osteogenic differentiation ability, consistent with the impaired expression of ALP and other bone regeneration-related genes.
ALP is downregulated through impairment of wnt/β-catenin signaling through high levels of Dkk-1
Previous studies have demonstrated that ALP plays a central role in calcification of the extracellular matrix [33]. The wnt/β-catenin signal pathway is known as a regulator of ALP expression [34]. Activation of the wnt/β-catenin signal pathway causes an accumulation of β-catenin that acts as a transcriptional coactivator of transcription factors that belong to the T-cell factor/lymphoid enhancer factor family [13]. Therefore, we hypothesized that lower activation of wnt/β-catenin signaling might cause the impairment of β-catenin accumulation and lower ALP expression. To test this hypothesis, we examined the protein expression of β-catenin and found that β-catenin was downregulated in sAT-MSCs compared with nAT-MSCs (Fig. 3A).

Wnt signaling was suppressed by glucocorticoid treatment through high levels of Dkk-1.
Dkk-1 is a known wnt signal antagonist and the expression of Dkk-1 is upregulated by glucocorticoid treatment through a glucocorticoid responsive element located on the promoter region of Dkk-1 in osteoblasts [14,35]. In this study, we found that sAT-MSCs showed a higher protein level of Dkk-1 compared with nAT-MSCs (Fig. 3B). We next examined whether Dkk-1 plays a central role in impairing osteogenic differentiation potential in vitro using shRNA of Dkk-1 in sAT-MSCs. Dkk-1 shRNA (shDkk-1)-transfected sAT-MSCs showed a significant reduction in Dkk-1 mRNA expression during osteogenic differentiation (Day 0: 0.059 ± 0.013-fold; Day 7: 0.15 ± 0.053-fold, Day 14: 0.16 ± 0.070-fold vs. mock, Fig. 3C), whereas the expression of β-catenin was upregulated by Dkk-1 knockdown on day 14 (2.4 ± 0.67-fold vs. mock) (Fig. 3D). During osteogenesis, shDkk-1-transfected sAT-MSCs showed recovered ALP gene expression and enzymatic activity, on the other hand, other osteogenic genes including Runx2, osteocalcin, and osteopontin were not changed by Dkk-1 knockdown (Fig. 3E, F, and Supplementary S4A). In addition, it should be noted that osteogenic ability was also rescued by Dkk-1 knockdown in sAT-MSCs (Fig. 3F, G).
Next, we examined whether dexamethasone-treated nAT-MSCs could have their osteogenic ability rescued by modulating wnt/β-catenin signaling. Interestingly, we found that dexamethasone treatment resulted in both downregulation of β-catenin and upregulation of Dkk-1 expression (Fig. 3H, I). Subsequently, we examined whether an inhibitor of Dkk-1, way262611, could rescue the impaired osteogenesis. We found the administration of way262611 (0.4 μg/mL) promoted the suppression of osteogenic differentiation (Supplementary Fig. S5A). Previous studies have reported that a wnt/β-catenin signal suppresses osteogenesis in MSCs, whereas the differentiation of osteoblasts is promoted through wnt/β-catenin signaling [36,37]. Therefore, the Dkk-1 inhibitor, way262611, might affect cell fate when the osteogenic linage has already committed to osteoblast. To test this hypothesis, we next examined whether the osteogenesis in dexamethasone-treated nAT-MSCs is rescued by adding way262611 at day 7 of osteogenic differentiation, after the expression of Dkk-1 would be already upregulated (Fig. 3I). Of note, the elevation of β-catenin protein expression was confirmed on day 14 in the presence of way262611 (Fig. 3J). We also examined ALP activity and osteogenic differentiation ability and found that both ALP activity and osteogenesis in dexamethasone-treated MSCs were recovered on day 21 in the presence of way262611 at the level of the control (Fig. 3K, L). On the contrary, the expression of other osteogenic related genes did not change in the presence of way262611 (Supplementary Fig. S5B).
Collectively, these data demonstrate that high expression of Dkk-1 in sAT-MSCs downregulates wnt/β-catenin signaling, resulting in the impairment of osteogenic ability. In addition, these inhibitory effects can be reversed with a Dkk-1 inhibitor after the MSCs commit to the osteoblast lineage.
Lowered bone regeneration capacity of sAT-MSCs is reversed by shDkk1
The impaired expression of osteogenesis-related and bone regenerative factors in sAT-MSCs suggested that sAT-MSCs might have less bone regenerative capacity than nAT-MSCs, possibly because of the overexpression of Dkk-1. We, therefore, examined the bone regenerative capacity of nAT-MSCs, sAT-MSCs, nAT-MSCs transfected with mock, sAT-MSCs transfected with mock, and sAT-MSCs transfected with shDkk-1, using the critical-sized calvarial defect mouse model [38]. As previously reported, nAT-MSCs and mock-transfected nAT-MSCs could facilitate bone formation, whereas sAT-MSCs showed impaired bone regenerative capacity [39,40]. In contrast, shDkk-1-transfected sAT-MSCs showed significant increases in bone regenerative capacity compared with the sAT-MSCs transfected with mock (Fig. 4A, B). Similarly, H&E staining of injured skulls showed that nAT-MSCs, mock-transfected nAT-MSCs, and shDkk-1-transfected sAT-MSCs enhanced new bone formation which could not be observed in mock sAT-MSCs (Fig. 4C). To prove the presence of transplanted AT-MSCs in the repaired bone regions, we performed immunohistological analyses using anti-hOPN antibody. Intriguingly, we found there were hOPN-positive cells in the repaired bone regions with nAT-MSCs, mock-transfected nAT-MSCs or shDkk-1-transfected sAT-MSCs. In contrast, no positive cells were observed in sAT-MSCs or mock-transfected sAT-MSCs at the transplanted regions (Fig. 4D).
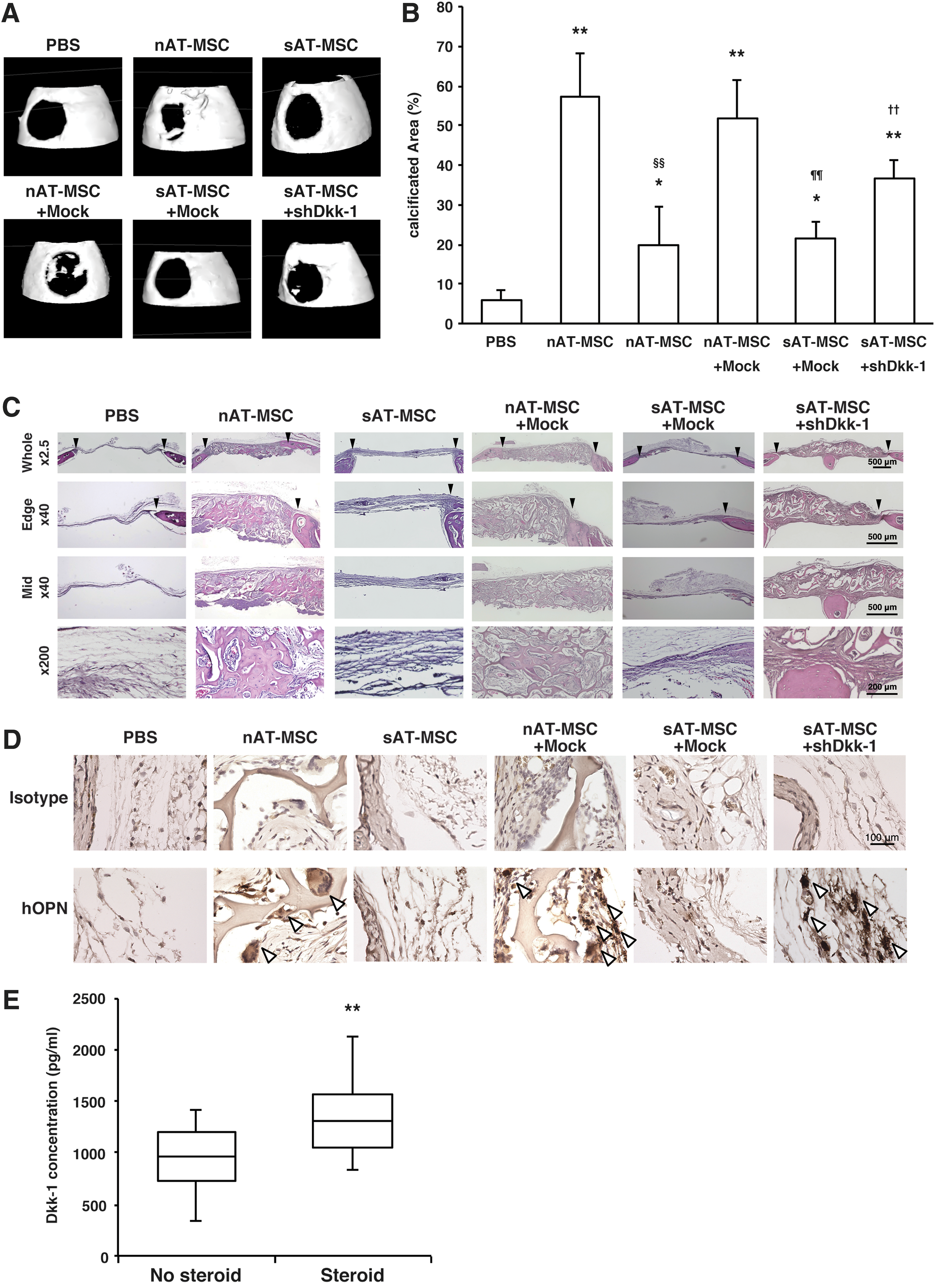
Lowered bone regeneration capacity of sAT-MSCs was reversed by shDkk1.
Thus, these data strongly indicate that continuous glucocorticoid therapy impaired the osteogenic and bone regenerative capacity of AT-MSCs because of upregulation of Dkk-1 expression.
We next investigated whether plasma Dkk-1 may reflect steroid-induced ONFH progression. Enzyme-linked immunosorbent assay analysis clearly showed increased level of Dkk-1 in steroid-induced ONFH patient plasma compared with nonsteroid-treated ONFH patients (nonsteroidal: 955.8 ± 328.6, steroidal: 1375.8 ± 397.4, 1.44-fold, Fig. 4E). Thus, our data indicate that plasma Dkk-1 levels may likely predict side effects in steroid-induced ONFH patients. However, more precise analysis is required to clarify the mechanism how Dkk-1 affects the overall condition of steroid-induced ONFH patients.
Discussion
In this study, we demonstrated that sBM-MSCs showed less self-renewal and proliferative ability compared with those of traumatic ONFH patients. Chronic glucocorticoid treatment impaired the osteogenic and bone regenerative capacity of AT-MSCs because of high upregulation of Dkk-1 expression, which antagonizes wnt/β-catenin signaling. The reduced expression of ALP was also observed in sAT-MSCs, possibly because of the impaired expression of β-catenin. Consistent with these findings, higher expression of Dkk-1 was measured in the plasma from steroid-induced ONFH patients versus traumatic ONFH patients (Fig. 5).

Study summary schematic. Glucocorticoid treatment reduces the number and the proliferative activity of BM-MSCs. The proliferative activity of AT-MSCs is not affected by glucocorticoid treatment; however, osteogenic ability is impaired by downregulation of ALP through Dkk-1/wnt/β-catenin signaling. The levels of Dkk-1 were not only upregulated in AT-MSCs, but also in the plasma derived from steroid-induced ONFH patients. BM-MSC, bone marrow-derived mesenchymal stem cell. Color images available online at
ONFH is characterized by bone tissue ischemia, which leads to death of osteocytes [18,19]. Approximately 51% patients with ONFH have received glucocorticoid therapy, and long-term or pulsed glucocorticoid treatments for severe respiratory syndrome, rheumatoid diseases, and organ transplantation increase the risk of ONFH [41]. Autologous BM transplantation has been reported as a novel approach to treat ONFH [27]. In this therapy, transplanted whole BM-derived cells, including MSCs, promote bone regeneration and this therapeutic outcome is thought to be correlated with the number of transplanted MSCs [42 –44]. In this study, we demonstrated that the number and proliferative ability of sBM-MSCs were impaired compared with traumatic ONFH patients and cultured sBM-MSCs displayed unstable growth as previously reported [30,45]. Thus, autologous cell therapy using sBM-MSCs may not achieve critical cell thresholds due to their impaired proliferative activity.
As an alternative cell source, AT-MSCs are attractive because of their abundance, proliferative activity, and lower invasiveness compared with BM-MSCs. As shown in Supplementary Fig. S1, BM-MSCs derived from steroid-induced ONFH patients patients have less proliferative activity, suggesting that cell therapy using AT-MSCs would be appropriate for steroid-induced ONFH patients. Consistent with these results, previous reports showed that AT-MSCs derived from steroid-induced ONFH patients have higher proliferative and osteogenic potential compared with BM-MSCs [31]. Therefore, AT-MSCs are a more robust source for cell therapy rather than BM-MSCs. However, no study has performed the comparative analysis of AT-MSCs derived from both traumatic and steroid-induced ONFH patients. Intriguingly, sAT-MSCs showed the same proliferative ability as nAT-MSCs (Fig. 1B, C). On the other hand, nAT-MSCs treated continuously by dexamethasone (mimicking the microenvironment of sAT-MSCs patients) showed a concentration-dependent proliferative ability (Fig. 2B, C), indicating that glucocorticoid treatment does not affect AT-MSCs proliferative ability. In addition, sAT-MSCs did not show any significant difference in adipogenesis compared with nAT-MSCs (Fig. 1G, H, and Supplementary Fig. S2B), although previous studies demonstrated that glucocorticoid treatment upregulated the expression of adipogenic genes, including peroxisome proliferator-activated receptor gamma (PPARγ) and lipoprotein lipase (LPL) and facilitated adipogenesis in vitro [11]. However, other previous study showed that steroidal ONFH patient-derived BM-MSCs did not show significant change in adipogenesis compared with osteoarthritis patient-derived BM-MSCs as consistent with our results (Fig. 1G, H, S2B) [46]. Therefore, it would be speculated that chronic glucocorticoid treatment to patients might not affect the adipogenic differentiation ability of MSCs.
It has been reported that several signaling pathways, such as p38/MAPK signaling and wnt/β-catenin signaling, are involved in chondrogenesis, osteogenesis, and regulation of ALP expression [34,47 –50]. In particular, the wnt/β-catenin signaling pathway is associated with osteogenesis and ALP expression independent of Runx2 and osteocalcin expression [34]. Moreover, several studies have reported that wnt antagonists, including WIF-1, SOST, and Dkk-1 were upregulated by glucocorticoids [15,23]. Based on these findings, we found that wnt/β-catenin signaling was suppressed by highly expressed Dkk-1 during osteogenesis (Fig. 4). In addition, the expression of bone regenerative cytokines, such as BMP-2, was reduced compared with the control, the expression of BMP-2 was not recovered, and even the expression of Dkk-1 was diminished (Fig. S4B, S5C), clearly indicating that BMP-2 expression was not related with Dkk-1 expression.
ALP is also known to play a central role in mineralization of the extracellular matrix [51]. ALP knockout mice display skeletal defects of infantile hypophosphatasia and osteoblasts derived from ALP knockout mice showed no calcification ability [52,53]. In addition, previous studies have reported that ALP activity in serum was correlated with bone healing processes [54 –57]. In this study, we demonstrated that the downregulation of ALP in sAT-MSCs caused reduced osteogenic differentiation ability and sAT-MSCs transfected with Dkk-1 shRNA showed ALP upregulation and restored osteogenic differentiation (Figs. 1N and 3E–G). Thus, the expression of ALP may be critical for not only osteogenesis in vitro, but also bone healing in vivo. Further analysis is therefore required to understand the precise physiological mechanisms of ALP during bone regeneration.
In addition to ALP, BMP-2 is also known as an important factor for osteogenesis and bone repair [58]. During osteogenic differentiation, BMP-2 regulates key transcription factors, such as Runx2 and Osterix that induce downstream gene expression of osteocalcin and type 1 collagen [59]. In sAT-MSCs, the expression level of BMP-2 was decreased and knockdown of Dkk-1 expression did not affect BMP-2 expression even if osteogenic ability was restored (Supplementary Fig. S2B), suggesting that in osteogenic differentiation medium, dexamethasone, β-glycerophosphate, and ascorbic acid induce the expression of Runx2 and type 1 collagen in the absence of BMP-2 [60]. In fact, osteoblasts derived from BMP-2 knockout mice showed less osteogenic ability, but still they possess osteogenic potential [61]. In sAT-MSCs, reduced BMP-2 expression showed impaired osteogenic ability, however, the expression level of Runx2 and osteocalcin was not significantly changed compared with nAT-MSCs (Fig. 1K). These results suggest that the major reason for reduced osteogenic differentiation ability in sAT-MSCs is reduced ALP expression, resulting in calcification inhibition. On the other hand, previous studies demonstrated that secretion of BMP-2 by AT-MSCs promotes bone repair by stimulating endogenous osteoblasts and MSC differentiation [62,63]. As shown in Fig. 4D, transplanted AT-MSCs differentiated into mature osteoblasts in vivo; however, hOPN-positive osteoblasts were not abundant in the bone defect region. These results suggest that both beneficial effects, such as differentiation into osteoblasts and cytokine secretion (including BMP-2) would be required for effective bone repair. Thus, supplementation of BMP-2 in addition to the diminished expression of Dkk-1 in sAT-MSCs may result in more effective bone repair.
Dkk-1 is a wnt signaling antagonist often associated with bone physiology, and Dkk-1-overexpressing mice are reported to show severe osteopenia [64]. In addition, several studies have clearly showed that high levels of Dkk-1 in plasma are associated with rheumatoid arthritis, osteoarthritis, bone mineral density, and osteolytic lesions in multiple myeloma [65 –68]. In this study, it was noted that the upregulation of Dkk-1 was observed in plasma derived from steroid-induced ONFH patients versus nonsteroid-treated patients (Fig. 4E). Excess glucocorticoid treatment also caused increased expression of Dkk-1 in murine BM cells and osteoblasts [14,69], suggesting that highly expressed Dkk-1 would be involved in the pathogenesis of steroid-induced ONFH.
In conclusion, we demonstrated that glucocorticoid treatment impairs the proliferative potential of BM-MSCs, whereas proliferation in sAT-MSCs was not affected. On the other hand, the osteogenic ability of sAT-MSCs was affected through the downregulation of wnt/β-catenin signaling because of highly expressed Dkk-1. Long-term treatment with glucocorticoids may therefore reduce the therapeutic function of AT-MSCs. In addition, it was noted that higher levels of Dkk-1 were also observed in the plasma from steroid-induced ONFH patients, suggesting that Dkk-1 expression would be key promoter of AT-MSCs as a useful therapy source instead of BM-MSCs. Taken together, in the treatment of steroid-induced ONFH, transplantation of BM-MSCs is not effective due to lower cell numbers with lower proliferative activity, whereas AT-MSCs would be more useful if combined with the downregulation of Dkk-1 expression.
Footnotes
Acknowledgments
The authors thank the Japanese Ministry of Education, Culture, Sports, Science, and Technology (MEXT) for their support. We thank Ms. Chizuko Fujisawa for excellent work on the colony formation assay. We thank Dr. Bryan Mathis, Medical English Communication Center, University of Tsukuba, for proofreading.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.