
Abstract
Spinocerebellar ataxia type 3 (SCA3) is caused by an abnormal expansion of the cytosine-adenine-guanine (CAG) triplet in ATXN3, which translates into a polyglutamine (polyQ) tract within ataxin-3 (ATXN3) protein. Although the pathogenic mechanisms remain unclear, it is well established that expression of mutant forms of ATXN3 carrying an expanded polyQ domain are involved in SCA3 pathogenesis, and several strategies to suppress mutant ATXN3 have shown promising potential for SCA3 treatment. In this study, we described successful clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9-mediated deletion of the expanded polyQ-encoding region of ATXN3 in induced pluripotent stem cells (iPSCs) derived from a SCA3 patient, and these patient-specific iPSCs retained pluripotency and neural differentiation following expanded polyQ deletion. Furthermore, the ubiquitin-binding capacity of ATXN3 was retained in the neural cells differentiated from the corrected iPSCs. For the first time, this work provides preliminary data for gene editing by CRISPR/Cas9 in SCA3, and demonstrates the feasibility of using a single-guide RNA pair to delete the expanded polyQ-encoding region of ATXN3, suggesting the potential efficacy of this method for future therapeutic application.
Introduction
S
Because polyQ expansion leads to a gain of toxic protein function, several strategies that aim to suppress causative genes have been tested and show good treatment potential [5 –7]. For example, single-stranded silencing RNA, based on RNA interference principles, can achieve allele-specific downregulation of mutant ATXN3 [8,9], while modified antisense oligonucleotides (ASOs) successfully bind to expanded CAG repeats in vitro, resulting in the reduction of mutant ATXN3 mRNA and/or its translation [10,11]. Additionally, ASOs are designed to induce an in-frame exon skipping within the pre-mRNA, resulting in a modified ATXN3 protein lacking the expanded polyQ tract, whereas total ATXN3 protein levels are unaltered and its functional domains remain intact [12]. However, major obstacles associated with the use of these molecules involve their inadequate affinity for their intended target sequences and off-target effects. Furthermore, the effect of gene downregulation is not permanent.
The clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated nuclease 9 (Cas9) have recently been developed and implemented to target and mutate specific genomic regions [13 –15]. In this system, a short guide RNA (gRNA), which is complementary to a specific site in the genome, is used to target the Cas9 nuclease and induce double-stranded breaks (DSBs), which can be repaired through homology-directed repair (HDR) or nonhomologous end joining (NHEJ). Genetic modifications through NHEJ have been applied in genetic diseases, including Duchenne muscular dystrophy and Fragile X syndrome [16,17]. NHEJ can be used to induce nucleotide deletions at the targeted region of the Huntingtin gene, which mediates gene silencing of the mutant Huntingtin [18]. Furthermore, as expanded CAG repeats cause some neurological diseases, contracting them with or without flanking sequences by CRISPR/Cas9 can be achieved [19,20]. However, the deletion of the expanded CAG repeats by application of CRISPR/Cas9 in SCA3 has not yet been reported.
In this study, we generated SCA3 patient-specific induced pluripotent stem cells (iPSCs) and then applied CRISPR/Cas9 for excising expanded CAG repeats in mutant ATXN3. Following gene modification, modified iPSCs retained full property of pluripotency and potential for neural differentiation, expressing both the wild-type ATXN3 and truncated ATXN3 without the expanded polyQ tract. Furthermore, in iPSCs-differentiated neural cells (NCs), we found the normal ubiquitin-binding capacity of ATXN3 was retained. Altogether, our study provides a CRISPR-based strategy to genetically treat SCA3.
Materials and Methods
Ethics statement
All participants in this study provided written informed consent for donating blood. The Ethics Committee of The Third Affiliated Hospital of Guangzhou Medical University (Guangzhou, China) approved experiments using human cells and mice. All methods were performed in accordance with the approved guidelines.
iPSCs generation and culture
To generate iPSCs, a blood sample was collected from a 31-year-old female SCA3 patient genotyped as harboring 26/78 CAG repeats (Supplementary Fig. S1; Supplementary Data are available online at
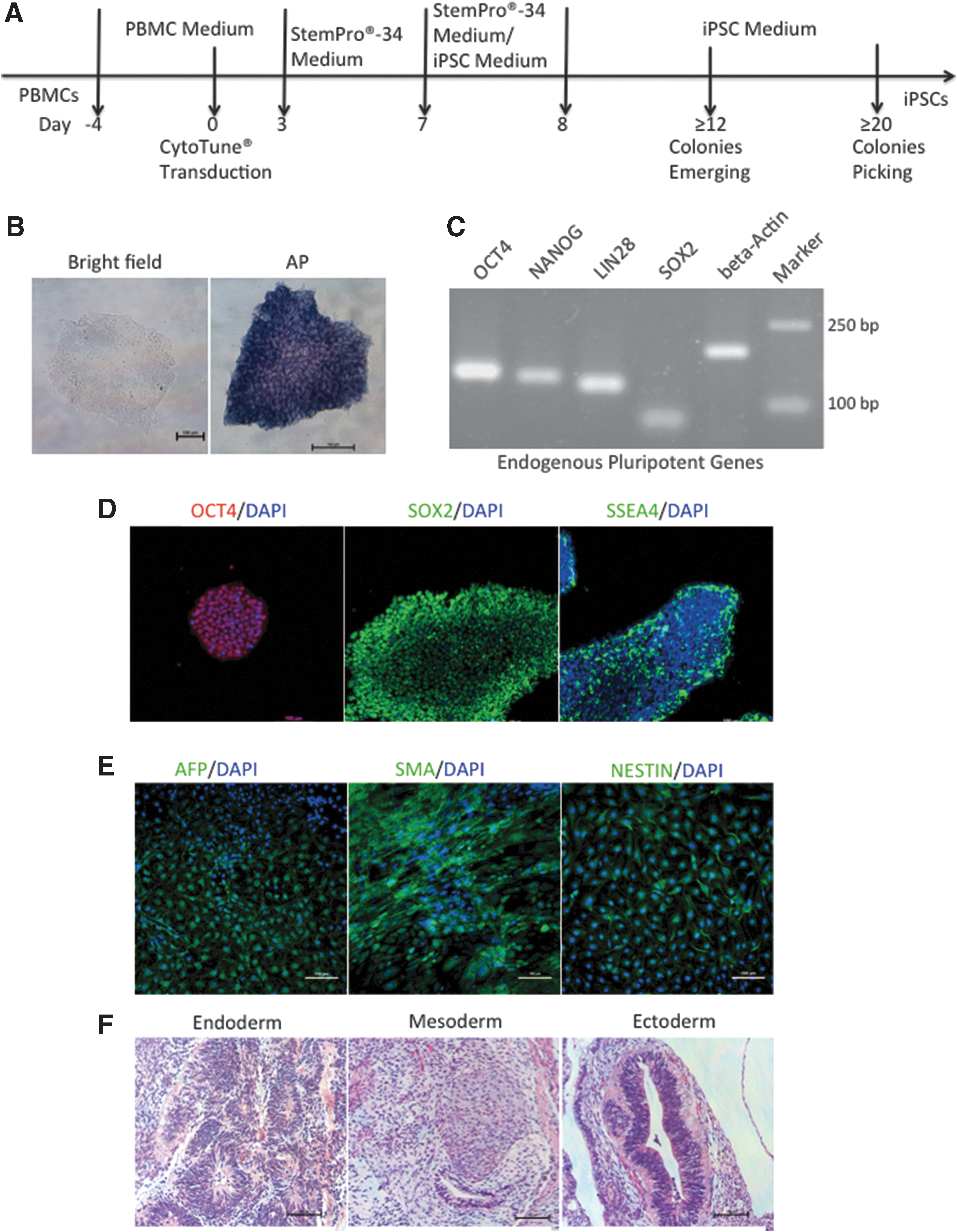
Characterization of iPSCs derived from PBMCs of an SCA3 patient.
HEK293T cells (ATCC, Manassas, VA) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM
Construction of CRISPR plasmids
The web tool (
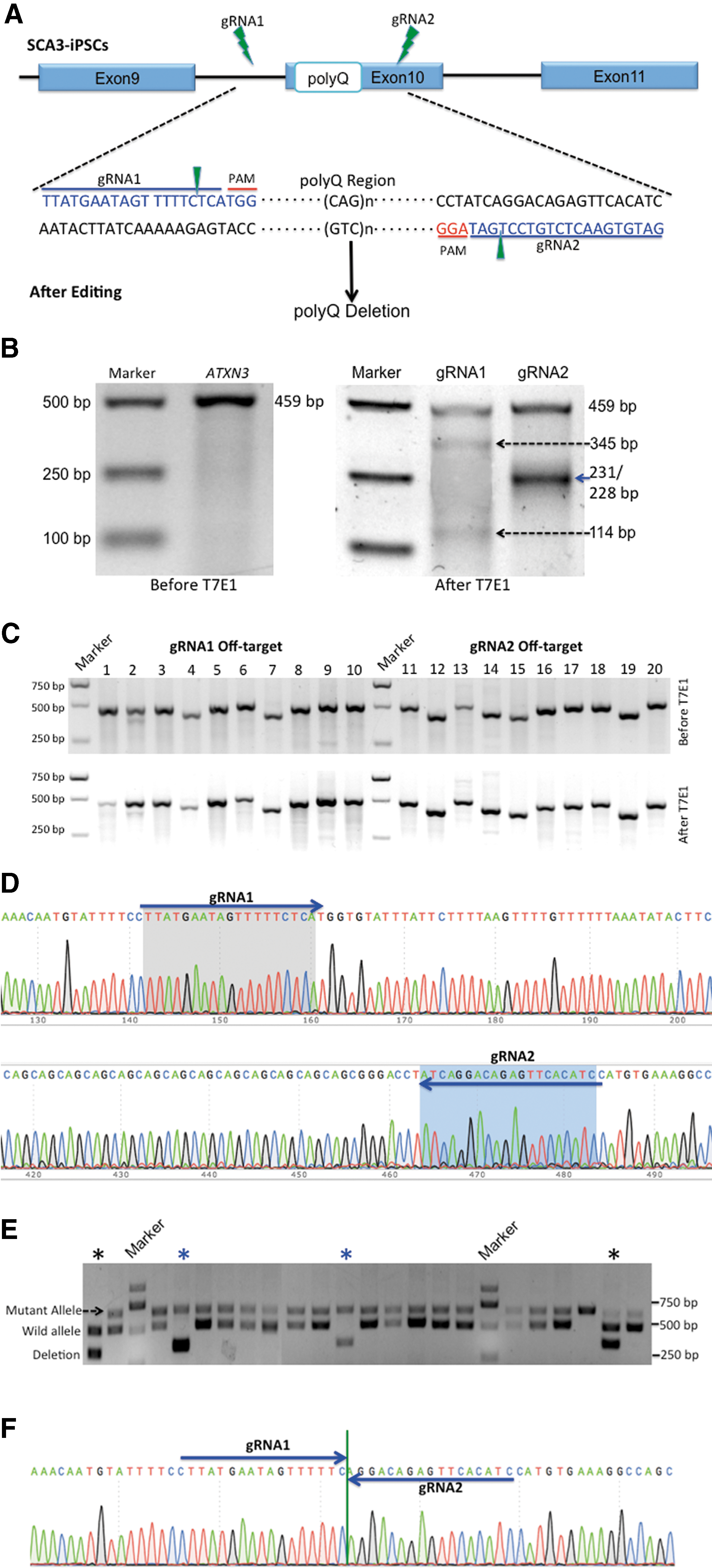
CRISPR/Cas9-mediated deletion of the polyQ-encoding region in SCA3-iPSCs.
T7 endonuclease I assay for on-/off-target analysis
HEK293T cells were transfected with CRISPR/Cas9-gRNA1 or CRISPR/Cas9-gRNA2 and incubated for 2 days. Then, genomic DNA from the cells was isolated using the DNA Extraction Kit (Tiangen, Beijing, China) according to the manufacturer's instructions. The on- or off-target sites for each gRNA were amplified by polymerase chain reaction (PCR). We purified the PCR products and detected heteroduplex formation using T7 endonuclease I (T7E1) (New England BioLabs) as described previously [22]. The products were separated by electrophoresis on a 2% agarose gel, and analyzed with ImageJ software (National Institutes of Health, Bethesda, MD). The following formulas were used to calculate the percentage of fraction cleaved:%fraction cleaved = 100 × sum of the cleavage product peak/(cleavage product + parent peak) [22].
Electroporation and fluorescence-activated cell sorting
For gene editing, we used 1 × 106 iPSCs for electroporation along with 5 μg CRISPR/Cas9-gRNA1, 5 μg CRISPR/Cas9-gRNA2, and the Neon Transfection System (Thermo Fisher Scientific, Waltham, MA), with two pulses of 1,200 V for 20 ms each.
After electroporation, we recovered the cells in mTeSR1 medium supplemented with 10 μM Rho-associated protein kinase inhibitor (Y-27632; Sigma-Aldrich, St. Louis, MO) for 24 h. At 48 h posttransfection, we collected green fluorescent protein (GFP)-positive cells by fluorescence-activated cell sorting (FACS) (AriaIII flow cytometer; BD Biosciences), and plated them in 6-cm dishes, culturing for ∼10 days until the clones were large enough for detection.
PCR screening and sanger sequencing
Portions of each clone were chosen to isolate DNA for PCR screening. Colonies were lysed in 20 μL of lysis buffer as previously described [23]. A portion of each sample was used for PCR (20 μL total volume) to detect modifications. PCR primers are listed in Supplementary Tables S1 and S2. PCR products were analyzed by agarose gel electrophoresis, targeted bands were purified and cloned into a pMD19-T vector (Takara, Shiga, Japan) according to the manufacturer's instructions, and the modified sequences were verified by Sanger sequencing.
Alkaline phosphatase staining and immunocytochemistry
Cells were treated with an Alkaline Phosphatase (AP) Staining Kit (Beyotime, Nanjing, China) according to the manufacturer's instructions. Immunocytochemistry (ICC) was performed as previously described [24], and 4′, 6-diamidino-2-phenylindole was used for nuclear staining. Cells were imaged using an inverted confocal microscope (Eclipse Ti, Nikon, Japan). The primary antibodies used in this study were as follows: rabbit anti-OCT4 (1:100; ab19857, Abcam, Cambridge, United Kingdom), mouse anti-SOX2 (1:100; AM2048a; Abgent, San Diego, CA), mouse anti-stage-specific embryonic antigen 4 (SSEA-4; 1:100; ab16287, Abcam), mouse anti-alpha1 fetoprotein (AFP; 1:200; ab3980, Abcam), mouse anti-Nestin (1:100; ab6142, Abcam), rabbit anti-smooth muscle actin (SMA; 1:200; ab5694, Abcam), mouse anti-Tubulin βIII (TUBB3; 1:200; 05-559, Millipore), mouse anti-microtubule-associated protein 2 (MAP2; 1:200; MAB3418, Millipore), and mouse anti-NeuN (1:200; MAB377, Millipore). The secondary antibodies used in this study were as follows: goat anti-mouse IgG (H+L) cross-adsorbed secondary antibody (Alexa Fluor® 488; 1:1,000; A11001, Thermo Fisher Scientific); goat anti-rabbit IgG (H+L) cross-adsorbed secondary antibody (Alexa Fluor 488; 1:1,000; A11034, Thermo Fisher Scientific), and donkey anti-rabbit IgG (H+L) cross-adsorbed secondary antibody (Alexa Fluor 594; 1:1,000; A21207, Thermo Fisher Scientific). To test the characteristics of neural stem cell (NSC), the Human NSC ICC Kit (Life Technologies), including NSC markers, such as paired box protein 6 (PAX6), SOX1, NESTIN, and SOX2, was used according to the manufacturer's instructions.
Reverse transcription–PCR
The TRIzol Kit (Invitrogen, Carlsbad, CA) and the PrimeScript RT Reagent Kit (TaKaRa) were used to isolate total RNA from cells and synthesize cDNA from 1 μg of RNA according to the manufacturer's instructions. Reverse transcription–PCR (RT-PCR) was performed to detect the expression of the SeV genome, transgenes, and endogenous pluripotency genes. Additionally, exon sequencing of ATXN3 was performed after RT-PCR.
In vitro and in vivo differentiation
To generate embryoid bodies (EB), iPSCs were incubated with 1 mg/mL dispase for 4 min and mechanically dissociated. The clumps of cells were transferred to ultralow attachment plates in human embryonic stem cells (hESC) culture medium without basic fibroblast growth factor (bFGF) supplementation. After 1 week of culture, EBs were transferred to gelatin-coated 24-well plates and cultured in the same medium for another week. Three germ layer-specific markers, AFP (endoderm), SMA (mesoderm), and NESTIN (ectoderm), were analyzed using immunofluorescent staining.
iPSCs from confluent 60-mm dishes were treated with 1 mg/mL dispase for 4 min, harvested by mechanical dissociation, resuspended in hES-qualified Matrigel matrix (Matrigel; 354277, Corning), and injected subcutaneously into the groin of a 6-week-old mouse with severe combined immunodeficiency. Eight weeks later, the tumors were dissected, fixed overnight in 4% (m/v) paraformaldehyde, embedded in paraffin, sliced, and stained with Hematoxylin and Eosin (H&E). The three germ layer-derived structures were then checked under a light microscope.
Neuronal differentiation
PSC neural induction medium (NIM; Thermo Fisher Scientific) was used to differentiate iPSCs to NSCs. iPSCs were treated with (NIM) containing 98% neurobasal medium (Thermo Fisher Scientific) and 2% PSC neural induction supplement (Thermo Fisher Scientific) for 7 days to obtain passage 0 (P0) NSCs. The NSCs were transferred to dishes coated with Matrigel and then cultured using neural expansion medium containing 49% Advanced DMEM/F-12 (Thermo Fisher Scientific), 49% neurobasal medium (Thermo Fisher Scientific), and 2% PSC neural induction supplement. Cells were dissociated with Accutase (Stem Cell Technologies) and passaged upon confluence. After dissociation of P0 to P4, NSCs were treated with Y-27632 (5 μM) overnight, which was required for plating. After obtaining P1 and later NSCs, NSCs were differentiated to NCs. NSCs were plated on dishes coated with poly-L-ornithine and laminin in StemPro NSC serum-free medium (SFM) complete medium containing 97% KnockOut DMEM/F-12, 2% StemPro neural supplement, 2 mM GlutaMAX-I supplement, 20 ng/mL bFGF, and 20 ng/mL epidermal growth factor at 2.5 × 104 cells/cm2. After 2 days, the culture medium was changed to neural differentiation medium (NDM) containing 97% neurobasal medium, 2% B-27 serum-free supplement, and 2 mM GlutaMAX-I supplement. The NDM was refreshed every 3–4 days until NCs maturation.
Western blot analysis
Proteins were extracted from cells lysed with RIPA buffer (Sigma-Aldrich) supplemented with protease inhibitor (Millipore) according to the manufacturer's instructions. Protein concentrations were determined using the BCA Protein Assay Kit (TaKaRa). Equal amounts of protein (50 μg) were separated by polyacrylamide gel electrophoresis and blotted onto nitrocellulose membranes. After blocking with 5% (w/v) milk, the membranes were then probed with rabbit anti-Ataxin 3 (1:1,000; ab175265, Abcam), mouse anti-poly-glutamine (polyQ; 1:1,000; MAB1574, Millipore), rabbit anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:1,000; AP0063, Bioworld), or rabbit anti-Vinculin (1:1,000; ab129002, Abcam) antibodies at 4°C overnight. The membrane was then incubated with anti-rabbit or anti-mouse IgG horseradish peroxidase-linked secondary antibody (1:10,000; LK2001 or LK2003; Sungene Biotech, Tianjin, China) for 1 h at room temperature. The blots were visualized using the Enhanced Chemiluminescence Detection Kit (Millipore), and protein expression was quantified using ImageJ software. The web tool (
Immunoprecipitation and ubiquitin-binding assay
Cells were lysed with IP lysis/wash buffer (Thermo Fisher Scientific) with protease inhibitor (Millipore). Whole cell lysates obtained by centrifugation were incubated with 3–5 μg of anti-ATXN3 antibody (13505-1-AP; Proteintech, Chicago, IL) overnight at 4°C followed by 2 h incubation with protein A sepharose beads (GE Healthcare). The precipitates were then washed with IP lysis buffer for three times and analyzed by western blot using anti-ubiquitin (1:1,000; 10201-2-AP, Proteintech).
Excitatory stimulation and solubility analysis
The iPSCs were differentiated to NCs (SCA3-NCs, N-NCs, and C-NCs-1/2) as described above. Then the cells were treated with L-glutamate (100 μM, Sigma) for 30 min left to recover for 30 min in differentiation media followed by a second 30 min, and subsequently cultured in differentiation media for 24 h until analyzed. After treatment, the cell protein was extracted into three fractions, including Triton X-100-soluble, sodium dodecyl sulfate (SDS)-soluble, and SDS-insoluble fraction according to previous study [24]. To that end, lysates of neuronal cultures were fractionated to identify ATXN3 species with different detergent solubility by Simple Western system (Protein Simple) according to the user manual and other study [25,26].
Statistical analyses
All statistical analyses were performed using ANOVA test with Bonferroni correction and graphically visualized using GraphPad Prism Software v.6.0. Results are expressed as mean ± standard error of the mean. Unless otherwise stated, all data shown represent the results of at least three independent experiments and P < 0.05 was considered significant.
Results
Characterization of iPSCs from human PBMCs of an SCA3 patient
We obtained nonintegrative iPSCs after 24 days from PBMCs collected from the SCA3 patient (SCA3-iPSCs) (Fig. 1A). We observed hESC-like morphology by inverted microscopy, and verified that the cells were positive for AP staining (Fig. 1B). RT-PCR analysis verified expression of endogenous pluripotency genes, such as OCT4, NANOG, LIN28, and SOX2 (Fig. 1C), and ICC analysis verified positivity for SOX2, OCT4, and SSEA4 expression (Fig. 1D). To verify that the SCA3-iPSCs were free of the SeV genome and transgenes, RT-PCR analysis found the absence of SeV, KOS, Klf4, and c-Myc in the target region (Supplementary Fig. S2A). To confirm the differentiation potential of SCA3-iPSCs further, we detected markers for three germ layers in vitro and analyzed the layer structure in vivo. Immunofluorescence examinations revealed AFP positivity in endoderm, SMA positivity in mesoderm, and NESTIN positivity in ectoderm (Fig. 1E). Additionally, a significant change was observed in teratoma formation in SCID mice at 8 weeks postinjection of SCA3-iPSCs. H&E staining confirmed that the teratomas contained various tissues comprising all three germ layers (endoderm, mesoderm, and ectoderm) (Fig. 1F).
CRISPR/Cas9-mediated deletion of the polyQ-encoding region of ATXN3 in the SCA3-iPSCs
To delete the polyQ-encoding region of ATXN3, we designed two gRNAs surrounding this region and inserted them into pSpCas9 (BB)-2A-GFP vectors (Fig. 2A). First, we evaluated the targeting efficiencies of each CRISPR/Cas9-gRNA using T7E1 in HEK293T cells. The lengths of PCR products were predicted by using bioinformatics analysis combined with agarose gel electrophoresis. We found that gRNA1 spliced the 459-bp PCR products into 345-bp and 114-bp bands, with a targeting efficiency of 36.0%. gRNA2 produced 231- and 228-bp bands, showing 32.3% efficiency (Fig. 2B). We then assessed off-target mutagenesis associated with each gRNA by the T7E1 assay and evaluated them against the top 10 potential off-target sites (Supplementary Table S3) predicted by a web tool (
gRNA1 binds to a region 61-bp upstream of the polyQ-encoding sequence, whereas gRNA2 binds 8-bp downstream of the sequence (Fig. 2D). Because the vectors contain GFP, we applied FACS to analyze the GFP-positive iPSCs at 2 days posttransfection, with GFP-positive cells accounting for ∼26.9% of the total cells (data not shown). After ∼10 days of incubation on Matrigel-coated dishes, GFP-positive colonies were large enough to be screened by PCR. SCA3-iPSCs were derived from a patient harboring a mutant allele consisting of 78 CAG repeats and a wild allele containing 26 CAG repeats. According to gel electrophoresis results, the mutant allele measured 651-bp, and the wild allele measured 495-bp.
After 213 clones were chosen for DNA extraction and PCR, cleaved products induced by gRNA revealed 340-bp bands in 24 clones, among which 11 contained no mutant allele (Fig. 2E). We chose these 11 clones harboring a wild allele that measured 495-bp accompanied with an edited allele that measured 340-bp. In two of these clones, we found that the expanded CAG repeats located between the two DSBs, which were cleaved 3-bp upstream of the protospacer-adjacent motif (PAM) sequence by either Cas9-gRNA1 or gRNA2, were deleted and the remaining sequences were joined together, as verified by Sanger sequencing (Fig. 2F and Supplementary Fig. S3). We chose these two edited clones, named as corrected iPSCs-1 (C-iPSCs-1) and corrected iPSCs-2 (C-iPSCs-2) for subsequent experiments.
C-iPSCs retain pluripotent characteristics
We applied similar methods to investigate C-iPSCs retention of the same pluripotent characteristics observed in parental SCA3-iPSCs. We observed hESC-like morphology in the bright field of an inverted microscope, and the cells were positive for AP staining (Fig. 3A). ICC staining for a panel of stem cell markers also showed uniform expression of SOX2, OCT4, and SSEA4 (Fig. 3B), and RT-PCR for endogenous pluripotency genes also returned positive bands similar to those seen in SCA3-iPSCs, with no respective bands observed in parental PBMCs (Fig. 3C). Immunostaining of EBs derived from the clones revealed that cells differentiated into endoderm, mesoderm, and ectoderm (Fig. 3D). Furthermore, histological examinations of the teratomas formed by the C-iPSCs showed various tissues comprising all three germ layers (endoderm, mesoderm, and ectoderm) (Fig. 3E).
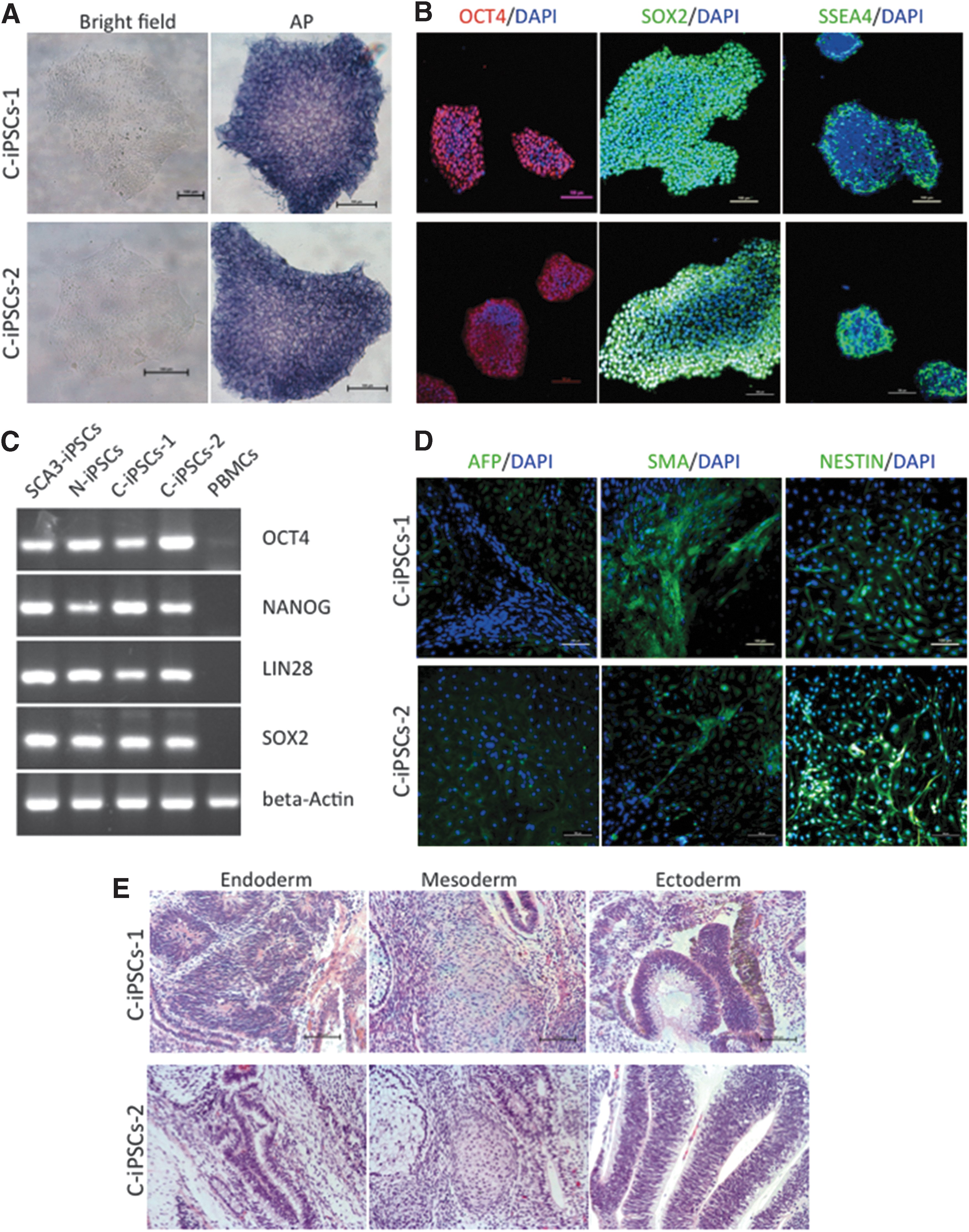
Characterization of C-iPSCs.
iPSCs differentiate to NSCs and NCs
SCA3-iPSCs, C-iPSCs, and N-iPSCs were differentiated to NSCs and NCs according to the protocol illustrated in Fig. 4A. Primitive NSCs were derived from iPSCs cultured with NIM after 1 week and verified by the expression of NSC markers, such as SOX1, PAX6, NESTIN, and SOX2, as detected by ICC staining (Fig. 4B). P0 NSCs were then obtained, and P1 or later NSCs were capable of differentiation to NCs in NDM culture, followed by transfer to StemPro NSC SFM complete medium. Subsequently, mature NCs were observed after one month. ICC staining confirmed the presence of classic neuronal markers, including TUBB3, MAP2, and NeuN (Fig. 4C), which indicated that SCA3-iPSCs, C-iPSCs, and N-iPSCs were capable of differentiation to both NSCs (SCA3-NSCs, C-NSCs, and N-NSCs) and NCs (SCA3-NCs, C-NCs, and N-NCs).
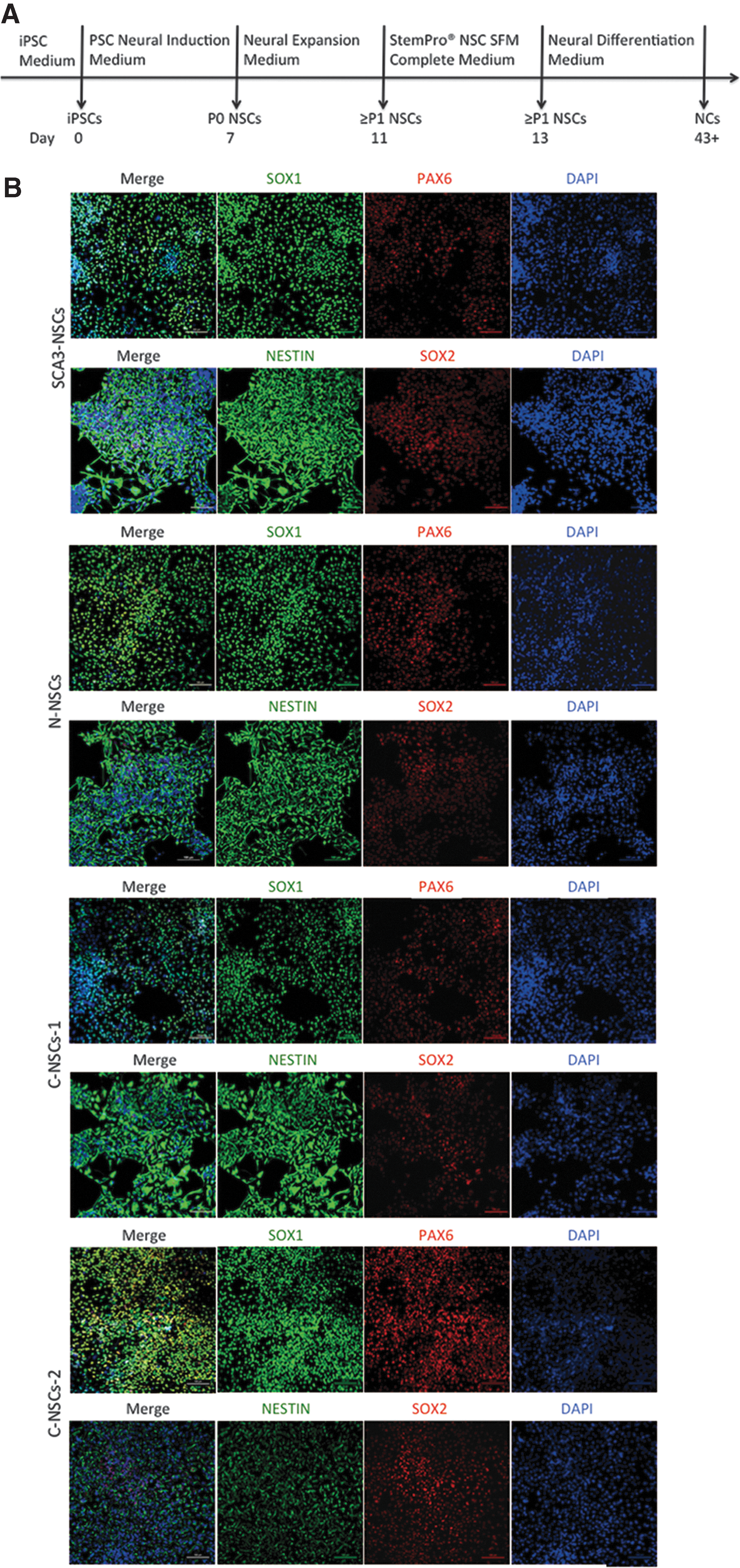
NSCs and NCs differentiation from SCA3-iPSCs and C-iPSCs.

CAG-repeat stability
To validate whether CAG-repeat lengths in ATXN3 were altered during reprogramming, passage, and differentiation, we performed PCR and fragment analysis to determine their lengths in the corresponding cells. All cells, including SCA3-PBMCs, SCA3-iPSCs, and SCA3-NCs showed stable CAG repeats by capillary electrophoresis and agarose gel electrophoresis (Fig. 5A and Supplementary Fig. S4A), which were also observed among NCs differentiated from C-iPSCs-1, C-iPSCs-2, and N-iPSCs (Fig. 5A).
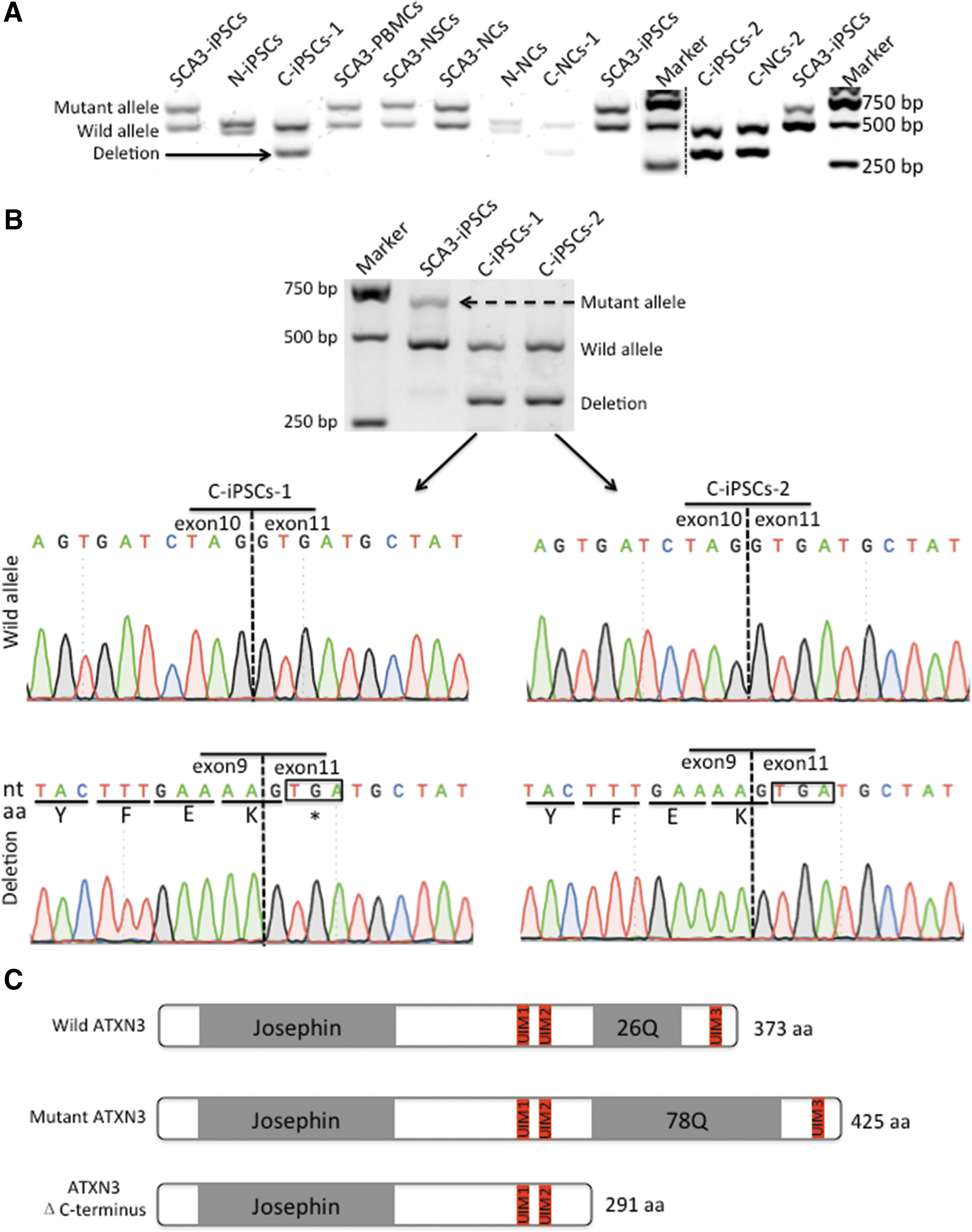
Altered ATXN3 expression and ubiquitin-binding capacity after expanded polyQ repeat deletion.

Exon sequencing of ATXN3
The polyQ-encoding region is located in exon 10 of ATXN3. To validate whether exon sequences were altered after gene editing, we performed RT-PCR and Sanger sequencing. After gene editing, the mutant allele measuring 613-bp changed into a length of 301-bp and the wild allele remained at a length of 457-bp in C-iPSCs-1/2 (Fig. 5B). The modified sequences were verified by Sanger sequencing. The wild allele retained the same sequence as the normal allele, without inverted sequences. The modified allele sequences were a deletion of exon 10, whereas exon 9 and exon 11 sequences joined together without change (Fig. 5B and Supplementary Fig. S4B–F).
Alterations in ATXN3 expression in gene-edited cells
Exon sequencing of ATXN3 showed that removal of exon 10 from the pre-mRNA resulted in a novel stop codon at the start of exon 11 (Fig. 5B). As a consequence, the truncated ATXN3 protein consisting of 291 amino acids with a predicted mass of ∼34 kDa lacked the C-terminal region containing the toxic polyQ repeat (Fig. 5C). Western blot analysis showed ATXN3 derived from SCA3-iPSCs and SCA3-NCs revealed two bands, including one encoded by the wild allele (∼42 kDa) and the other by the mutant allele (∼50 kDa). C-iPSCs1/2 and their differentiated NCs (C-NCs1/2) showed one wild allele (∼42 kDa) accompanied by a truncated allele (ΔC-terminus, ∼34 kDa) rather than the mutant allele. N-iPSCs and their differentiated NCs (N-NCs) showed the wild allele. Furthermore, the normal ATXN3 expression level showed no significant difference among SCA3-iPSCs, N-iPSCs, C-iPSCs, or their differentiated NCs (Fig. 5D).
For proteins involved in polyQ-linked neurodegenerative disorders, the anti-polyQ antibody represents a more effective method of detecting pathological proteins containing polyQ expansions as compared with antibodies targeting the wild variants [27]. Western blot results using the anti-polyQ antibody were positive for SCA3-iPSCs and SCA3-NCs, but there were no bands associated with proteins derived from C-iPSCs, N-iPSCs, C-NCs1/2, and N-NCs (Fig. 5E).
Modified ATXN3 maintains its ubiquitin-binding capacity
Immunoprecipitation was adopted to investigate whether the ubiquitin-binding capacity of ATXN3 in modified cells was still intact. Results showed that ATXN3 coprecipitated polyubiquitinated protein size was mainly larger than 25 kDa. The mutant ATXN3 in SCA3-NCs and wild ATXN3 in N-NCs could readily bind ubiquitin, indicating that the mutant ATXN3 retained the capacity to bind ubiquitin. Modified ATXN3 is consisting of a wild allele accompanied by a truncated allele without the third ubiquitin-interacting motifs (UIM), and it also bound ubiquitin comparable with full-length ATXN3 in C-NCs-1/2. Although the fact that short bands (<25 kDa) corresponding to mono-ubiquitin or di-ubiquitin were different among cells, including C-NCs-1 and C-NCs-2 may be related to clonal variance, these short bands only took a small proportion in the total ATXN3 coprecipitated ubiquitin. Therefore, the main ubiquitin-binding capacity after gene editing was still intact (Fig. 5F).
No ATXN3 aggregation induced by glutamate
SDS-insoluble ATXN3 aggregates were considered as potential hallmark of SCA3 pathology. Our study showed that the Triton X-100-soluble fraction revealed specific ATXN3 bands with or without
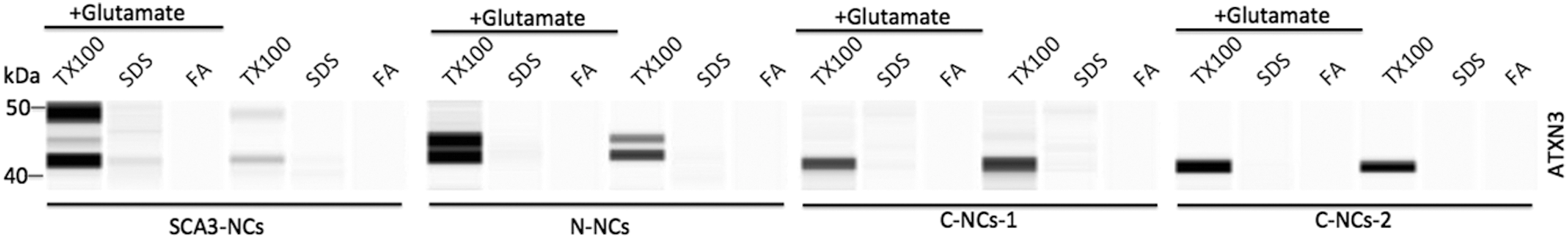
Detection of SDS-insoluble ATXN3 aggregates in NCs. After treatment with
Discussion
The polyQ expansion in exon 10 of ATXN3 leads to a gain of toxic protein function in SCA3. We successfully removed the expanded polyQ tract from SCA3 patient-specific cells by CRISPR/Cas9. Furthermore, the ubiquitin-binding capacity of ATXN3 was retained in NCs differentiated from corrected iPSCs.
Previous studies targeting mutant ATXN3 by silencing of specific genes and transcripts were based on transgenic animals and patient-derived fibroblast models [12,28]. We used SCA3 patient-derived iPSCs as a cellular model because of the advantages of iPSCs having the same genomic background, limitless self-renewal, and differentiation properties [29]. In our study, the nonintegrative SCA3-iPSCs maintained genomic stability during reprogramming, proliferation, differentiation, and gene editing (Supplementary Fig. S5), and it offered an opportunity to evaluate modified proteins in multiple cell types affected in SCA3. However, one of the challenges of human iPSCs-based disease modeling is the variability of function due to variations in genetic background [29]. Even if N-NCs showed higher oxygen consumption rates (OCR) than SCA3-NCs, we should add one more normal control line to support the conclusion in future (Supplementary Fig. S6). It also should be noted that although gene correction might reduce the phenotypic variability related to genetic background, variability due to clonal differences remains [30]. In our study, although the result showed no significant difference (P > 0.05) between C-NCs-1 and C-NCs-2 in the mitochondrial function test (Supplementary Fig. S6), multiple clones should be assessed even when using isogenic controls in future work.
In contrast to HDR, NHEJ occurs at a much higher frequency and requires no DNA repair template in the CRISPR/Cas9 system [31]. In addition, the intervening region can be deleted or inverted through NHEJ when a pair of gRNAs flanking a genomic region is used [32]. Therefore, we designed a pair of gRNAs to target sites in ATXN3, resulting in the deletion of the expanded CAG repeats that translated to the polyQ tract in mutant ATXN3 in our study. Additionally, sequencing of C-iPSCs showed that DNA sequences between two predicted cleavage sites were deleted without other insertions or deletions, indicating that NHEJ achieved precise genetic modifications using the gRNA pairs, in agreement with previous reports [17,31,33]. Off-target cleavage is a serious concern for CRISPR/Cas9-mediated genome engineering, and remains the main obstacle for clinical application [34]. In this study, we showed that predicted gRNA off-target sites were free of unintended mutations according to T7E1 assays. Additionally, we verified the genome stability of the gene-modified cells through chromosomal microarray and karyotype analyses (Supplementary Fig. S5C–E). Furthermore, we observed that C-iPSCs successfully differentiated into NCs, confirming the retention of differentiation capability similar to that observed for parental SCA3-iPSCs and N-iPSCs (Supplementary Fig. S7).
Although we obtained corrected cells that harbored the wild ATXN3 allele accompanied with the truncated allele without expanded CAG repeats, the process was time consuming and tedious. Focusing on improving efficiency and specificity, an allele-selective CRISPR/Cas9 strategy, based on PAM-altering SNPs at target patient-specific CRISPR/Cas9 sites, was used to completely inactivate the mutant allele without impacting the normal allele [35]. Recently, CRISPR/Cas9 has been used to reduce mutant Huntingtin expression selectively in vitro and in vivo [36]. We should use an allele-selective CRISPR/Cas9 strategy to reduce mutant ATXN3 in the future work.
ATXN3, a conserved and ubiquitous protein, which is known to bind polyubiquitin chains and to function as a deubiquitinating enzyme, contains an N-terminal Josephin domain that displays ubiquitin protease activity and a C-terminal tail with two or three UIMs, depending on the isoform [3]. Previous studies showed that the interaction between ATXN3 and K48-linked polyubiquitin chains is dependent on the first two UIMs, whereas the third UIM located C-terminally of the polyQ stretch appears dispensable for this process [12,37,38]. In this study, we excised expanded CAG repeats in exon 10, to remove the toxic polyQ repeat that is located between UIM2 and UIM3. Even if a partial DNA sequence of exon 10 remained, the total exon 10 was removed after RNA splicing, and the exon 10-skipping transcript resulted in a truncated ATXN3 protein lacking the third UIM due to the novel stop codon at the start of exon 11.
The ATXN3 protein (ΔC-terminus) lacks the third UIM while still containing the main known functional domains, including the catalytic Josephin domain (amino acids 1–180), the first two UIMs, and the valosin-containing protein-interacting motif (amino acids 257–291) [39]. Previous experiments showed that this truncated protein retained a ubiquitin chain-binding capacity similar to that of full-length ATXN3 [40]. Our modified ATXN3, which contained a normal allele accompanied by a truncated allele (ΔC-terminus), bound ubiquitin comparably to full-length ATXN3, indicating that gene editing did not negatively impact the ubiquitin-binding capacity.
Several potential pathogenic pathways triggered by expanded polyQ repeat are shared among the polyQ diseases [41]. Mitochondria are organelles that play crucial roles in the maintenance of cellular homeostasis and a clear link between mitochondrial dysfunction and polyQ diseases, such as Huntington disease and SCA3, has been demonstrated [30,42 –45]. It has been demonstrated that the mitochondrial respiration was significantly reduced in the SCA3 cells in contrast to normal control cells, and was improved in the SCA3 cells after FIR treatment [44]. Our mitochondrial respiration tests showed preliminary data that the basal respiratory, as well as maximal respiratory and adenosine-triphosphate (ATP) production were significantly reduced in the SCA3-NCs in contrast to N-NCs (Supplementary Fig. S6B–D). Otherwise, the basal respiratory, as well as maximal respiratory and ATP production were improved in the two corrected NCs as compared with uncorrected SCA3-NCs (Supplementary Fig. S6B–D), suggesting that elimination of expanded CAG repeats may result in higher mitochondrial respiration. However, as few studies showed mitochondrial dysfunction in SCA3-derived iPSCs and its differentiated cells, more cell lines, including normal control lines and SCA3 cell lines, should be used in the mitochondrial functional test in the future.
Apart from retaining ubiquitin-binding capacity, an important concern is that the ATXN3 protein (ΔC-terminus) should not result in a gain of toxic function. As
Interestingly, a recent study showed that truncated ATXN3 (ΔC-terminus) was not toxic to fibroblasts or U2OS cells, and overt signs of toxicity based on bodyweight and locomotor activity were not observed in mice expressing ATXN3 ΔC-terminus. Furthermore, an ATXN3 protein of similar size to the ΔC-terminus protein in untreated SCA3 animals was found perhaps indicating that this protein was also expressed in the transgenic mouse brain [40]. Coincidentally, we also found similar size bands (∼34 kDa) corresponding to the ΔC-terminus in untreated cells (SCA3-iPSCs, N-iPSCs, and their differentiated NCs), and their band intensity showed lower density than gene-modified cells (C-iPSCs-1/2, C-NCs-1/2) in our study. In addition, CCK8 assay showed that there were no cell viability differences among SCA3-NCs, N-NCs, and C-NCs-1/2 with or without glutamate treatment (Supplementary Fig. S8). So we speculated that the truncated ATXN3 alleles that can be induced from gene editing might be a naturally occurring isoform or cleavage fragment and are likely not a toxic variant. As a result, future studies are necessary to expand on the findings presented in this study to assess the cytotoxicity of modified ATXN3, and to evaluate whether the modified protein is fully functional and not toxic in vitro and in vivo.
In summary, we adopted CRISPR/Cas9 to allow permanent excision of expanded CAG repeats from mutant ATXN3 alleles in SCA3 patient-derived iPSCs. Deletion of the expanded polyQ-encoding region using a gRNA pair resulted in deletion of mutant ATXN3 in both genetically modified iPSCs and NCs. Furthermore, this modified protein without expanded polyQ repeat retained its ubiquitin-binding capacity of ATXN3 in gene-modified NCs. Our findings provide preliminary data for gene editing by CRISPR/Cas9 in SCA3, demonstrate the feasibility of using a single CRISPR/Cas9-gRNA pair to delete the expanded CAG-repeat region present in SCA3 patients, and could promote future research and strategies for SCA3-specific therapeutic applications.
Footnotes
Acknowledgments
This study was supported by the Frontier and key technology innovation special grant from the Department of Science and Technology of Guangdong Province (2016B030229008),the Natural Science Foundation of Guangdong Province (2014A030312012), the International Cooperation Project of Science and Technology Planning Project of Guangdong Province (2013B51000087), and the Science and Information Technology of Guangzhou Key Project (201508020258, 201400000003-4, and 201400000004-4).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.