
Abstract
Tissue healing is a highly complex process involving a cascade of biochemical and cellular events. Excessive inflammation can impair the healing response. Previous in vitro studies have shown that mesenchymal stromal cells can modulate macrophage-induced inflammation and, therefore, are promising candidates for cell-based therapies aimed at promoting tissue repair. Recently, cell sheets were introduced as a new method of delivering stromal cells to the repair site. The goal of the current study was to compare the effect of different types of stromal cell sheets on the inflammatory state of macrophages in vitro. We compared the effects of adipose tissue-derived stromal cell (ASC) sheets, bone marrow derived stromal cell (BMSC) sheets, and fibroblast sheets on macrophage functional phenotype using flow cytometric analysis, gene expression, as well as cell sheet protein secretion. This was evaluated with and without inflammatory stimulation. Viability and senescence for the different types of sheet were also evaluated. Macrophages cultured in ASC sheet conditioned medium (CM) displayed a higher fluorescence intensity of the anti-inflammatory CD206 surface marker than when cultured in BMSC sheet CM and expressed more CCL18 and IL1RA than when cultured in fibroblast sheet CM. Moreover, ASC sheets had higher cell viability and less senescent cells than BMSC sheets and fibroblast sheets. Taken together, ASC and BMSC can stimulate the anti-inflammatory macrophage (M2) phenotype to a better extent than fibroblasts. It is suggested that ASC sheets might outperform BMSC sheets in an inflammatory situation since ASC sheet CM induced-macrophages have more M2 characteristics, and ASC in the sheet was more viable.
Introduction
E
Emerging evidence suggests that macrophage dysfunction plays an important role in impaired tissue healing [5]. Modulation of macrophage phenotype and function could therefore be a targeted approach to resolve excessive tissue inflammation during healing [3,6].
We and others have shown that mesenchymal stromal cells (MSCs) can secrete an array of growth factors and cytokines [7,8]. Several studies indicate that these secreted soluble factors can influence macrophage function [9 –12]. Therefore, cell based therapies using MSCs may offer a future alternative to the current therapies for resolving excessive inflammation and promoting tissue healing.
Next to injecting the MSC as a suspension, cell sheets are investigated as a delivery method of stromal cells to the tissue that needs repair. Sheets have been shown to be superior over injection of single cells because of reduction in cell loss and better engraftment [13,14]. However, cell sheets from different cell sources are not well investigated and compared regarding their immunomodulatory capacities. Comparing cell sheets from different cell sources may enhance understanding to select the most suitable cell source to make cell sheets for different clinical applications.
Although the ability to regulate macrophage phenotype and function is described for several MSCs such as adipose tissue-derived stromal cells (ASCs) [15,16] and bone marrow derived stromal cells (BMSCs) [10,17,18], differences in regulation between these types of adult mesenchymal stromal stem cells and differentiated MSCs such as fibroblasts are actually unknown. Similarly, whether culturing of MSCs in multilayered cell sheets instead of single cell cultures affects macrophage regulation has not been tested yet.
Therefore, we investigated the following questions: (1) Can MSC sheets influence macrophage phenotype and is there a difference between sheets from ASC, BMSC, and fibroblasts? And (2) Does inflammatory stimulation of MSC sheets affect their regulation of macrophage phenotype? To investigate this, we generated conditioned medium (CM) from unstimulated and inflammatory stimulated multilayered cell sheets of ASC, BMSC, and fibroblasts and used these conditioned media to evaluate their effect on macrophage phenotype.
Materials and Methods
ASC, BMSC, and fibroblast isolation
Human subcutaneous abdominal adipose tissue was obtained as waste material (mean age 45 ± 11 years, all female) with approval of the Medical Ethics Committee of the Erasmus Medical Center, Rotterdam (MEC-2014-092). ASC was isolated as previously described [7,19] and expanded in a density of 8,000 cells/cm2 in low glucose Dulbecco's modified Eagle's medium (LG-DMEM; Gibco, Life Technologies, The Netherlands) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco), 50 μg/mL gentamycin, 1.5 μg/mL amphotericin B (All Invitrogen), 1 ng/mL fibroblast growth factor-2 (FGF2; R&D Systems, Minneapolis, MN), and 25 μg/mL
BMSC was isolated as previously described [20] from heparinized femoral-shaft marrow aspirates of patients undergoing total hip arthroplasty (mean age 49 ± 9 years, all male) with informed consent after approval by the Medical Ethics Committee of the Erasmus Medical Center, Rotterdam (protocol METC-2004-142). BMSC was expanded in Minimum Essential Medium-Alpha (Gibco) supplemented with 10% FBS (Gibco), 50 μg/mL gentamycin, and 1.5 μg/mL amphotericin B, in a density of 2,300 cells/cm2 supplemented with 1 ng/mL FGF2 and 25 μg/mL ascorbic acid-2-phosphate. With these isolation and culture methods, our group has previously shown that ASC and BMSC have their characteristic cell surface markers and tri-lineage differentiation ability [7,21,22].
Human dermal fibroblasts adult (HDFa) were purchased (Gibco; mean age 37 ± 1 years, male and female) and expanded in a density of 5,000 cells/cm2 in GlutaMAX DMEM (Gibco) supplemented with 10% FBS (BioWhittaker®; Lonza, Verviers, Belgium), 50 μg/mL gentamycin, and 1.5 μg/mL amphotericin B.
All cells were trypsinized at 85%–90% confluence with 0.25% trypsin EDTA (Gibco) and frozen with 10% DMSO (Sigma-Aldrich) in expansion medium before further use.
Preparation of CM from cell sheets and ASC in low density
Frozen cells were thawed, expanded, and used at passage 3–6. ASC, BMSC, and HDFa were seeded at a density of 400,000 cells/cm2 in a 12-well plate to create cell sheets. ASC was also seeded at a lower density of 50,000 cells/cm2 in a 12-well plate. After overnight adherence in culture medium specific for each cell type, cells were refreshed with 750 μL LG-DMEM supplemented with 1% FBS (Gibco) in the presence or absence of inflammatory stimuli, 10 ng/mL TNFα (PeproTech), and 25 ng/mL interferon γ (IFNγ; PeproTech, Rocky Hill, NJ). After 48 h, the medium was removed, and cells were washed once with phosphate-buffered saline (PBS; Gibco) before adding 750 μL/well X-VIVO™15 (Lonza) medium supplemented with 20% FBS (Lonza), 50 μg/mL gentamycin, and 1.5 μg/mL amphotericin B, as this medium promotes the growth and differentiation of macrophages [3,23]. Culture was continued for 24 h before harvesting this CM, to be used for monocyte culture at a later time point (Fig. 1A). The medium was collected and centrifuged at 250 g for 8 min to remove cell debris, and supernatant was stored at −80°C before further use. For each cell type and density, CM from three separate donors was used.
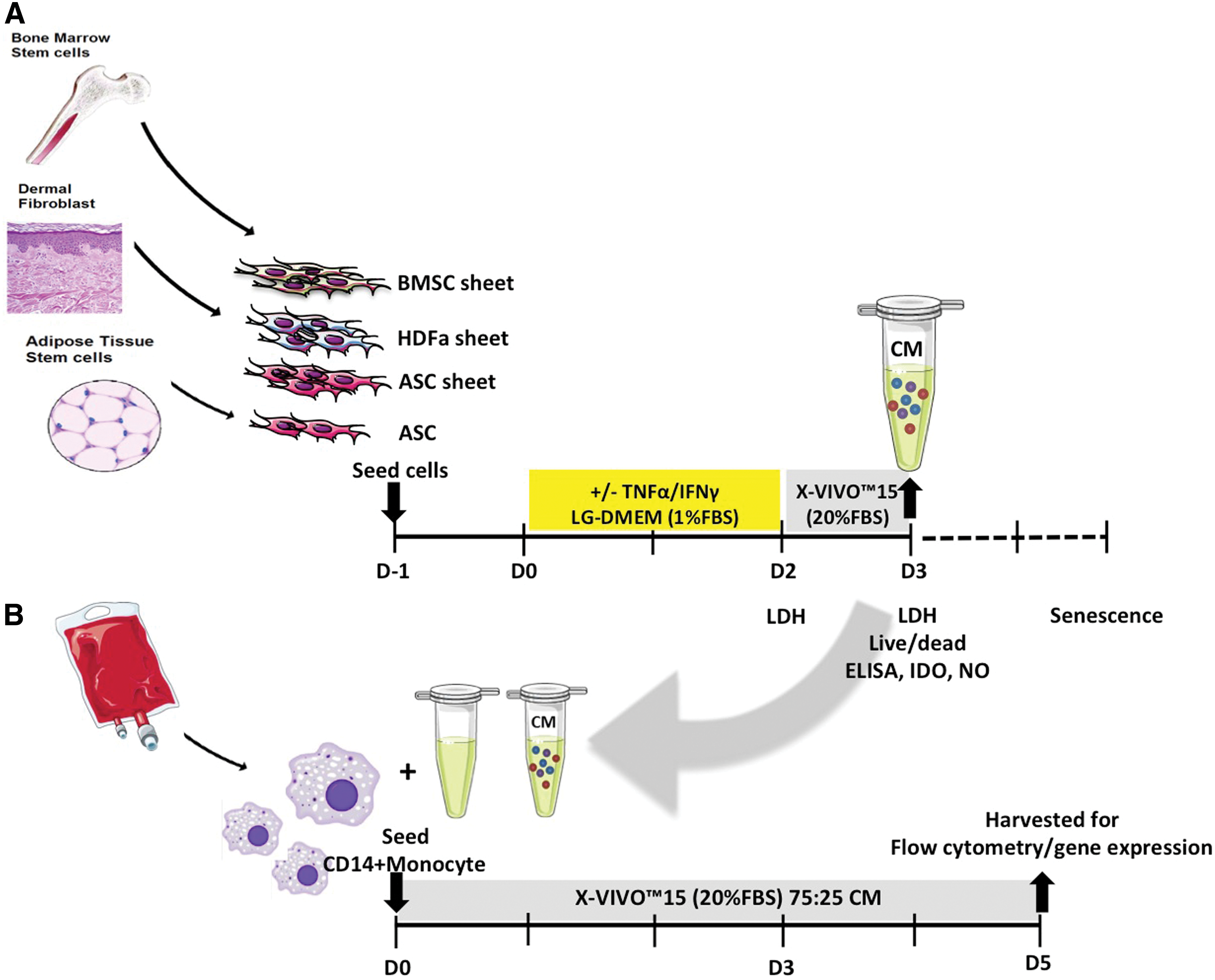
Diagram of cell sheet culture for preparation of CM and macrophage culture with CM.
Monocyte isolation and culture
Monocytes were isolated from a total of six buffy coats (all males, mean age 61 ± 11 years; Sanquin blood bank, Amsterdam, the Netherlands) using Ficoll (GE Healthcare, Little Chalfont, United Kingdom) density gradient separation and CD14 magnetic-activated cell sorting microbeads (Miltenyi, Bergisch Gladbach, Germany) as previously described [24]. Monocytes were seeded in 24-well temperature responsive culture plates (UpCell; CellSeed, Tokyo, Japan) for flow cytometry and in normal tissue culture polystyrene (TCPS) plates (Costar®; Corning, Inc.) for RNA isolation at 500,000 monocytes/cm2 and cultured in X-VIVO™15 containing 20% FBS (Lonza), 50 mg/mL gentamicin (Gibco), and 1.5 mg/mL amphotericin B (Gibco) at 37°C and 5% CO2 (Fig. 1B) to differentiate them toward macrophages.
To characterize the phenotype of macrophages, CD14+ monocytes of each donor were seeded separately and stimulated with 10 ng/mL IFNγ and 10 ng/mL TNFα to obtain pro-inflammatory macrophages, from now on referred to as M(IFNγ+TNFα) macrophages. The anti-inflammatory M2a subtype was obtained after stimulation with 10 ng/mL IL4 (PeproTech), from now on referred to as M(IL4). The anti-inflammatory M2c subtype was acquired by stimulation with 10 ng/mL IL10 (PeproTech), from now on referred to as M(IL10) as done previously [3]. Monocytes were exposed to each cytokine for a total of 5 days before harvesting. The culture medium was refreshed with fresh cytokines at day 3.
Collected CM of ASC, BMSC, and HDFa multilayer sheets (seeded at 400,000 cells/cm2) and of ASC single cell culture (seeded at 50,000 cells/cm2) was used to culture macrophages. Macrophages from CD14+ monocytes of three buffy coats were pooled (mean age 42 ± 21 years, all male), seeded in 500,000 cells/cm2, and stimulated with 25% CM. For each batch of CM (three donors per cell type and density), macrophages were cultured in triplicate. Macrophages were refreshed (with prewarmed medium in the case of the temperature responsive plates) at day 3 before harvesting at day 5 for further analysis.
Flow cytometric analysis
Macrophages cultured with stimulating factors or CM were analyzed using FACSJazz™ (BD Biosciences). Macrophages were nonenzymatically detached from the temperature responsive culture plates by reducing the temperature from 37°C to room temperature. Reducing the temperature of this culture plate below 32°C results in a hydrophilic surface, which in turn results in spontaneous detachment of the cells. After detachment, the macrophages were collected by pipette aspiration. After spinning for 5 min at 390 g and removal of the supernatant, cells were resuspended in 100 μL FACSFlow (BD Biosciences) with 2 × 105 cells per sample. Cells were incubated with conjugated antibodies (all BD Biosciences) against CD14 (allophycocyanin-H7, APC-H7; BD561384), CD64 (APC; BD561189), CD80 (PE-Cy™7; BD561135), CD163 (Peridinin-Chlorophyll Protein Complex, PerCPCy™5.5; BD563887), and CD206 (Fluorescein isothiocyanate, FITC; BD551135) for 15 min at room temperature in the dark. After washing twice with FACSFlow and PBS, samples were fixed in 200 μL of 1.8% paraformaldehyde in PBS for 20 min in the dark followed by washing and filtering through a 70 μm filter into FACS tubes before analysis. Flow cytometry was used with compensation with CompBeads (BD Biosciences) according to the manufacturer's protocol. Values were expressed as median fluorescence intensity (MFI) compared with an unstained control and CD14-only control to identify gating boundaries in FlowJo software version 10.0.7 (Tree Star, Inc.).
Gene expression analysis for macrophage phenotype
Adhering macrophages were harvested from the TCPS plate by resuspending them in 500 μL RNA-Bee™ (TEL-TEST, Friendswood, TX) per well. Total RNA was extracted according to the manufacturer's guidelines for mRNA isolation. Briefly, RNA was extracted with chloroform followed by precipitation with isopropanol and washing with 80% and 70% ethanol. Total RNA was quantified using a NanoDrop™ ND-8000 spectrophotometer (Thermo Scientific, Wilmington, DE), and 200 ng RNA was reverse transcribed into complementary DNA (cDNA) using RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). The mRNA levels of Glyceraldehyde-3-phosphate dehydrogenase (GAPDH; fw: GTCAACGGATTTGGTCGTATTGGG, rev: TGCCATGGGTGGAATCATATTGG) were used as housekeeper gene and analyzed with TaqMan® Gene Expression Assays (Applied Biosystems, Foster City, CA). Expression of IL1β (IL1B; fw: CCCTAAACAGATGAAGTGCTCCTT; rev: GTAGCTGGATGCCGCCAT), IL6 (fw: TCGAGCCCACCGGGAACGAA; rev: GCAGGGAAGGCAGCAGGCAA), TNFα (TNFA; fw: GCCGCATCGCCGTCTCCTAC; rev: AGCGCTGAGTCGGTCACCCT), CCL18 (CCL18; fw: GCACCATGGCCCTCTGCTCC; rev: GGGCACTGGGGGCTGGTTTC), and IL1RA (fw: AACAGAAAGCAGGACAAGCG, rev: CCTTCGTCAGGCATATTGGT) (all from Eurogentec, Liege, Belgium) was assessed using qPCR™ MasterMix Plus for SYBR® Green I (Eurogentec) as they can be used to discriminate between different macrophage phenotypes [3]. The amplification efficiency of all primers was between 0.90 and 1.05. Quantitative polymerase chain reaction was performed on a Bio-Rad CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA). Relative gene expression was calculated using the 2−ΔCT method.
CM analyses
Human TNFα, IL1RA DuoSet® (R&D Systems, Abingdon, United Kingdom), and Prostaglandin E2 (PGE2) (Enzo life Sciences BVBA, Antwerp, Belgium; ADI-930-001) ELISA Kits were used according to the manufacturer's instructions. Indoleamine 2,3-dioxygenase (IDO) activity in CM was measured in an
Cell sheet viability
Cell sheet viability was evaluated using a live/dead staining (Invitrogen; Life Technology, Foster City, CA). Briefly, adhered ASC sheets, BMSC sheets, and HDFa sheets were washed with PBS after removal of CM and incubated for 30 min in calcein AM dye 1 μL/mL and ethidium bromide dye 1.5 μL/mL at 37°C in a humid atmosphere with 5% CO2. The sheets were washed twice followed by analysis with fluorescence microscopy using Olympus IX71 inverted microscope. Images were captured with Cell F Imaging Software (Olympus, Hamburg, Germany, 2008). Second, lactate dehydrogenase (LDH, Cytotoxicity Detection Kit; Roche, Mannheim, Germany) was measured to determine the percentage of cytotoxicity, according to the manufacturer's protocol as previously described [7].
Cell sheet senescence
Cell sheets were evaluated for senescence by determining β-galactosidase expression. Senescent cell cytoplasm will stain blue to light blue after incubation with the substrate X-gal [26]. After harvesting day 3 CM, cell sheets were trypsinized to obtain single cells. These cells were plated in 24-well plates at 2,000 cells/cm2. After overnight adherence at 37°C in a humid atmosphere with 5% CO2, cells were washed twice with PBS and fixed with 1% formaldehyde/0.5% glutaraldehyde in Milli-Q for 15 min at 4°C. After rinsing with Milli-Q, 500 μL of β-galactosidase staining solution, consisting of 400 mM citric acid/sodium phosphate, 1.5 M NaCl, 20 mM MgCl2, 500 mM potassium ferrocyanide, and 20 mg/mL X-gal (Roche Diagnostics; in dimethylformamide), was added to the cells. Cells were then incubated with this staining solution for 24 h in a dry incubator at 37°C. After washing, cells were counterstained with 4′, 6-Diamidine-2′-phenylindole dihydrochloride (DAPI; Sigma-Aldrich) 1:10,000 in Milli-Q for 5 min at room temperature. The images were captured at bright and dark field on a Nikon Eclipse fluorescent microscope using 10 × objective with 0.38 μm per pixel. Images were blinded before counting blue β-galactosidase positive cells and total cells to determine the percentage of senescent cells.
Statistical analyses
Data are presented as mean ± standard deviation (SD) and analyzed with Kruskal–Wallis one-way analysis of variance followed by Dunn-Bonferroni post hoc tests for multiple comparison. Values with P < 0.05 or less were considered significant. All statistical analyses were done using SPSS 21.0 (IBM, Inc., Armonk, NY).
Results
Macrophage subtype surface marker expression
Median fluorescent intensity (MFI) of CD64 was highest in the M(IFNγ+TNFα). Likewise, CD80 MFI was higher in M(IFNγ+TNFα) than in M(IL10). CD163 MFI, however, was highest in M(IL10), and CD206 MFI was highest in M(IL4) (Fig. 2A). The same trend was seen for each marker regarding the percentage of positive cells (Fig. 2B).
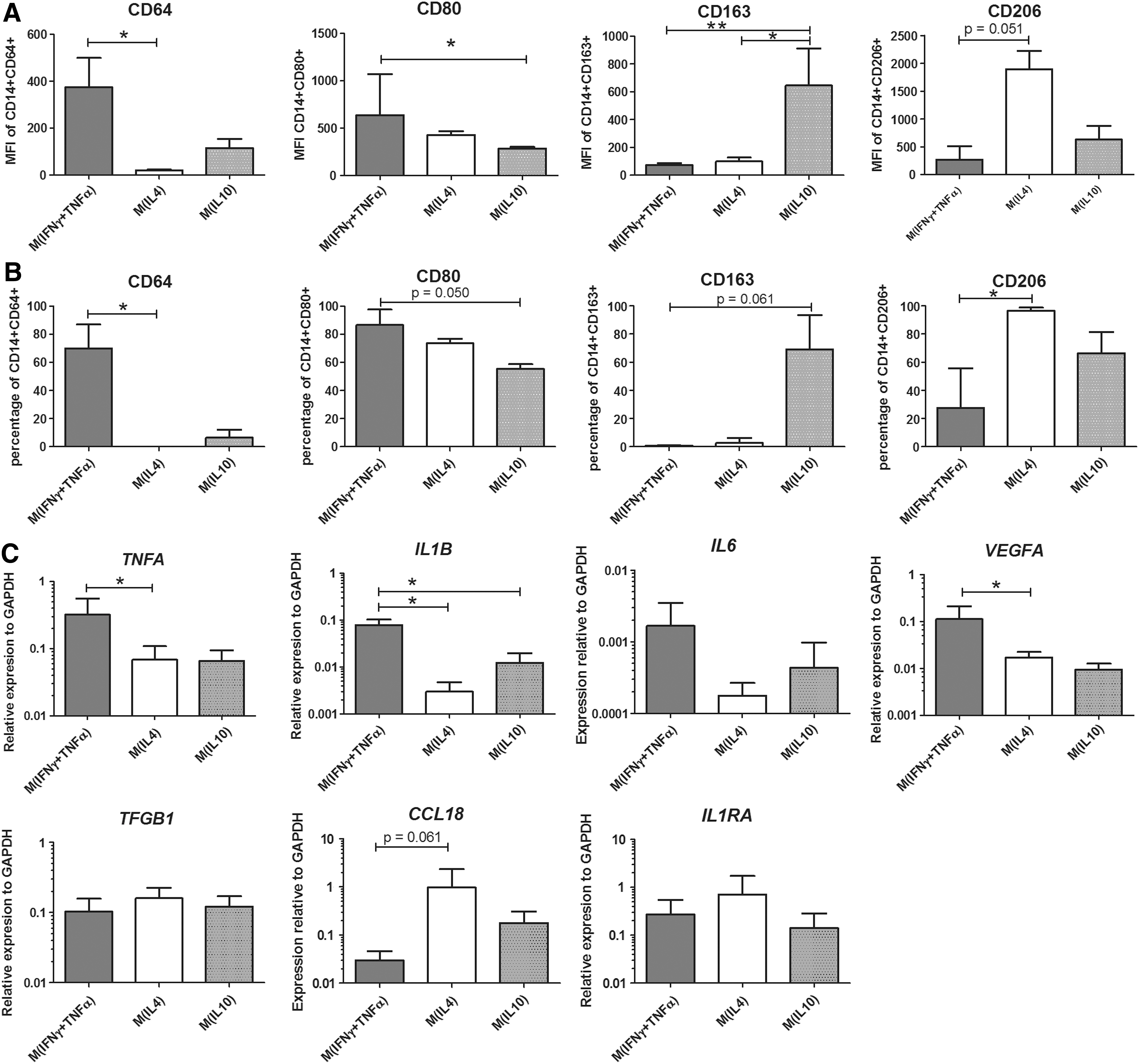
Macrophage subtype characteristics.
Macrophage subtype gene expression
Expression of genes encoding for pro-inflammatory cytokines TNFA and IL1B and the angiogenic factor VEGFA was significantly higher in pro-inflammatory M(IFNγ+TNFα) than in the anti-inflammatory M(IL4) and M(IL10) macrophages, while expression of IL6, CCL18, TGFB1, and IL1RA was not significantly different between the three conditions (Fig. 2C).
Effect of CM on macrophage surface marker expression
The MFI of CD64, CD80, and CD163 was not different between macrophages cultured in CM from ASC-, BMSC-, or HDFa sheets. CD206 MFI was higher in macrophages cultured in CM from TNFα+IFNγ stimulated ASC sheets than from TNFα+IFNγ stimulated BMSC sheets (Fig. 3A).
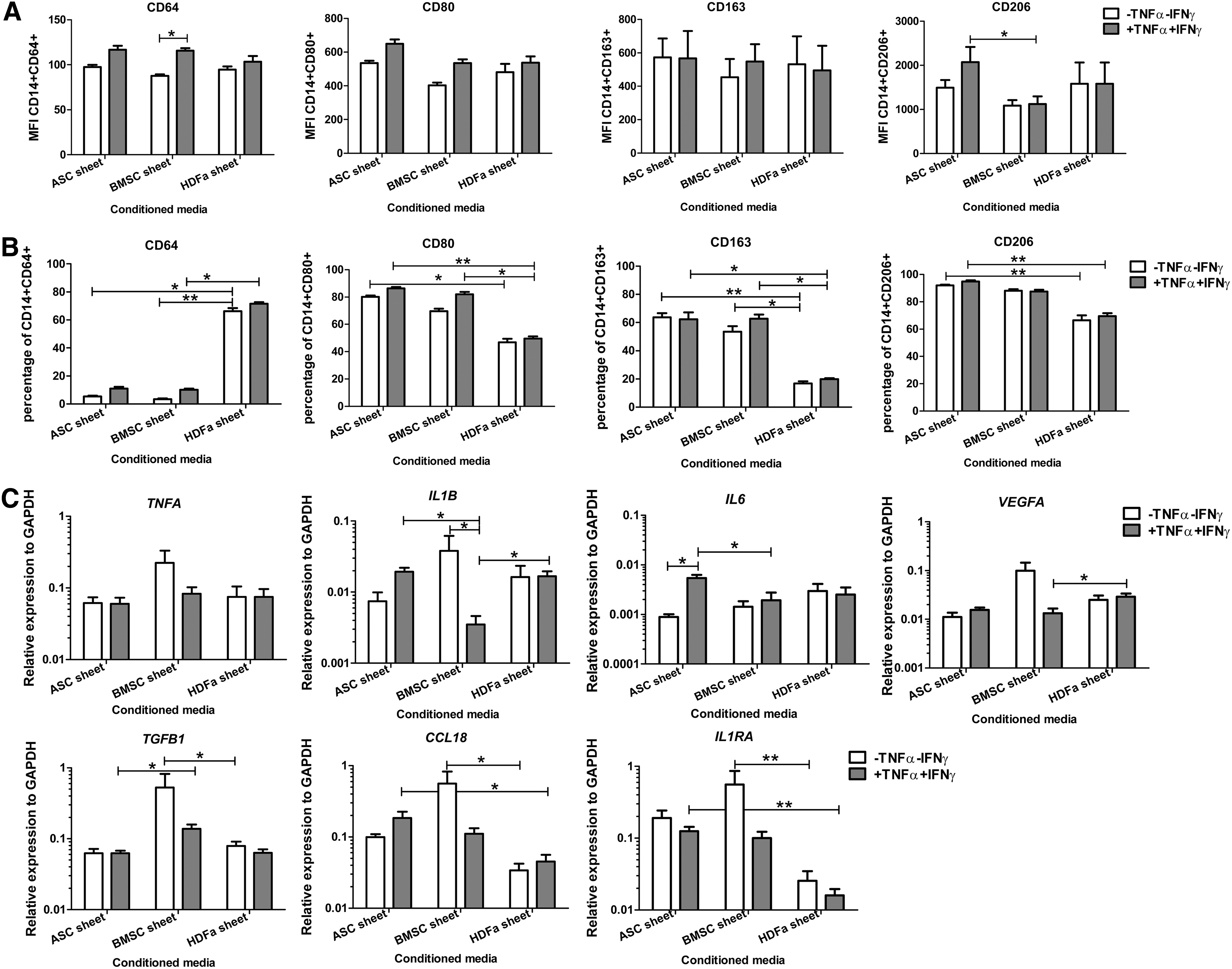
Effect of cell sheet CM on macrophage characteristics. Macrophages were cultured in 25% CM. Before making CM, cell sheets were either cultured in the absence (white bars) or presence of TNFα+IFNγ (gray bars).
In addition, CD64 surface marker expression was higher in macrophages cultured in CM from TNFα+IFNγ stimulated BMSC sheets than in CM from unstimulated BMSC sheets. Although the MFI of CD64, CD80, and CD163 was not different between macrophages cultured in CM from the different sources, significantly higher percentages of positive macrophages were seen for the surface marker CD64 and significantly less for CD80 and CD163 after culture in CM from HDFa sheets than CM from ASC sheets and BMSC sheets. Especially for CD206, those macrophages cultured in CM from HDFa sheets have lower percentages of positive cells than macrophage culture in CM from ASC sheets (Fig. 3B).
Effect of CM on macrophage gene expression
TNFA in the macrophages was unaffected by any of the sheet-conditioned media. Comparison among cell sheets revealed that macrophages cultured in CM from unstimulated BMSC sheets have higher TGFB1, CCL18, and IL1RA expression than macrophages cultured in CM from HDFa sheets.
When sheets were stimulated with TNFα+IFNγ, expression of IL1B was lowest in macrophages cultured in CM from BMSC sheets. Moreover, IL6 macrophage expression was higher in response to CM from ASC sheet than in CM from BMSC sheet. Besides that, macrophages expressed higher levels of VEGFA when cultured in stimulated CM from HDFa sheets than in CM from stimulated BMSC sheet. In contrast, TGFB1 was higher in macrophages cultured in CM from stimulated BMSC sheet than in CM from stimulated HDFa sheets.
Finally, expression of CCL18 and IL1RA was higher in macrophages cultured in CM from stimulated ASC sheet compared to CM from stimulated HDFa sheets (Fig. 3C).
Effect of cell source on cell sheet secretory profile
When cultured without TNFα+IFNγ, PGE2 production was not different between the conditioned media from the different cell sheets. When cultured with TNFα+IFNγ, PGE2 production was similarly increased in the conditioned media from all types of cell sheets.
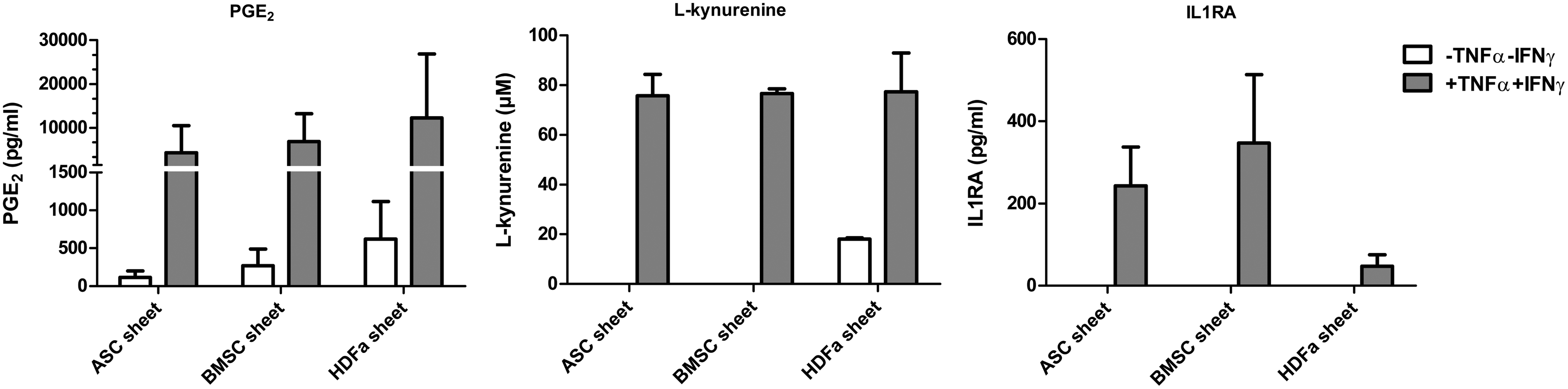
Levels of PGE2,
BMSC sheets are less viable than ASC sheets and HDFa sheets
As seen with live/dead staining, all cell sheets remained viable (green) at day 3 after harvesting CM, although some dead cells (red) were seen (Fig. 5A). At day 2 and 3, BMSC sheets had a significantly higher percentage of LDH release than ASC sheets and HDFa sheets. Changing sheet culture medium to macrophage culture medium containing 20% FBS lowered the LDH release ∼10 times in every condition.
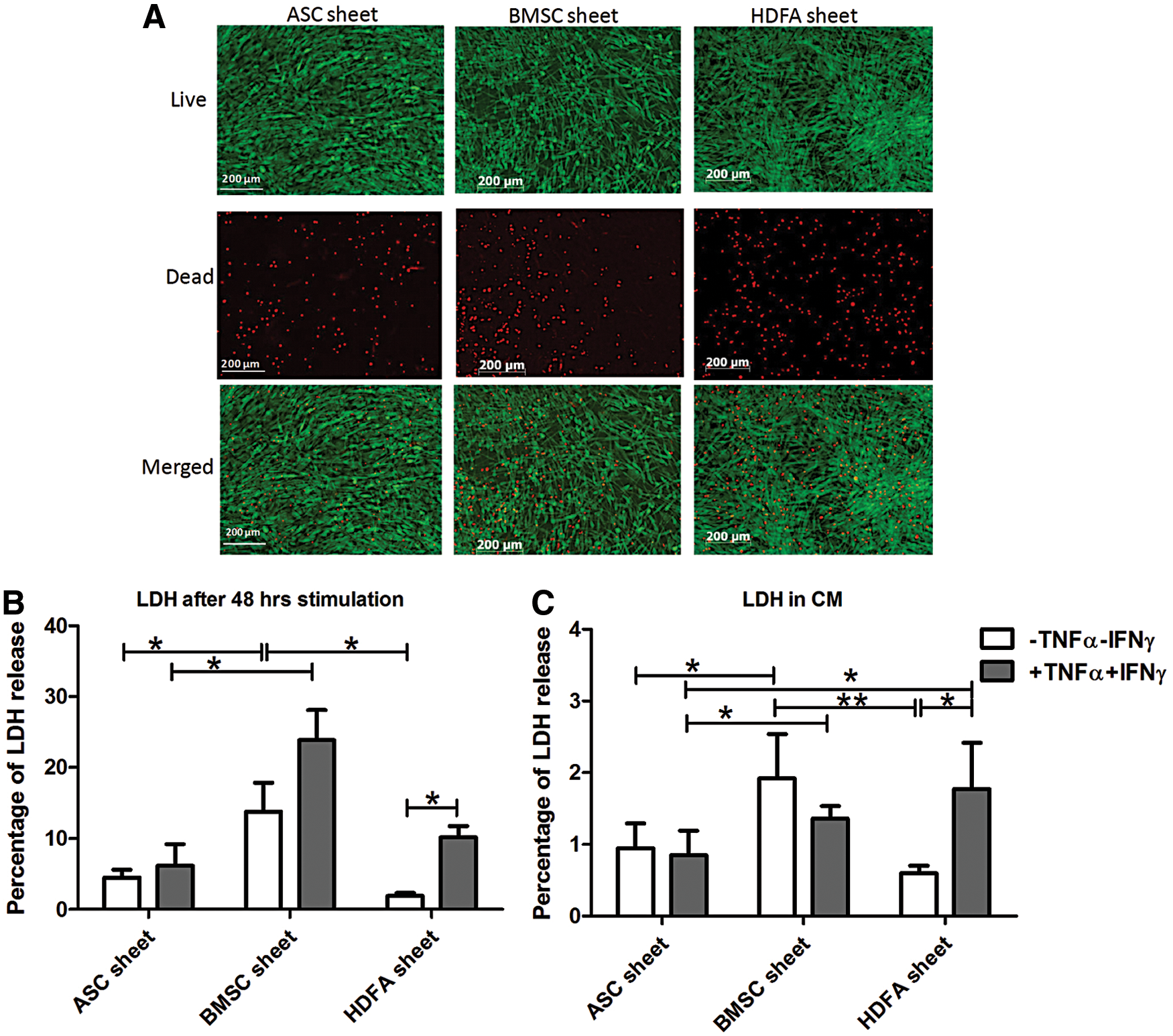
Cell sheet viability.
Stimulation of the sheets with TNFα+IFNγ did not change LDH release in ASC or BMSC sheet, but did increase LDH release in HDFa sheets (Fig. 5B, C).
ASC is less senescent than BMSC or HDFa in cell sheets
ASC sheets have the lowest percentage of senescent cells compared to BMSC and HDFa sheets, especially after TNFα+IFNγ stimulation. Exposure to TNFα+IFNγ lowered the percentage of senescent cells in both ASC and BMSC sheets (Fig. 6).
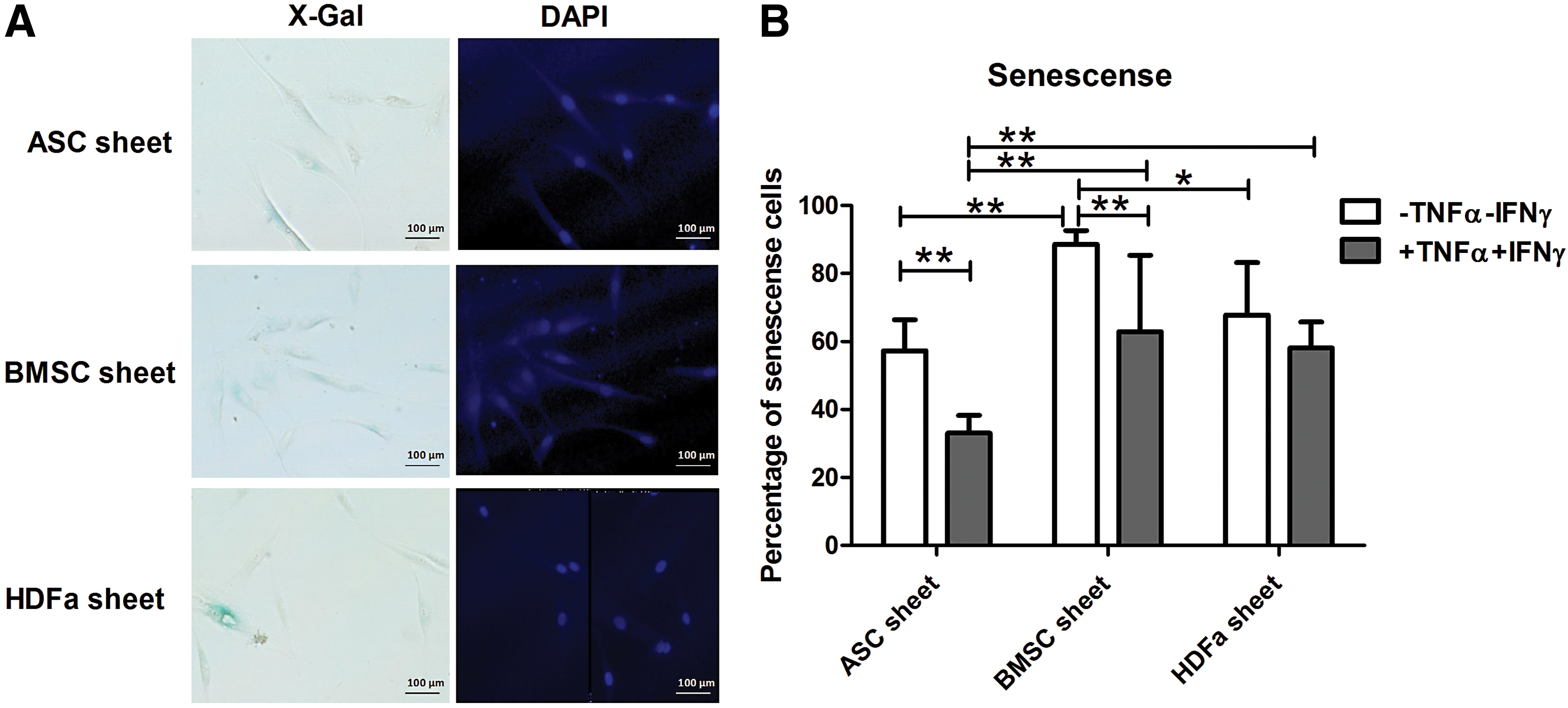
Cell sheet senescence.
Effect of ASC density on macrophage characteristics
Macrophages cultured with CM from ASC sheets (400,000 cells/cm2) have a higher MFI for CD64, CD80, CD163, and CD206 than when cultured in CM from single culture ASC (50,000 cells/cm2), independent of whether the ASC was cultured in the presence of TNFα+IFNγ (Fig. 7A). Same trends were seen regarding the percentage of positive cells for each of the marker sets, although almost all CD14 positive cells were positive for CD206 (Fig. 7B).

Effect of CM from ASC cultured in 50,000 and 400,000 cells/cm2 on macrophage characteristics.
Gene expression for TNFA, IL1B, IL6, VEGFA, CCL18, and IL1RA was not different between macrophages exposed to CM from ASC sheets or single culture. Only TGFB1 was lower in macrophages cultured in CM from TNFα+IFNγ stimulated ASC sheet compared to culture in CM from stimulated ASC in single culture (Fig. 7C). Stimulation of ASC in single culture before making CM resulted in a higher CD64/CD14 MFI, percentage of CD64+CD14+ cells, percentage of CD80+CD14+ cells, and increased expression of TNFA and IL6 by macrophages exposed to this CM.
Effect of cell density on ASC secretory profile
PGE2,

Secretion of PGE2,
Discussion
Stromal cell sheets in tissue healing have promising features, but detailed information on the effects of stromal cell sheets on immune cell phenotype and function is currently lacking. We investigated the effects of three different types of stromal cell sheets—ASC, BMSC, and fibroblast sheets—on the phenotype of macrophages that are key immune cells in tissue healing. Indeed, CM of the three different types of stromal cell sheets differentially influenced macrophage phenotype. Macrophages cultured in stimulated ASC sheet CM expressed more CD206 on their surface and more IL1B and IL6 and lower TGFB1 than macrophages cultured in stimulated BMSC sheet CM. These macrophages also expressed more CCL18 and IL1RA than macrophages cultured in HDFa sheet CM. Finally but not less important, ASC sheets had a higher viability and a lower percentage of senescent cells than BMSC sheets and HDFa sheets.
The different macrophage phenotypes, M(IFNγ+TNFα), M(IL4), and M(IL10), are characterized based on surface protein expression and gene expression, in which M(IFNγ+TNFα) can be used as a model for M1 macrophages, M(IL4) for M2a, and M(IL10) for M2c macrophages [27]. Although macrophages are plastic and their phenotypes are not black and white, especially in vivo, distinctions could be made between these phenotypes as shown before [3,28].
Based on the number of positive CD163 and CD206 cells and the expression quantity (MFI) as determined by flow cytometric analysis, macrophages seem to differentiate toward an M2 phenotype in response to CM of ASC sheets and BMSC sheets, irrespective of whether the sheets were stimulated with IFNγ and TNFα or not.
Looking at the expression quantity with MFI, macrophages cultured in CM of stimulated ASC sheets express the highest levels of CD206. This is similar to a previous study in which ASC increased CD206 after a 5 day coculture with macrophages [29]. The differentiation toward M2 is supported by more CCL18 and IL1RA expression in macrophages cultured in stimulated ASC sheet CM than in stimulated HDFa sheet CM. When comparing flow cytometric and gene expression data, ASC sheets and BMSC sheets seem to influence macrophages quite similarly. However, viability of stimulated ASC sheets was the highest and senescence the lowest compared to both BMSC and fibroblast sheet, making ASC sheets more promising candidates to modulate macrophages toward a M2 phenotype in an inflamed environment.
HDFa sheets had the opposite effect on macrophages; more cells were positive for CD64, an M1 surface protein, and had the lowest gene expression for CCL18 and IL1RA. In addition, LDH release from HDFa increased in response to IFNγ and TNFα, indicating that HDFa sheets might be less suitable for implantation in an inflamed area.
Cell sheet viability was measured in our standard sheet medium stimulated with IFNγ and TNFα (consisting of LG-DMEM with 1% FBS) and in the medium normally used for macrophage cultures (X-VIVO with 20% FBS). The macrophage medium conditioned by the three different stromal cell types was used for macrophage culture. Switching to macrophage medium containing 20% FCS instead of 1% FCS dramatically reduced the LDH release in all stromal cell sheets by 10-fold. Compared to BMSC and fibroblast sheet, however, ASC sheets had the lowest percentage of LDH release in both culture media, representing lowest cell death.
Initially, HDFa sheets had relatively low cell death but after stimulation with IFNγ and TNFα, LDH release increased. This might imply that HDFa sheets might be less suitable for implantation in an inflamed environment. Similar to LDH release, cell senescence was lower in ASC sheet compared to BMSC and fibroblast sheets, especially after stimulation with IFNγ and TNFα. Senescent MSC has lower ability to proliferate, differentiate, and migrate and has increased pro-inflammatory cytokine secretion [30,31]. Moreover, senescent cells also influence polarization of nearby macrophages toward M1 [32]. This might explain why ASC sheets more strongly drive macrophage differentiation toward the M2 phenotype than BMSC and fibroblast sheets.
To look into the mechanism behind the effect of stromal cells on macrophage differentiation, we measured PGE2 and
Besides viability and senescence, cell number can also influence the level of cytokine production. However, we previously found that cytokine production did not solely increase because of an increase in cell number. In addition, a higher production of cytokine per cell was observed within cell sheets [7]. In addition, since cells will be applied as a sheet, we believe that the total production of factors is more important than the production of factors per cell, and therefore, we did not correct the production of factors for the number of cells or DNA.
From our comparisons, ASC seemed the most promising stromal cells in sheets not only with regard to modulation of the macrophage phenotype but also regarding viability and senescence. We previously found that the ASC density has an effect on cytokine production, endothelial cell proliferation, and fibroblast migration [7], but the effect of different densities on macrophages was not investigated. Therefore, we decided to investigate the effect of two different ASC densities on the macrophage phenotype. Although PGE2,
Many previous studies investigated the relationship between MSC and macrophages using coculture of these cells. For practical reasons, we chose to use CM since we aimed to discriminate between what was expressed by the macrophages and what was expressed by the stromal cells. This makes it therefore difficult to compare our results to previous results. Using CM from stromal cells to culture macrophages allowed us to focus on the effect of the secretory profile from the stromal cells. A coculture would contain secretory factors from both cell types. Even if factors would have been measured in the CM after removal from the sheets and after culture of the macrophages in the stromal cell CM, distinguishing which factor is made by which cell will be difficult since natural decay of the factor is unknown. We therefore analyzed CM-stimulated macrophages for the expression of genes encoding several relevant cytokines to evaluate the effect of stromal cell CM on macrophage phenotype. Gene expression and protein secretion are however not necessarily correlated. Discrepancies may be due to (post-) transcriptional or the secretory level of the protein under investigation. Nonetheless, we previously showed that expression of IL6, CCL18, and CD163 corresponded to their respective release by stimulated macrophages, suggesting that stimulated macrophage gene expression and protein secretion follow a similar trend [3].
In the current setup, macrophages were cultured with the different CMs, without taking a condition along in which macrophages were cultured without CM. As we have seen earlier [33], plating out monocytes in monolayer might be a stimulus itself. In addition, in vivo, macrophages will also always have certain stimuli around them. We therefore believe that comparing the effect of the different CMs with unstimulated macrophages might be less relevant. Availability of stromal cell donors was unfortunately limited and especially for the BMSC we had limited freedom to match both age and gender. We therefore aimed to include patients with approximately the same age.
To conclude, ASC and BMSC sheets stimulate the anti-inflammatory macrophage (M2) phenotype more than fibroblasts. These findings suggest that ASC sheets are most suitable for promoting tissue healing since ASC sheets more strongly drive macrophage differentiation toward the M2 phenotype than BMSC and fibroblast sheets and also have a lower senescence and cell death, especially in an inflammatory environment. For example, we can envision the use of ASC sheet for therapies in chronic inflammatory wounds. This information might allow people to choose the suitable cell source to make a cell sheet for each clinical application.
Footnotes
Acknowledgments
The authors express their gratitude to Prof. Dr. S.E.R. Hovius, Dr. M.A.M. Mureau, and all surgeons of the department of Plastic Surgery for the collection of subcutaneous adipose tissue. The authors also thank Johannes Lehmann for supplying the β-galactosidase staining protocol, Andrea Lolli for blinding the images taken after β-galactosidase staining, and Caoimhe Kiernan and Eric Farrell for their help with setting up the flow cytometric protocol. P.S. is supported by a grant from the Netherlands Fellowship program (NFP-12/435), during the conduct of the study. Y.M.B.-J. is supported by a grant from Dutch Arthritis Association (LP11).
Author Disclosure Statement
No competing financial interests exist.