
Abstract
Angiogenesis is a complicated and sequential process that plays an important role in different physiological processes. Mesenchymal stem cells (MSCs), which are pluripotent stem cells, are widely used for the treatment of ischemic and traumatic diseases, and exosomes derived from these cells can also promote angiogenesis. Therefore, we aimed to uncover mechanisms to improve MSC exosome-mediated angiogenesis. For this study, we isolated human adipose-derived MSCs (hAD-MSCs) and assessed differentiation ability and markers. Cells were divided into hypoxia-treated MSCs (H-MSCs) and normoxia-treated MSCs (N-MSC), and exosomes were extracted by ultrafiltration. Exosomes (100 μg/mL) from H-MSCs and N-MSCs were added to human umbilical vein endothelial cells (HUVECs). Exosome uptake and the ability of endothelial cells to form tubes were detected in real time. Protein samples were collected at different time points to detect the expression of inhibitors (Vash1) and enhancers (Angpt1 and Flk1) of angiogenesis; we also assessed their related signaling pathways. We found that exosomes from the hypoxia group were more easily taken up by HUVECs; furthermore, their angiogenesis stimulatory activity was also significantly enhanced compared to that with exosomes from the normoxia group. HUVECs exposed to exosomes from H-MSCs significantly upregulated angiogenesis-stimulating genes and deregulated angiogenesis-inhibitory genes. The expression of vascular endothelial growth factor (VEGF) and activation of the protein kinase A (PKA) signaling pathway in HUVECs were significantly increased by hypoxia-exposed exosomes. Moreover, a PKA inhibitor was shown to significantly suppress angiogenesis. Finally, we concluded that hypoxia-exposed exosomes derived from hAD-MSCs can improve angiogenesis by activating the PKA signaling pathway and promoting the expression of VEGF. These results could be used to uncover safe and effective treatments for traumatic diseases.
Introduction
I
Exosomes are important active substances involved in the paracrine effects of MSCs. Studies have shown that these structures are widely distributed in different types of cells. It is known that almost all types of cells (leukemia cells, pericytes, MSCs, dendritic cells, and tumor cells, among others) can secrete exosomes under both physiologic and pathologic conditions. Moreover, active molecules such as proteins, mRNA, and miRNA that are characteristic of the source cells can be transported to target cells for the transmission and exchange of signals among cells [9]. When applied to ischemic diseases and diseases associated with tissue repair, exosomes can also exhibit therapeutic effects consistent with MSCs such as reducing myocardial ischemia/reperfusion injury, promoting neurological recovery after stroke, increasing skin cell survival, and promoting wound healing, among others [10,11]. In recent years, exosomes have become incredibly important to the scientific community, providing new concepts for the treatment of clinical injuries and ischemic diseases. In addition, exosome extraction methods have become relatively refined. Exosomes are easy to store and have stable biological function, which demonstrates that they represent an ideal substitute for stem cell therapy.
In recent years, researchers have become committed to using exogenous means to promote angiogenesis and improve ischemia and injury [12,13]. At present, MSCs are widely used for such diseases, and thus improving their ability to promote angiogenesis has become a hot topic of research. Oxygen concentration plays an important role in the self-renewal, proliferation, and differentiation of MSCs. However, these cells are generally exposed to normoxic conditions (21% O2) during in vitro culture. However, in fact, there are big differences in the concentration of oxygen in the body under natural physiological conditions. Most MSCs exist in an environment of 2%–8% oxygen (or even lower) in the body. It has been reported that hypoxia promotes the differentiation of adipose-derived MSCs into cartilage, which is mediated by HIF-1α, as knockdown of this protein inhibits chondrogenesis [14,15]. In addition, hypoxia activates the AkT signaling pathway, upregulates c-Met and hepatocyte growth factor, and increases vascular endothelial growth factor (VEGF) production [15]. We hypothesized that the angiogenesis-promoting effect of MSC-derived exosomes might be enhanced by culture in hypoxic conditions. Therefore, we exploited the principle that hypoxia favors angiogenesis and extracted exosomes from hypoxia-treated MSCs (H-MSCs). We then compared the proangiogenic effect of exosomes derived under conditions of normoxia to that of exosomes from H-MSCs; we also sought to determine the possible associated mechanisms.
Materials and Methods
Human adipose tissues and human umbilical cords were obtained with informed consent and all experiments were approved by the Ethics Committee at the Chinese Academy of Medical Sciences and Peking Union Medical College, and all clinical investigations have been conducted according to the principles expressed in the Declaration of Helsinki.
Isolation and culture of human adipose-derived MSCs (hAD-MSCs): Adult fat samples were collected from plastic surgery hospitals after obtaining informed consent from donors. Adipose tissue was washed twice with D-Hanks' buffer containing two antibiotics (penicillin and streptomycin) and centrifuged at 1,000g for 3 min. After washing, the lower layer was aspirated with a pipette, and the adipose tissue was transferred to a new 50-mL centrifuge tube. Then, 0.2% collagenase P (Life Technology Corporation) was added for digestion, and the sample was incubated at 37°C for 30 min; the digested adipose tissue was filtered with a 100-μm cell strainer by adding the appropriate amount of D-Hanks' solution. Undigested tissue was removed and the sample was centrifuged at 1,500g for 10 min. After discarding the supernatant, cells were resuspended in D-Hanks' solution and washed once with phosphate-buffered saline (PBS). Next, 2 × 106 cells were seeded in T75 flasks, and incubated at 37°C and 5% CO2 in a cell incubator. Forty-eight hours later, the upper layer of unattached cells was removed and culture medium (Life Technology Corporation) was exchanged every 2–3 days. When the confluence of cells reached 80%, they were passaged or frozen.
Isolation and culture of umbilical vein endothelial cells: Under sterile conditions, find the venous cavity, which is the umbilical vein from a neonatal umbilical cord (length, 1,520 cm) with a thin wall and a large venous cavity in the fractured surface, and clamp both ends of the umbilical cord with the hemostatic clamp mark in an ultraclean bench. A syringe was gently inserted into the venous cavity, which was then fixed with tweezers. Washing of the venous cavity was repeated with PBS until the liquid was clear. Next, 0.1% collagenase P was injected into the venous cavity with a syringe until the umbilical vein refilled; both ends were then clamped with a hemostat. After digesting at 37°C for 12 min, the hemostatic force was taken off from the venous cavity, and the umbilical vein was washed with 10% fetal bovine serum (FBS)-containing medium (Life Technology Corporation); the medium was collected in a centrifuge tube at the same time. The sample was centrifuged at 300g for 5 min, and the supernatant was removed. Subsequently, cells were seeded in 10-cm Petri dishes with M200 medium (Life Technology Corporation) and cultured in a 5% CO2 incubator at 37°C. After 24 h, the medium was changed and subsequently changed every 3–4 days until the cells grew to 80% confluence. The original medium was discarded under sterile conditions, the cells were washed twice with PBS, and 5 mL of 0.05% trypsin was added per dish; digestion proceeded for 2–3 min until most of the cells appeared to shrink and round up. The same amount of serum was then added to stop the digestion. The cell suspension was collected and centrifuged at 300g for 5 min. The supernatant was then discarded, and cells were resuspended in M200 medium at a 1:2 ratio of cell seeding. Cells were incubated at 37°C, in a 5% CO2 in cell incubator; after 24 h, the culture medium was changed.
Identification of hAD-MSC immunophenotype
Passage 4 (P4) hAD-MSCs at 70% −80% confluency were washed twice with D-Hanks' solution and digested by adding 0.25% trypsin (containing 0.01% EDTA) (Life Technology Corporation). Cells were counted and centrifuged at 1,200g for 5 min. After discarding the supernatant, cells were resuspended with an appropriate amount of PBS to obtain a concentration of 2 × 105/mL; cells were dispensed into a 1.5 mL microcentrifuge tube (1 mL/tube). They were then centrifuged at 1,200g for 5 min. Intracellular and surface antigen detection: Cells were fixed and permeabilized by first incubating them in BD Cytofix/Cytoperm cell fixation/permeabilization (BD Biosciences) at 4°C for 20 min and washed with 1 × BD Perm/Wash buffer (BD Biosciences) twice. Then, cells were resuspended in 50 μL of a 1:50 dilution of anti-human primary antibody (CD73, CD31, CD34, CD44, CD105, CD29, CD90, HLA-DR) (Peprotech), mixed well, and incubated at 4°C for 30 min. Next, 1 mL PBS was added, and the sampled was centrifuged at 1,200g for 5 min and washed twice. The supernatant was discarded and 50 μL of a 1:50 dilution of FITC-goat anti-mouse IgG secondary antibody (Millipore) was added, and cells were incubated at 4°C for 30 min. PBS (1 mL) was added, and the sample was centrifuged at 1,200 rpm for 5 min; these two steps were repeated twice. The supernatant was discarded and cells were fixed with 200 μL of 4% PFA at room temperature. Cells were analyzed by using the BD Accuri C6 (BD Biosciences).
Extraction and identification of exosomes secreted by hAD-MSCs
hAD-MSC culture medium was replaced with Dulbecco's modified Eagle's medium (DMEM)/F-12 medium (Life Technology Corporation) without FBS 36 − 48 h before exosome extraction. Supernatants were harvested after culture, centrifuged at 1,500g for 10 min to remove dead cells and cell debris, and the medium was filtered with a 0.22-μm microporous membrane filter to remove residual cell debris and large vesicles. The filtrate was transferred to the ultrafiltration apparatus (Life Technology Corporation) equipped with a 100,000 molecular weight ultrafiltration membrane with nitrogen pressurization ultrafiltration. As a result, exosomes remained on the ultrafiltration membrane. Then, after appropriate washing with D-Hanks' solution and two rounds of nitrogen pressurization ultrafiltration (Millipore), the ultrafiltered inner suspension was collected to a centrifuge tube. The sample was ultracentrifuged at 700,000g at 4°C for 40 min, and the supernatant was discarded. Exosomes were resuspended in DMEM, and the suspension was filtered with a 0.2-μm microporous membrane filter, dispensed in 1.5-mL sterile microcentrifuge tubes, and preserved at −80°C.
Transmission electron microscopy identification of Exosomes: The purified exosomes were appropriately diluted and dropped onto a copper mesh for 5 min for precipitation. Then, filter paper was used to absorb excess liquid, and the sample was air-dried. Subsequently, 3% phosphotungstic acid in water was used to counterstain the sample for 2 min, and the specimen was air-dried. Finally, exosomes were observed by transmission electron microscopy (Olympus, Japan) and photographed.
Exosome uptake
1'-Dioctadecyl-3,3,3',3-tetramethylindocarbocyanine perchlorate (Dil) (Life Technology Corporation) is a lipophilic carbon cyanine dye that binds lipoproteins in a manner similar to phospholipids and is embedded in the membrane of the biomass and oriented within the membrane. Diffusion movement can be used to observe cell-bound or endocytic lipoproteins under a fluorescence microscope, and this allows for semiquantitative analysis. Purified exosomes were added to 1 μM Dil and stained for 10 min. After ultracentrifugation at 40°C and 700,000g for 40 min, twice, the supernatant was discarded, and the labeled exosomes were resuspended in DMEM and incubated with endothelial cells for 6 h. Then, the cells were washed with PBS three times and fixed with 4% paraformaldehyde for 10 min. Nuclei were stained with Hoechst 33342 (1:1,000 dilution) for 5 min at room temperature and washed with PBS three times. The cells were observed under a fluorescence microscope (OLYMPUS) and photographed.
Matrigel plug experiment
Matrigel in vitro tube formation: For this, 200 μL of Matrigel (Sigma-Aldrich) glue was added to each well of a precooled 24-well plate (to avoid bubbles); then, the 24-well plate was incubated at 37°C for 15 min, and cells were seeded at 1 × 105/well, and then incubated at 37°C for 8–10 h, Cells were observed using an inverted microscope and photographed.
In vivo Matrigel plug experiment: P2 generation endothelial cells were collected and resuspended at 3 × 106 endothelial cells per 200 μL of precooled PBS (on ice). Cells were mixed with the same amount of Matrigel with a precooling syringe and subcutaneously injected into nude mice [16]. Tissues were removed after 8 days, and paraffin sections were obtained to observe the tubular structure.
Western blot analysis
Cells were harvested in RIPA lysis buffer (Beyotime) with 1 mM phenylmethanesulfonyl fluoride; lysates were quantified using a BCA Protein Assay Kit (Beyotime), separated by 10% SDS-PAGE, and transferred onto polyvinylidene fluoride membranes (Millipore). The membranes were blocked with 5% nonfat milk in TBST for 1 h, and incubated with primary antibodies [HSP70/90, CD63, ANGPT1, GAPDH, FLK1, VASH1 (1:500) from Abcam] overnight at 4°C and then in horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature. Results were observed with an immobilon western chemiluminescent HRP substrate (Millipore) and detected using an ImageQuant LAS 4000 mini imaging system (GE Healthcare).
RNA extraction
Cell culture medium was discarded, and 1 mL of TRIzol was added to 1 × 106 cells at room temperature for 5 min. Repeated pipetting with an enzyme-free pipette was performed to completely lyse the cells. The lysate was transferred to an EP-free tube and stored at −20°C or used immediately to extract RNA. For extraction, 200 μL of chloroform was added to each tube, and samples were shaken vigorously for 15 s and mixed well at room temperature for 5 min. Then, lysates were centrifuged at 12,000 g at 4°C for 15 min. The upper aqueous phase was aspirated to a new, enzyme-free EP tube, and an equal volume of isopropanol was added; the tube was inverted gently for mixing and incubated at room temperature for 10 min. The samples were then centrifuged at 4°C at 12,000 g for 15 min, and the supernatant was discarded. Then, 1 mL of precooled 75% ethanol (anhydrous ethanol plus enzyme-free DEPC water) was added to each tube, upside down, to clean the sediment (without disrupting the pellet), and the sample was centrifuged at 7,500 g for 5 min. This step was then repeated, and the supernatant was discarded. An appropriate amount of enzyme-free DEPC water was then added to solubilize the RNA precipitate.
Real-time PCR
cDNA was reverse transcribed (40 μL) and treated according to the protocol recommended by TaKaRa M-MLV reverse transcriptase (TaKaRa). Real-time polymerase chain reaction (PCR) experiments were performed following the procedure recommended by the manufacturer for the TaKaRa SYBR® Premix Ex TaqTM kit. Reactions were performed in triplicate and independent experiments were repeated three times.
Statistical analysis
All data are expressed as mean ± standard deviation (SD), and two-tailed t-tests and one-way analysis of variance (ANOVA) were performed. P < 0.05 was considered significant. Each experiment was repeated at least three times to obtain a P value and to control for systematic errors.
Results
Phenotype of hAD-MSCs
Passage three of hAD-MSCs, which were 70%–80% confluent, were assessed for their differentiation ability; they were found to have adipogenic, osteogenic, and chondrogenic differentiation ability based on Oil Red O staining, Alizarin red staining, and alkaline phosphatase staining, respectively. These positive results are depicted in Fig. 1A–D. Further analysis of the immunophenotypes of MSCs treated with normoxia and hypoxia showed that cells were positive for CD90, CD29, and CD73 (positive rate >95%); moreover, cells were HLA-DR and CD34 negative (positive rate <5%; Fig. 1E).

Characterization of bone marrow-derived MSCs and their differentiation capacity and surface marker expression.
Separation and identification of exosomes derived from human adipose MSCs
To investigate the effect of exosomes on endothelial cell angiogenesis, we isolated and purified exosomes derived from human adipose MSCs (adMSC-Exos) by ultrafiltration. Electron microscopy showed that a large number of exosomes were extracted and their diameters were ∼30–100 nm. Normoxia and hypoxia produced consistent phenotypes (Fig. 2A). Western blotting also showed that exosomes expressed HSP70, HSP90, and CD63 in both normoxia and hypoxia groups (Fig. 2B).
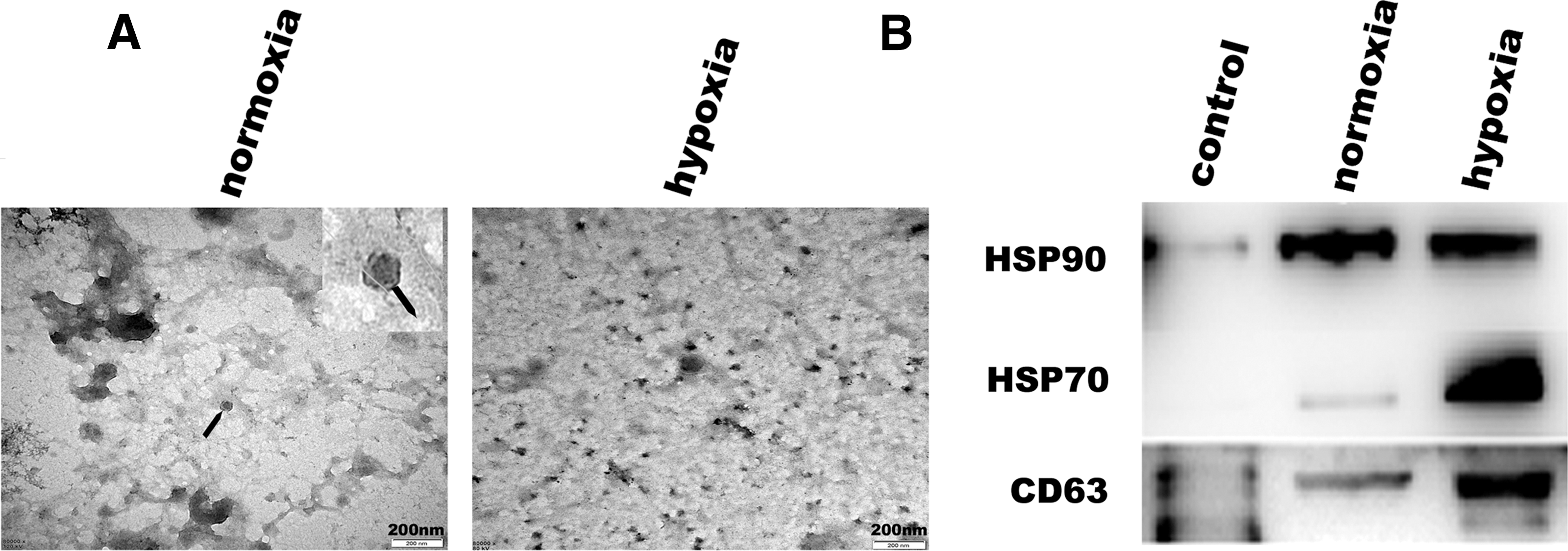
Expression of specific markers in adMSC-Exos and morphology assessed by electron microscopy.
Differential endothelial cell exosome uptake based on hypoxic and normoxic conditions
To examine whether exosomes were taken up differentially by human umbilical vein endothelial cells (HUVECs), based on whether they were derived under normoxia or hypoxia, we used Dil dye to label adMSC-Exos and then cocultured them with HUVECs for 24 h. Fluorescence microscopy was used to monitor the rate of exosome uptake by HUVECs in real time. The results showed that the rate of adMSC-Exo uptake in the hypoxic exosome group was higher than that in the normoxic group. Figure 3A shows the proportion of Dil-labeled-positive cells at different time points, 2, 10, and 22 h after exosome phagocytosis by HUVECs. Figure 4B shows the statistical results from the different groups. The results showed that there was statistically significant difference between the two groups (P < 0.05) after 12 h (Fig. 3A, B), suggesting that adMSC-Exos derived under hypoxic conditions are more easily taken up by endothelial cells.
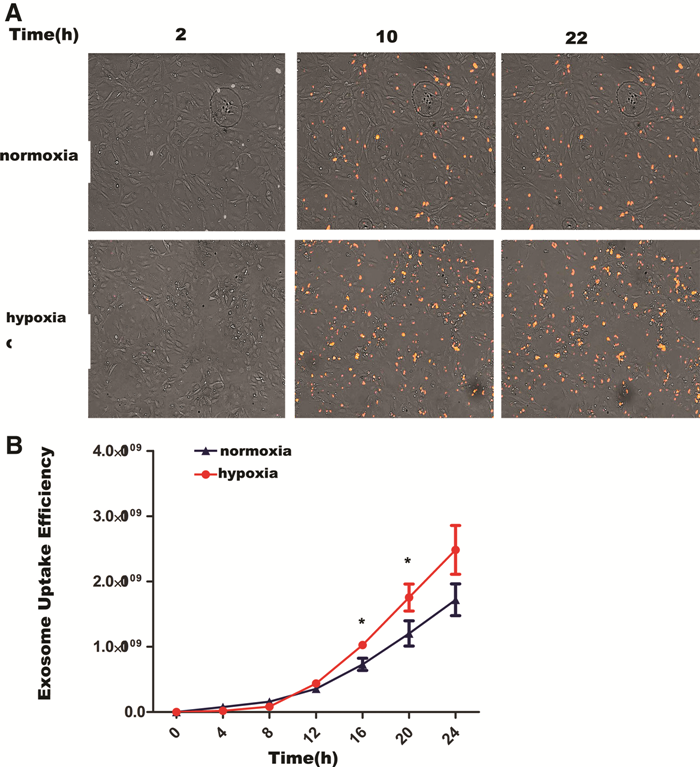
Dil (1′-Dioctadecyl-3,3,3′,3-tetramethylindocarbocyanine perchlorate)-labeled adMSC-Exos uptake by HUVECs.
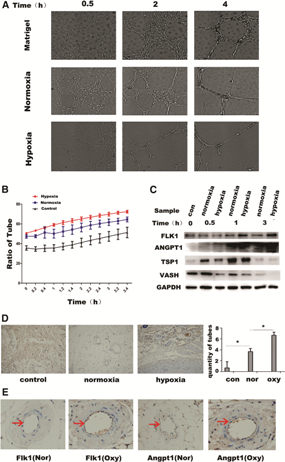
Hypoxia-derived exosomes promote angiogenesis in HUVECs.
Exosome-mediated angiogenesis in HUVECs
To further investigate the effect of adMSC-Exos on HUVEC angiogenesis, after derivation under hypoxic and normoxic conditions, we performed Matrigel tube-forming experiments using HUVECs. The results showed that hypoxia-treated exosomes significantly improved tube forming, compared to that in the normoxia group at 0.5, 2, and 4 h after exosome administration (Fig. 4A). Figure 4B shows the statistical analysis of tube area calculation in different groups at different time points (0–4 h, recorded every half hour); regarding the promotion of tube formations, exosomes derived under hypoxic and normoxic conditions were better than the control group, whereas those from the hypoxic group were superior to those from the normoxic group, and this difference was significant (P < 0.05). After administering normoxia- and hypoxia-derived exosomes to HUVECs, we further analyzed HUVEC gene expression profiles at different time points. Expression of the angiogenesis inhibitory gene Vash1 were significantly lower with hypoxia-derived exosomes than with those derived under conditions of normoxia. Moreover, angiogenesis-promoting genes Angpt1 and Flk1 were higher in the hypoxic group than in the normoxic group after 1 h (Fig. 4C). To further verify the different effects of adMSC-Exos on angiogenesis, we used endothelial cells that were treated with 100 μg/mL of MSCs-Exos, and subcutaneously injected these cells, with Matrigel, into mice. After 8 days, samples were collected; immunohistochemical staining for CD31, a marker of endothelial cells, showed that endothelial cells formed more tubular structures when exposed to hypoxia-derived exosomes, compared to that after exposure to exosomes from the normoxia group (Fig. 4D; P < 0.05). Immunohistochemistry was further performed for Angpt1 and Flk1. The results showed that the expression of both was significantly increased in the hypoxia group, compared with that in the normoxia group (Fig. 4E).
Hypoxia enhance up-expression of VEGF in adMSCs and their exosomes
To further investigate the molecular mechanism of hypoxia-induced angiogenesis, we collected hypoxia-stimulated MSC samples at different days and found that HIF-1α (Fig. 5A, B) and VEGF expression was significantly increased 6 days after hypoxia stimulation (Fig. 5C, D). The protein kinase A (PKA) signaling pathway in MSCs was significantly activated with hypoxia after 4 days (Fig. 5E). The expression of VEGF at different time points in the exosomes was also significantly increased after 6 days (Fig. 5F), consistent with changes in MSCs.
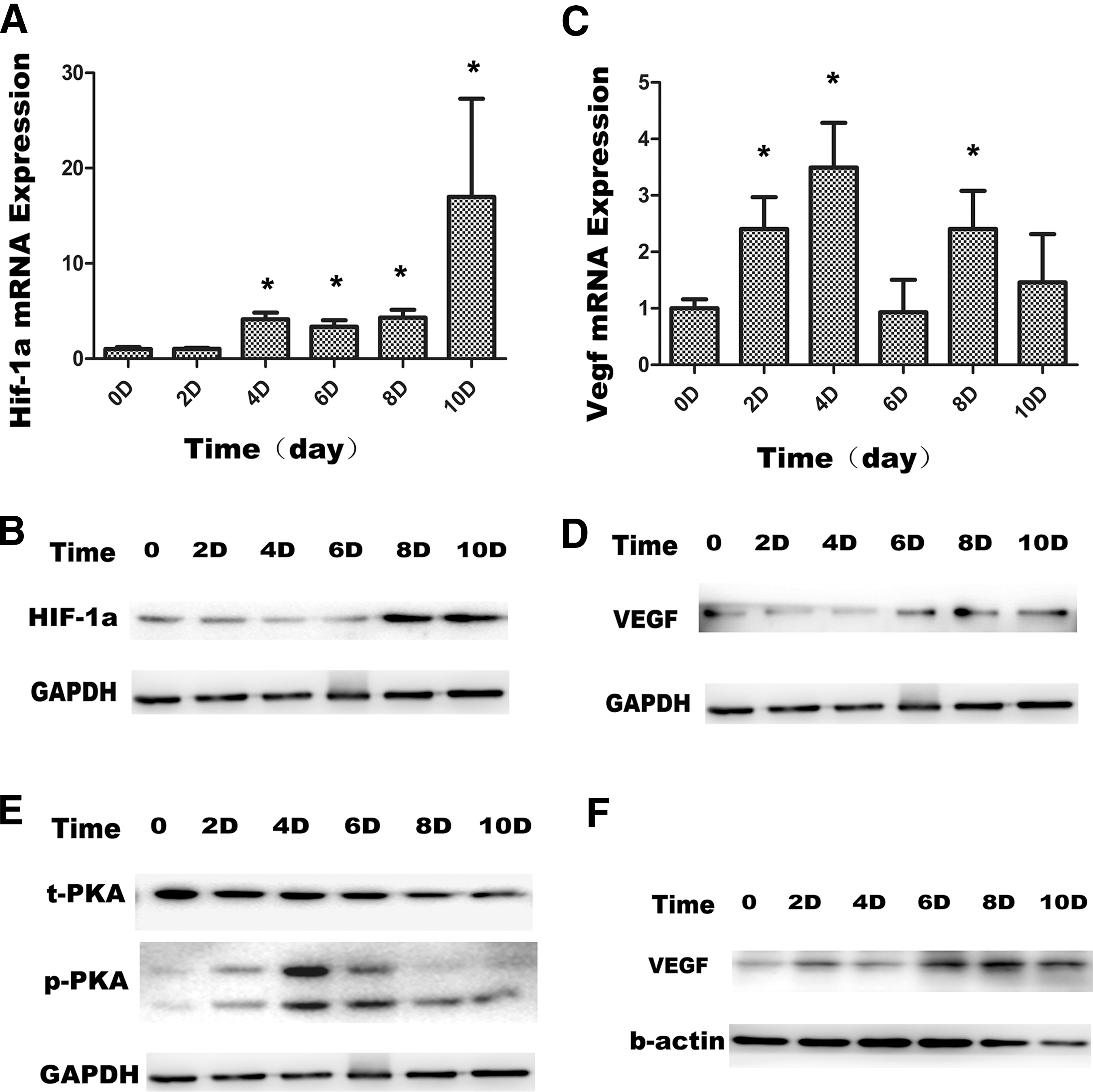
Hypoxia-treated MSC increased VEGF expression and PKA phosphorylation.
Hypoxia-treated exosomes promote VEGF expression in HUVECs through activation of PKA signaling
We next compared the effect of hypoxia- and normoxia-derived on PKA-related signaling pathways in HUVECs. The results showed that exosome (100 μg/mL) treatment activated PKA signaling after 0.5 h, as assessed by PKA phosphorylation (Fig. 6A). VEGF, VASH, and ANGPT1 were also detected. The results showed that VEGF expression in HUVECs stimulated with hypoxia-derived exosomes after 2 h was higher than that in the normoxia exosome group. Moreover, the expression of the proangiogenic gene ANGPT1 was increased, whereas that of the angiogenesis inhibitory gene VASH was decreased (Fig. 6B). Further, the phosphorylation of PKA was significantly inhibited after HUVECs were treated with H89H 2HCL (PKA inhibitor), but the expression of total PKA was not affected (Fig. 6C). Moreover, the addition of PKA inhibitors significantly inhibited the expression of angiogenic genes ANGPT1 (Fig. 6D). Moreover, the expression of vascular-related genes angpt1 and flk1 were decreased (Fig. 6E, F). These findings reveal that hypoxia-derived exosomes enhance angiogenesis, and this is mediated by the PKA signaling pathway.

Hypoxia-treated exosomes induce increased VEGF expression and angiogenesis in HUVECs through the PKA signaling pathway.
Discussion
A large number of studies have reported that MSC-derived exosomes can promote angiogenesis [1]. Our laboratory also reported that HUVECs treated with MSC-derived exosomes can promote the expression of proangiogenesis genes Angpt1 and Flk1 [16]. Therefore, exosomes have potential therapeutic value for the treatment of traumatic diseases. To further explore the effect of exosomes derived under hypoxic conditions, we conducted a series of experiments to verify that exosomes excreted by MSCs under hypoxic conditions have a stronger proangiogenic effect.
In our study, we treated HUVECs with exosomes derived under conditions of hypoxia (1% O2) and normoxia; adMSC-Exos derived under conditions of hypoxia were more easily taken up by endothelial cells. It has been reported that exosomes are taken up by different cellular pathways, including endocytosis, membrane fusion, and receptor binding. In this study, exosomes secreted by MSCs after exposure to different conditions might have affected the efficiency of uptake through receptor binding, but the detailed mechanism needs to be further studied.
In this study, we chose vein endothelial cells for in vitro tube formation as a model. This model is well-established and defined [16]. Angpt1 and Flk1 are key genes that promote tube formation, whereas Vash1 is key gene that inhibits tube formation. We found that hypoxia-treated exosomes promoted proangiogenesis gene expression, downregulated antiangiogenic genes, and promoted tube formation. In addition, we subsequently implanted in exosomes mixed with Matrigel in to mice subcutaneously. In vivo results assessing vascular formation were similar to vitro cell model results, and these results suggest that hypoxia-treated exosomes can indeed enhance angiogenesis.
Angiogenesis and human growth and development, inflammation, tissue repair, ischemia, tumor migration, and invasion, as well as other physiological and pathological occurrences are closely related [17 –19]. Angiogenesis involves two important regulatory pathways: one is vascular through VEGF and its receptor, and the other is via angiopoietin (Ang) and its receptor. These two pathways have a synergistic effect, and together promote blood vessel formation. A large number of studies have reported that under conditions of hypoxia and inflammation, most cells can secrete VEGF, which can specifically act on vascular endothelial cells, promote cell proliferation, and induce blood vessel formation in vivo. Our study also found that H-MSCs upregulate the expression of early HIF-1α and subsequently induce increased expression of VEGF. In addition, levels of VEGF in H-MSC-derived exosomes were also significantly higher than those from the normoxia groups. It has been reported that VEGF secreted by the cystic biliary epithelium activates the PKA signaling pathway [20]. Furthermore, activation of the PKA signaling pathway in mice with Pkd1KO and Pkd2KO liver defects has been found to increase VEGF expression and after this pathway was suppressed, VEGF expression was significantly reduced [21]. Therefore, we examined hypoxia-stimulated MSCs and found that phosphorylation of PKA was significantly increased 4 days after hypoxic stimulation, which might be related to VEGF secretion. Subsequently, we examined the PKA signaling pathway in HUVECs after exosome treatment. We found that PKA signaling was activated, which further promoted endogenous VEGF expression in HUVECs, and this synergistically regulated the expression of downstream proangiogenic genes Angpt1 and Flk1, while decreasing vasculogenic genes Vash, thereby promoting angiogenesis. After HUVECs were treated with the PKA inhibitor H89H 2HCL, exosomes significantly inhibited PKA phosphorylation, but did not affect total PKA protein expression. Moreover, the addition of PKA inhibitors significantly inhibited the expression of proangiogenic genes ANGPT1. These findings reveal that hypoxia-derived exosomes enhance angiogenesis, and that this is mediated by the PKA signaling pathway.
In summary, our results show that hypoxia-treated exosomes markedly enhanced angiogenesis. The associated mechanism was also deciphered; specifically, a high concentration of VEGF can activate PKA signaling in endothelial cells, thereby inducing endogenous VEGF expression, and synergistically promoting angiogenesis. Therefore, hypoxic preconditioning of MSC-derived exosomes could represent a novel strategy for the clinical treatment of ischemic diseases with stem cell-derived products.
Footnotes
Acknowledgments
This study was supported by grants from National Natural Science Foundation of China (81672313, 81371344, 81473450); the National Key Research and Development Program of China (2016YFA0101000, 2016YFA0101003); CAMS Innovation Fund for Medical Sciences (2017-I2M-3-007); and Beijing Key Laboratory of New Drug Development and Clinical Trial of Stem Cell Therapy (BZ0381).
Author Disclosure Statement
No competing financial interests exist.