
Abstract
Paired box protein 6 (PAX6) is a master regulator of the eye development. Over the last past two decades, our understanding of eye development, especially the molecular function of PAX6, has focused on transcriptional control of the Pax6 expression. However, other regulatory mechanisms for gene expression, including alternative splicing (AS), have been understudied in the eye development. Recent findings suggest that two PAX6 isoforms generated by AS of Pax6 pre-mRNA may play previously underappreciated role(s) during eye development, especially, the corneal development.
Alternative Splicing of Pre-mRNA As an Important Regulatory Mechanism in Gene Expression
A
The regulation of AS is mediated by dynamic and coordinated interactions between cis-acting element on pre-mRNA, either as an enhancer or a repressor and trans-acting splicing regulators, and eventually catalyzed by a macromolecular complex called spliceosome [4]. Trans-acting splicing regulators include two classes of molecules; (i) development- and tissue-specific auxiliary factors, which are primarily the RNA-binding proteins (RBPs), and (ii) general splicing factors, components of the spliceosome [5]. A primary function of RBPs, which include heteronuclear ribonucleoproteins and Serine- and Arginine-rich (SR) proteins, is to bind to an exonic or intronic splice enhancer or repressor, thereby recruiting or blocking the spliceosome near the alternatively regulated splice site (SS) [4]. Several recent studies have provided evidence that general splicing factors can also regulate AS by potentially modulating the kinetics of the assembly and disassembly of the spliceosome during pre-mRNA splicing adding the complexity of the regulatory mechanisms by which AS is regulated [6,7].
AS has been implicated in many developmental processes, including cancer development and metastasis [8], neural synapses [9], and epithelial–mesenchymal transition (EMT) [10]. More recently, numerous studies have provided evidence on important functions of the splicing regulators for stem cell pluripotency and differentiation, and embryonic development [11 –15]. Traditionally, transcriptional network or chromatin landscape of the stem cells has been viewed as main regulatory mechanisms by which the cell fate specification and differentiation of stem cells are determined. However, there has been increasing evidence that AS adds another layer of regulatory mechanism to this dynamic process of stem cell differentiation [16].
Cornea Development, Differentiation, and Homeostasis
Animal cornea, intricately organized by specialized set of cell types, during embryonic development, and its transparency, wound repair and regeneration, and maintenance, are essential for normal vision. The complete cornea consists of five to seven layers of the superficial epithelial cell layers, Bowman's layer, corneal stroma, Descemet's membrane, and corneal endothelium. During embryonic development, animal cornea is derived from surface ectoderm and neural crest-derived mesenchymal stem cells. The outermost epithelial cell layers arise from the lens placode, which originally is derived from surface ectoderm and segregates into the corneal epithelial layers and lens pit during embryonic corneal development by signals from the underneath optic vesicle [17,18]. The epithelium eventually matures postnatally to two layers of basal epithelial cells and multiple layers of nonkeratinized squamous epithelial cells, tightly joined together through tight junction, which provide the protection and transparency of the cornea. The corneal stromal and endothelial layers are from the migration of ocular mesenchyme, which are originally derived from the neural crest. The corneal epithelial homeostasis is critical for maintaining a healthy cornea, especially after wounds, and vitally supported by the limbal epithelial stem cells; slow cycling and highly proliferating cells, proliferate and stratify basally after migrating from limbus to basal layer of the cornea, and eventually replace the dead or damaged superficial epithelial cell [19]. Opacification of the cornea, which may affect the vision of millions, are attributable to three primary causes; a thickened stromal layer often due to an infiltration of the inflammatory cells followed by neovascularization, a reduced proliferation of the corneal epithelial cells (CECs), and a slower turnover of the CECs after corneal wounds.
Paired Box Protein 6 Functions in Eye Development, Particularly Corneal Development
Importance of paired box protein 6 (Pax6) during eye development in metazoan from mouse to humans has been well documented [20 –22]. Many studies have demonstrated that the dosage effect of Pax6 is critical for the eye morphogenesis as both knockout and overexpression of Pax6 abrogated the normal eye development. In this review, we focus on the role of Pax6 in the development and homeostasis of the cornea. First, Davis et al. demonstrated the function of Pax6 in the corneal development and elucidated the thinning of the corneal epithelial layers due to adhesion abnormalities as reflected by reduced expression of desmoglein, β-Catenin, and γ-catenin in Pax6 heterozygous animals [23]. Second, animals containing chimeric cells, wild-type and mutant Pax6 cells, and heterozygous mutant mice (Pax6 +/−) have shown a delayed corneal epithelium formation, implicating the role of Pax6 in the proliferation of the corneal epithelium or cell fate specification of intermediate stages of the corneal differentiation [24,25]. Indeed, other studies showed that the proliferation and cell cycle progression of CECs seem to be inversely correlated to the Pax6 concentration [26,27]. Pax6 expression is restricted to the transparent corneal epithelium, not keratinized skin epidermis, and thus demarcating a genetic barrier between the corneal and epidermal epithelium during ectoderm differentiation. In fact, Pax6 KD converts CECs to epidermal epithelium, whereas overexpressing Pax6 in epidermal epithelial cells converts them into CECs [28,29]. Consistent with the notion, PAX6 expression is diminished or even absent in the keratinized corneal epithelium in severe ocular surface diseases [30]. KRT12 expression, which is directly regulated by Pax6, is dramatically reduced, whereas KRT10 expression is elevated, further supporting the keratinized phenotype of the corneal epithelium [30]. Additionally, Shalom-Feuerstein et al. showed that PAX6 expression can be modulated by a miRNA, miR-450b-5p, and thereby determining the cell fate between epidermal and epithelial cells [31]. The molecular functions of PAX6 during eye development, specifically corneal development, are summarized in Table 1.
BMP, bone morphogenetic protein; PAX6, paired box protein 6; OV, optic vesicle; EFTF, eye field transcription factor; NCC, neural crest cells; HH, Hamburger Hamilton.
PAX6 AS and Corneal Development
One of the early studies on the function of PAX6 AS on corneal development was carried out by Epstein et al., in which they analyzed a naturally occurring point mutation at −3 position from splice acceptor site of exon 5a. This single mutation (T to C) strengthens the 3′ SS and stimulates the inclusion of exon 5a [32]. This disrupts the balance in the ratio between two isoforms, PAX6a and PAX6b, as the longer isoform, PAX6b, is preferentially generated in the patient, and confers opaque cornea due to pannus formation. The same study demonstrated that these two isoforms would have two distinct consensus binding sites, implicating that differential transcriptional activities may modulate immune function and vascularization [33,34]. A genetic study by Favor et al. [35] provided an interesting clue with regard to the correlation of Pax6 AS with severity of the eye phenotypes. In this study, they generated several heterozygous deletion mutants spanning Pax6 locus with various flanking upstream and downstream genes and analyzed the eye phenotype. A deletion mutant line, Pax611neu/+, in which Pax6 and upstream gene Rcn1 were deleted, had much more severe eye phenotypes than Pax612neu/+, in which Pax6 and two downstream genes, Elp4 and Immp1L, were deleted [35]. Pax6 11neu/+ displayed an extreme microphthalmia and opaque cornea, compared with Pax6 12neu/+ mutant line. Surprisingly, however, the overall expression level of Pax6 was not different in these two lines. Instead, the ratio between two Pax6 isoforms was altered in the eyes of two mutant mice lines, suggesting two possibilities, a potential regulation of PAX6 AS by Rcn1 and an independent role of Rcn1 on eye development. A recent work by Sasamoto et al. demonstrated that two PAX6 isoforms can differentially and cooperatively regulate the gene expression specific for the structure and function of human CECs [36]. Isoform specific functions of Pax6 are summarized in Table 2. With more emerging evidence in terms of importance of AS in many biological functions, we examined literature on the functional role of two PAX6 isoforms and their potential role in eye and corneal development.
RPE, retinal epithelial cells; hESC, human embryonic stem cell.
Evolutionary Conservation of PAX6 AS
Pax6 is highly conserved evolutionarily through 400 million years of divergence [37]. PAX6 or its close homolog is present in Caenorhabditis elegans, nemertean, mollusks, cnidarians, annelids, and arthropods for its conserved role in eye and brain development [38 –43]. Human PAX6 still shares 90% and 96% sequence identity with Drosophila melanogaster and zebrafish, respectively [37,44]. More importantly, AS of PAX6 has been conserved in all vertebrates from zebrafish to human. In Drosophila, there are four PAX6 orthologs, ey and toy, and eyg and toe, each corresponding to PAX6a and PAX6b, respectively, and thought to be duplicated during evolution. The duplicated genes are thought to have redundant functions during eye development [45]. Ey and its paralog toy, the canonical form of PAX6 (PAX6a), are the key players in eye development and induce ectopic eyes when overexpressed mainly through specification and differentiation of eye cells [46]. However, the rescue of the eye-growth defects in Notch mutants is only made by eyg, equivalent to mammalian PAX6b, but not by ey or toy, indicating that eyg or PAX6b isoform plays an important role in eye growth, whereas ey and/or toy may play a role in eye specification [47]. These findings strongly suggest that two structurally similar yet distinct PAX6 isoforms play two functionally distinct roles by differentially activating or repressing target genes during eye development.
Molecular Mechanisms by Which Pax6 AS Modulates the PAX6 Functions
Epstein et al. provided a hint for the first time that AS may render a regulatory mechanism for normal eye development by producing two functionally distinct PAX6 isoforms with divergent consensus binding sites [32]. Two major alternative isoforms in mammals, PAX6a (− exon5a) and PAX6b (+ exon 5a), are generated from PAX6 gene by including or skipping a mutually exclusive exon, exon 5a (Fig. 1). The canonical PAX6 (PAX6a) is composed of 128 amino acids long DNA-binding PAIRED domain (PD), linked by 60 amino acids long DNA-binding Homeodomain (HD) and proline/serine/threonine-rich transactivation domain (PST) [48]. However, the second isoform (PAX6b) with 14 amino acid addition into the PD disrupts the structure of the PD and regulates the DNA binding mostly through HD [49]. PAX6 primarily carries out its function through DNA binding by PD and HD and transcriptional activation by PST domain. Previously, structural study by Xu et al. showed that 14 amino acids insertion of PAX6b is located between the second and third alpha helices of the PD (Fig. 2A) [49]. The insertion disrupts the conformation of the PD, specifically the interaction between PAI and RED domains, thereby modifying the DNA-binding activity [32,49]. It is hypothesized that two isoforms differing in 14 amino acids of the PAI domain may exert different biological function by differential transcriptional activities with unique target genes [32,50,51] or cooperatively act for normal development through a precise balance between two isoforms [52].
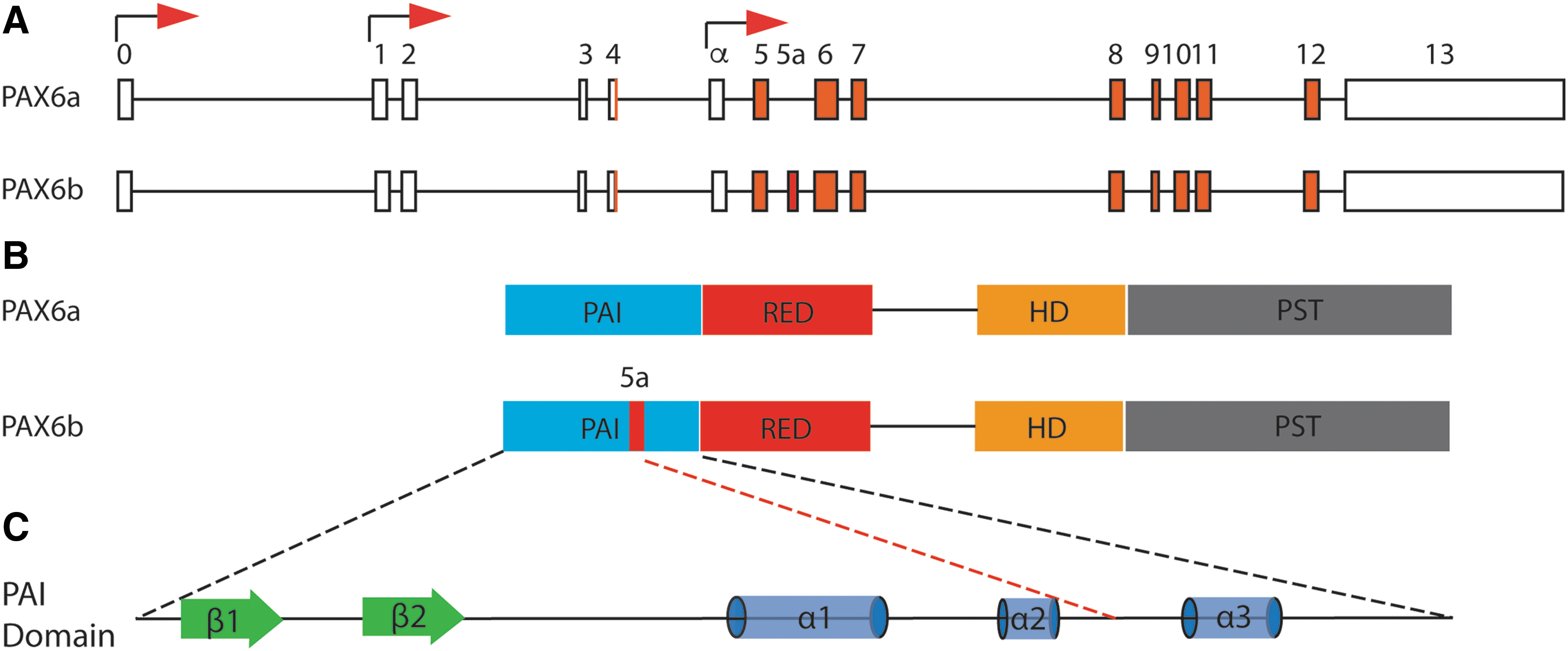
Genomic and protein structure of Pax6.
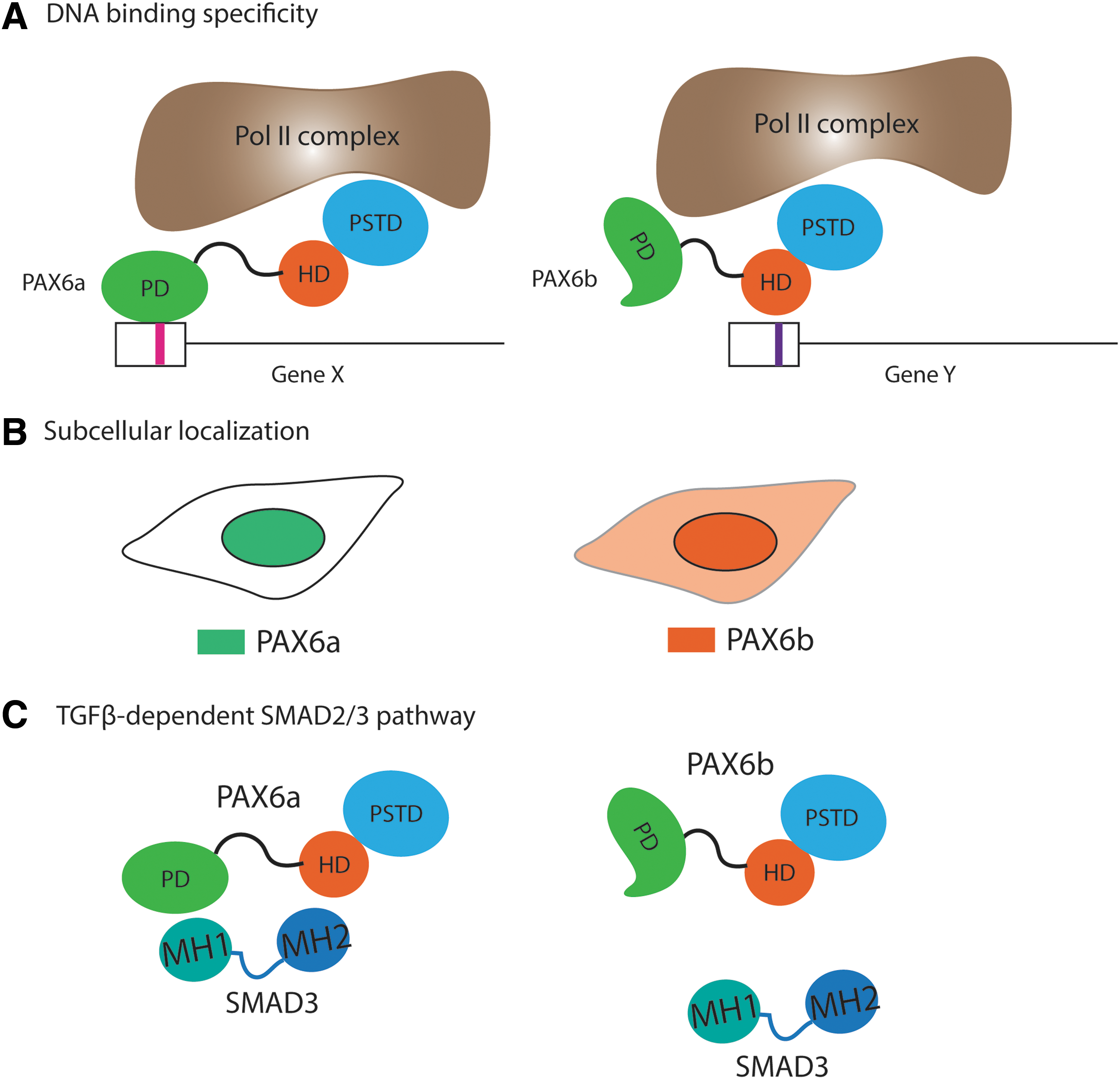
Three potential molecular mechanisms by which PAX6 alternative splicing regulates cellular phenotypes in metazoan.
Another possible regulatory mechanism by which AS of PAX6 can modulate biological activity is through its subcellular localization. Carriere et al. identified several Pax6 isoforms from screening cDNA library of quail neuroretina cells [53]. Their data show that the PAX6 isoform containing the alternative exon, annotated as exon 4a in quails, which is equivalent to PAX6b in murine and human, localizes both in nucleus and cytoplasm, whereas the isoform lacking exon 4a (PAX6a) was completely nuclear. Additionally, it was demonstrated that PAX6 localized in the nucleus inside the ganglion cell and inner nuclear layers of the retina, whereas a strikingly different subcellular localization, that is, cytoplasmic, in the photoreceptor and outer nuclear layer of the retina [54]. Since an antibody against the common epitope for PAX6a and b was used for the study, it is not clear whether a differential subcellular localization was due to AS or additional protein interacting partner(s) for such a dramatically different distribution with single cells. In the case of AS, a structural change due to the insertion of 14 amino acids at the PAI domain, may lead to masking the nuclear localization signal, or disrupting the protein–protein interaction for subcellular distribution (Fig. 2B). Altered subcellular localization of PAX6 isoforms in cell type or time-specific manner may consequently trigger differential transcriptional activation of the downstream targets, eventually determining the cellular properties.
Finally, protein–protein interaction between PAX6 isoforms and other proteins may contribute to modulating the differential cellular phenotypes. Biochemical studies by Grocott et al. demonstrated that PD of PAX6 directly interacts with MH1 domain of SMAD3 [55]. They determined that the PD of PAX6 is sufficient to bind to SMAD3, and more specifically the RED domain, the region between PAI and linker domain of the PAX6 protein, is responsible for the binding (Fig. 2C). Although whether PAX6b is capable of interacting with SMAD3 is yet to be determined empirically, it is highly possible that an insertion of a short stretch of peptide in PD disrupt the protein–protein interaction. Another study showed the physical interaction of PAX6 with secreted protein, acidic and rich in cysteine and transforming growth factor β, suggestive of the tetrameric complex involved in the nuclear–cytoplasmic shuttling [56]. Taken together, two Pax6 isoforms generated by AS can modulate the cellular phenotypes or cell fate determination by differential DNA-binding specificity, protein–protein interaction, and subcellular localization.
Spatial and Temporal Expression of PAX6 Isoforms During Eye and Corneal Development
Differential expression of Pax6 isoforms in ocular tissues have been demonstrated in many organisms from Drosophila to humans. Earlier works demonstrated that Drosophila Pax6 homologs, ey/toy and eyg/toe, are expressed in different regions of the Drosophila eye discs [57]. In addition to differential expression of the Pax6 isoforms, the ratio of the two appears to be tightly regulated. The ratio of two Pax6 isoforms appears to be temporally regulated in a tissue-specific manner during chick eye development [58]. Furthermore, the expression level of Pax6b is highly upregulated in posterior retinal tissues in the later part of the eye development, whereas Pax6a expression is predominant in most other tissues during development [58]. Jaworski et al. showed that retina and lens have preferential expression for Pax6a, whereas iris and cornea for Pax6b in bovine eye [59]. The same study demonstrated a highly conserved intronic sequence flanking exon 5a, including putative lariat sites, between Xenopus and bovine, further supporting the significance of isoform-specific expression in the eye. It appears that both isoforms of PAX6 were shown to be expressed in all epithelial layers, but at various levels in different areas of the corneal epithelial layer [36]. Sasamoto et al. [36] recently showed the differential expression of PAX6a and PAX6b in human corneal epithelium. A laser microdissection of the frozen section of the human corneal epithelium into four areas, central–apical, central–basal, limbal–apical, and limbal–basal, was followed by the determination of the expression level for each PAX6 isoform in each region. They demonstrated the differential expression of two isoforms in two areas of human corneal epithelium, where the relative expression ratios are quite different, ranging from 1:1 to 3:1, PAX6a to PAX6b, respectively [36]. In fact, isoform-specific expression of Pax6 in various parts of the eye is conserved all the way from fish to humans [58,60,61]. This tissue-specific enrichment of different isoforms may confer unique biological function through differential transcriptional control during eye development and provide mechanistic insight to corneal differentiation and maintenance.
Gain- and Loss-of-Function Studies Involving Two PAX6 Isoforms During Eye and Corneal Development
Davis et al. [23] overexpressed Pax6a in wild-type mice under a cornea-specific promoter (Aldh3a1) in mice, and observed opaqueness, erosion, and neovascularization of the cornea, and immune cell invasion in the stromal region. Overexpression of Pax6a drastically altered gene expression of many genes in the cornea, particularly the genes involved in angiogenesis and immune response, consistent with the corneal defective phenotypes. In addition, in severely affected animals, the Krt12 expression level was reduced as high as 50% compared with the wild-type animals. Duncan et al. demonstrated that the overexpression of human PAX6b in the lens of mice caused phenotypic defects as well as activating several important genes for cornea development, such as α5 and β1 integrins, paxillin, and p120 [62]. They further supported this argument that the gene expression of the abovementioned adhesion molecules regulated by Pax6b plays a critical role in cornea morphogenesis [23].
Overexpression of Pax6b in the eyes of the developing chicken embryos developed a well-differentiated retina-like structure, suggesting a specific role for Pax6b in retinal differentiation [58]. This notion was also supported by the same group that Pax6b expression is greater in posterior retina than anterior side, with the highest concentration in the foveal region of the retina [58]. Additional studies showed that a balanced expression ratio of Pax6 isoforms is required for normal development during brain and eye development [63,64]. As discussed earlier, AS modulates the ratio of isoforms to shape the cellular transcriptome or functional polymorphism. This idea was clearly demonstrated by Dora et al. in which a specific ratio of two Pax6 isoforms was also critical for controlling the cell fate or maintaining the homeostasis in the cornea [65].
An ectopic expression of human PAX6b induced strong overgrowth of the eye in D. melanogaster, whereas that of the canonical isoform, PAX6a, hardly did [47]. It has also been shown that an overexpression of Pax6b inhibited the cell proliferation without affecting the cell fate determination during neural development [66]. Furthermore, only PAX6a can trigger the neuroectoderm specification, whereas combination of both isoforms, PAX6a and 6b, was required to suppress the pluripotency of human embryonic stem (hES) cells to trigger the neuronal differentiation [67]. This indicates that two alternative splicing isoforms may exert similar but distinct functional activity during early neuroectoderm specification and eye development through differential transcriptional regulations. Based on these findings, one can hypothesize that PAX6a, the canonical form, modulates the transcriptional activity for differentiation or cell fate specification aspect, whereas PAX6b modulates the gene expression for proliferation by either antagonizing the activity of PAX6a or regulates the gene expression independent of PAX6a activity.
Carbe et al. created a transgenic mouse in which the full-length Pax6b cDNA replaced the Pax6 genomic locus by recombineering to determine the function of Pax6b isoform for lens development. In their study, they showed that Pax6 5a/5a could not replace endogenous Pax6a and Pax6b for lens induction and neural differentiation in retina, whereas it was sufficient to properly segregate corneal epithelial layer from lenticular layer [68]. This implicates that Pax6b plays an indispensable role during the detachment of corneal epithelium from lens placode in response to an interaction with optic vesicle.
Favor et al. generated several heterozygous Pax6 deletion mutant mice to analyze the function of Pax6 for the eye development [35]. One of which, Pax6 11neu/+ with the deletion of Pax6 locus along with the immediately upstream gene, Rcn1, resulted in severe eye phenotypes, ranging from extreme microphthalmia, a failure to separate the cornea from the lens, absence of the corneal stromal layer, coloboma in the retinal epithelial cells (RPE), and to an ectopic formation of RPE in the anterior part. However, another deletion mutant (Pax6 12neu/+), with deletion of Pax6 gene and two immediately downstream genes, Elp4 and Immp1L, had milder phenotypes. Interestingly, while there was no detectable difference in the expression levels for Pax6 in the developing eye of P1 mouse for both mutant strains, the ratio of two isoforms, Pax6a and Pax6b, was clearly altered. The level of Pax6b was higher than Pax6a in Pax6 11neu/+, compared with either wild-type or Pax6 12neu/+. This raises an intriguing possibility that a change in the ratio of two isoforms generated by AS or increased level of Pax6b are attributable for the severity of the several eye phenotypic defects, although the possibility that Rcn1 has an independent role during ocular development cannot be completely ruled out.
Single cell analysis by Sasamoto et al. [36] observed an interesting correlation between the expression levels for two classic CEC markers, KRT12 and KRT3, and two PAX6 isoforms, PAX6a and PAX6b. During the corneal development, KRT3 expression precedes KRT12 expression, converting embryonic epithelial cells (superficial, superbasal, and basal) to adult epithelial cells. Overexpression of PAX6a isoform in non-PAX6-expressing cell line, OKF6/TERT-1, induced the expression of KRT3, whereas that of PAX6b induced the expression of KRT12. This demonstrates that there may be two mutually exclusive populations of KRT3- and KRT12-positive cells. Interestingly, either mutation in KRT3 or KRT12 causes Meesmann corneal dystrophy in an autosomal dominant manner, indicating that both KRT3- and KRT12-positive cells may be necessary for normal CEC development and maintenance. The physiological significance of mutually exclusive KRT3- and KRT12-positive cells is not known. However, they further demonstrated that cells expressing KRT12 was promoted by only PAX6a, whereas KRT3 was only expressed by PAX6b.
Genetic Evidence PAX6 AS Pertaining to Specific Eye Phenotypes, Particularly, Corneal Defects
Several mutations have been identified in alternatively spliced exon5a and these mutations appear to be associated with several ocular defects, including congenital cataract and corneal opacity, implying the importance of exon 5a inclusion for normal eye development [69 –71]. Opacification of the cornea may be attributable to three potential mechanistic causes, a thickened stromal layer with or without infiltration of the inflammatory cells, followed by neovascularization, a reduced proliferation of the CECs, and a slower turnover of the CECs after corneal wounds. Therefore, it is plausible to think that PAX6b plays a critical role, at least in part, in the proliferation of the CECs during normal corneal development/differentiation.
More strikingly, a mutation in splice regulatory element on PAX6 exon 5a was discovered in family members with several ocular defects. A disruption of the inclusion of exon 5a, thus perturbing the ratio between two isoforms without affecting the total Pax6 mRNA level, caused abnormal iris, corectopia, and corneal opacity [32]. Another study showed that alternative spliced Pax6b isoform null mice displayed specific defects in cornea, lens, and retina, as well as iris hypoplasia, whereas transgenic Pax6a null mice exhibited severe central nervous system defects and lethality [72]. This finding is consistent with the fact that only PAX6a, not PAX6b, was capable of activating neuroectoderm specification of undifferentiated hES cells. More importantly, this suggests a notion that PAX6b may be an eye-specific isoform, whereas PAX6a may be a form, specifying the neuroectoderm cell fate. In addition, there are several single nucleotide polymorphisms (SNPs) in exon 5a of PAX6 gene conferring anterior segment dysgenesis based on human genome database. It is likely that altered ratio of two PAX6 isoforms, or transcriptional activity by these SNPs could result in ocular defective phenotype.
Potential Regulation of PAX6 AS
Common mechanisms by which AS is regulated is by cell-type or tissue-specific splicing regulators. Two potential trans-acting splicing regulators have been described for alternative pre-mRNA splicing of PAX6 in the context of human CECs. PININ (PNN), a SR-related protein, initially identified as an associating protein for desmosomes of the epithelium [73], is a component of the mRNA splicing-dependent exon junction complex as well as active spliceosomal complex C, which is the active catalytic complex involved in pre-mRNA splicing [74]. Pinin was first shown to have a role in murine corneal epithelia differentiation as Pax6-cre-mediated conditional Pnn deletion displayed the defects in the formation of the normal corneal epithelia, more epidermis-like, including the downregulation of corneal epithelial marker, Krt12, and upregulation of epidermal markers, Krt10 and Krt14 [75]. In addition, Pnn was shown to colocalize with several splicing factors in nuclear speckles and interact with SRp75, SRrp130, SRm300, and RNPS1 [76,77]. In humans, PNN was shown to have a repressive function in the inclusion of exon 5a in CEC line [78 –80].
Epithelial splicing regulatory protein 1 (ESRP1) is a pre-mRNA splicing factor, which is specifically expressed in epithelial cells and regulates the formation of epithelial cell-specific isoforms. A cell-type specific AS of FGFR2 pre-mRNA, by which one of the two mutually exclusive exons, 8 and 9, can be selected to produce FGFR2-IIIb or FGFR2-IIIc. By selecting one of the two mutually exclusive exons, FGFR2-IIIb or FGFR2-IIIc can modulate the cellular phenotype as FGFR2-IIIb is specifically expressed in epithelial cell types. In addition, ESRP1 controls the splicing of CD44, CTNND1, and ENAH pre-mRNA, which undergo changes in splicing during the EMT [81].
It acts by directly binding to GU-rich sequence motifs in the intronic sequence silence or enhancer. For example, ESRP1 binds to this cis-element regulatory region, which is present in the mRNA of FGFR2. Indeed, GU-rich sequence is found in the exon 5a and its flanking introns of exon 5a of PAX6 gene and conserved evolutionarily, raising a possibility that this region may be a binding site for ESRP1 and further recruiting other splicing regulators, including SR proteins for the spliceosomal assembly to facilitate the inclusion of exon 5a (Fig. 3). A functional analysis showed that PAX6 AS was affected by knockdown of either PNN or ESRP1, implicating that PAX6 is the common target of two splicing regulators or the target for cooperative action of both regulators [80].
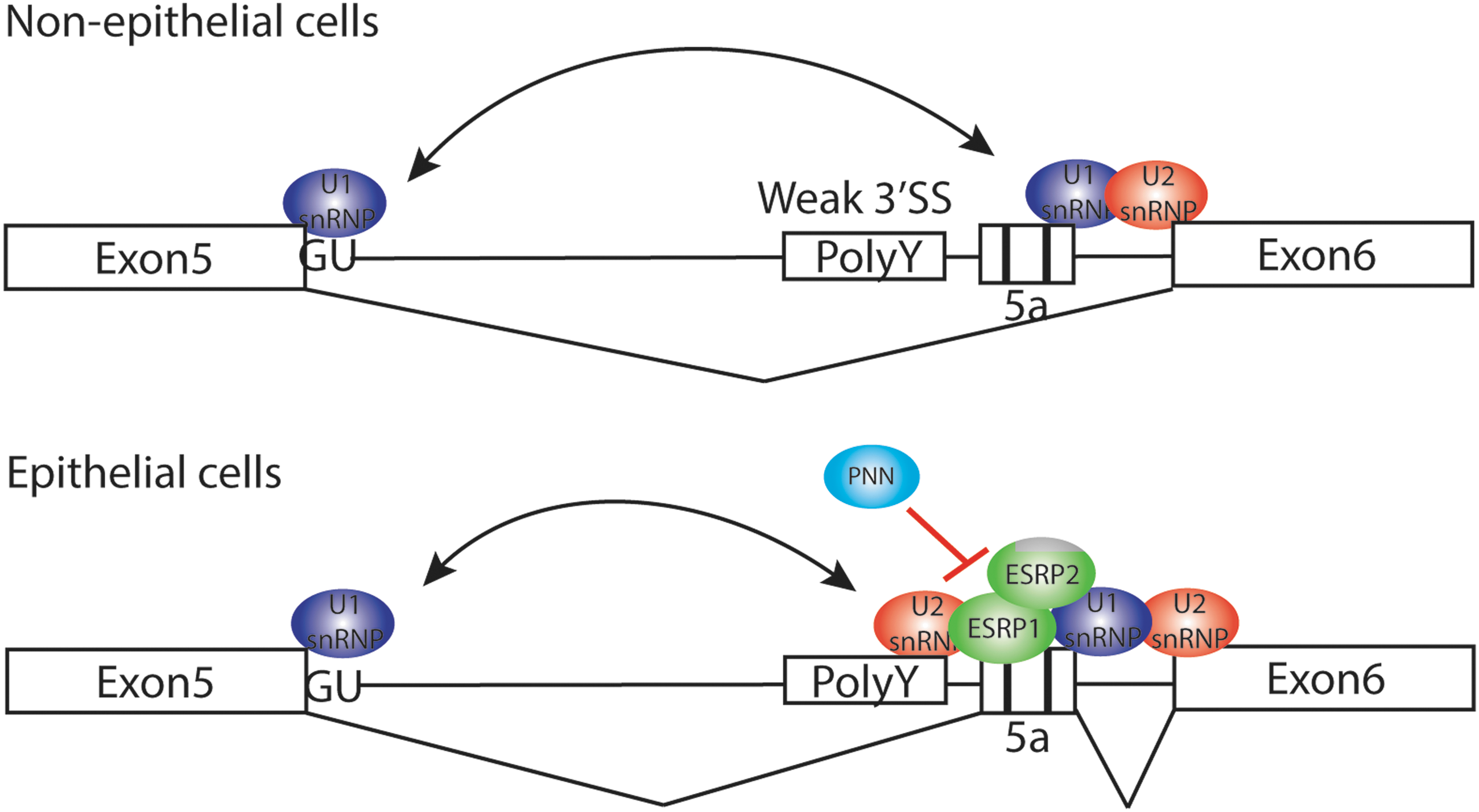
A regulatory mechanism of Pax6 alternative splicing. In nonepithelial cells, U2 snRNP binds to the stronger 3′ splice site upstream of exon 6, thus skipping exon 5a. In contrast, epithelial cell-specific ESRP1/2 binds to exonic splicing enhancer (GU-rich sequence) and recruit U2 snRNP to 3′ splice site adjacent to exon 5a in epithelial tissues, thus preferentially including exon 5a. PNN, also epithelial-specific splicing regulator without RNA-binding motif, may have an antagonistic action against ESRP1/2 for the inclusion of exon 5a. U2 snRNP, U2 small nuclear ribonucleoproteins.
Although the precise mechanism by which PNN and ESRP1 regulate PAX6 AS is not known, PNN and ESRP1 appear to perform opposite functions as a repressor and an activator, respectively, for the inclusion of exon 5a. PNN regulates the inclusion of exon 5a through an interaction with other SR proteins within the spliceosome and/or ESRP1, epithelial-specific splicing regulator. Since PNN is also a downstream target gene of PAX6 [80], a tight control of PNN level and Pax6 AS must be essential in regulating cell fate and differentiation potential of the CECs by forming a negative feedback loop.
Furthermore, a coimmunoprecipitation experiment to determine PAX6-interacting proteins in CECs identified several integral components of U2 small nuclear Ribonucleoproteins (U2 snRNP), including, SF3b1, 2, 3, 4, and U2SURP1, as bona fide binding proteins [36]. U2 snRNP, one of the five snRNPs, which constitute spliceosome, a macromolecular machinery, plays an important role in pre-mRNA splicing by binding to branch point and thereby defining 3′ SS. It has been shown clearly shown that modulation of the concentration of U2 snRNP components have a specific effect on AS, rather than global splicing defects [7]. Thus, it is highly plausible that PAX6 can autoregulate the inclusion of exon 5a by interacting with U2 snRNP during CEC development, either in specifying either corneal or epidermal epithelium fate. However, whether either PAX6 isoforms may regulate AS of other targets is not known at present.
Perspectives
From the literature findings, we propose a life cycle of the PAX6 isoforms during corneal development (Fig. 4). PAX6 pre-mRNA undergoes AS by tissue-specific splicing regulators, such as PNN and/or ESRP-1/-2 and may facilitate subcellular localization of developmentally important signaling molecules such as SMAD-3 through protein–protein interaction. In addition, many studies have demonstrated that PAX6a and PAX6b differentially activate the transcription of the proliferation- and cell fate specification-related genes, thereby shaping the corneal developmental program. Many questions remain to be answered, including molecular mechanism(s) by which each isoform can regulate the fate of early progenitor cells toward either a proliferating or differentiating state through differential transcriptional activities, protein–protein interaction, or subcellular localization of the two isoforms for corneal development.
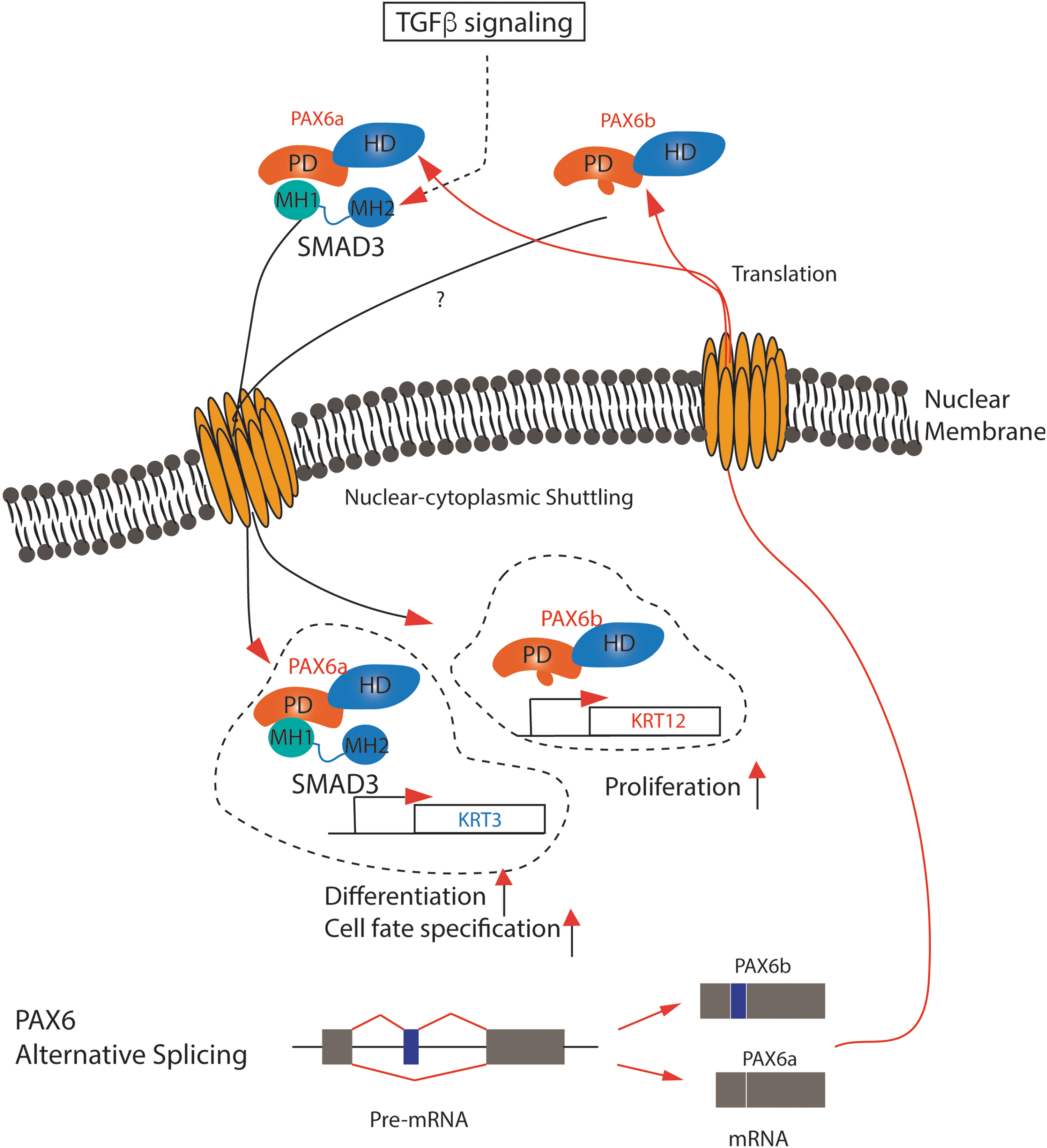
A life cycle of Pax6 isoforms. Two PAX6 mRNA isoforms are produced by alternative splicing and exported to the cytoplasm. Two protein isoforms modulate the cell fate specification or cellular phenotypes by differential DNA-binding specificity, protein–protein interaction, or nuclear–cytoplasmic shuttling.
Footnotes
Acknowledgment
This work was supported by University of Macau Research Committee funds MYRG #2015-00169-FHS, 2016-00070-FHS, 2017-00124-FHS and Macau Science and Technology Development Fund (FDCT) #128-2014-A3 and 028/2015/A1 to R.-H.X., and 2016-00249-FHS, 2017-00249-FHS to J.W.P.
Author Disclosure Statement
No competing financial interests exist.