
Abstract
Aggressive chemotherapy in childhood often results in testicular damage and consequently jeopardizes future fertility. The presence of spermatogonial cells (SPGCs) in the testes of prepubertal cancer patient boys (PCPBs) can be used to develop future strategies for male fertility preservation. In the present study, we examined the presence of SPGCs in testes of chemotherapy-treated PCPBs and their ability to develop spermatogenesis in vitro using a three-dimensional culture system. Seven testicular biopsies were obtained from chemotherapy-treated PCPBs and one from a patient with β-thalassemia major. Isolated testicular cells were cultured in a methylcellulose culture system (MCS)-containing StemPro enriched with growth factors for 5–15 weeks. The presence of premeiotic, meiotic, and postmeiotic cells was examined by immunofluorescence staining and/or reverse transcription-polymerase chain reaction (RT-PCR) analysis. We observed SPGCs in the examined testicular biopsies. Isolated testicular cells cultured in MCS developed into colonies and contained premeiotic, meiotic, and postmeiotic cells. Furthermore, we identified sperm-like cells that had developed from testicular cells of a PCPB. Our results demonstrate, for the first time, the presence of biologically active SPGCs in testicular biopsies of chemotherapy-treated PCPBs and their capacity to develop in vitro to different stages of spermatogenesis, including the generation of sperm-like cells. This study may open the way for new therapeutic strategies for fertility preservation of PCPBs and for azoospermic patients.
Introduction
C
Our hypothesis is that testicular biopsies from chemotherapy-treated prepubertal cancer patient males contain biologically active SPGCs. These cells could be induced in vitro using a 3D culture system to complete spermatogenesis. The introduction of intracytoplasmic sperm injection and in vitro fertilization techniques facilitates fertilization with only a very few mature sperm and future reproduction to prepubertal boys who undergo aggressive chemo/radio therapy from their own germ line.
Materials and Methods
Human testis material
Local Institutional (#5011) and National (#920100438) Ethics Committees approved the study. All patients' parents (or guardians) signed informed consent for the fertility preservation procedure and to use part of the testicular biopsy for research. Testicular biopsies were obtained from seven prepubertal cancer patients (age range 6–13 years) with recurrent ALL (n = 4), medulloblastoma of Cerebellum (n = 1), rhabdomyosarcoma (n = 1), and acute promyelocytic leukemia (n = 1) that were treated before the testicular biopsy with chemotherapy. As a control for chemotherapy-treated patients, we used patient #8 who is a thalassemia major patient, and required frequent regular blood transfusions and was never treated with chemotherapy before the testicular biopsy (Table 1). All patients were scheduled for aggressive chemotherapy before bone marrow transplantation. Tanner stage of development was evaluated for all patients, but the Johnsen score was performed on only three patients, for whom we had histology for their testicular biopsies (Pt#2, Pt#3, Pt#6). We also measured the diameters of the STs of these patients [presented as average of 35 STs ± SD]. Urogenital history of the patients was not remarkable.
In this table, we summarized clinical data related to the eight prepubertal patient boys who testicular biopsies were examined. The data included age of the patient at time of biopsy (years, Y), diagnosis (type of cancer and other disease for asking for a biopsy), type of treatment before taking the biopsy, duration of chemotherapy (chemo) treatment (months; M), accumulative dosage, time lapse between last chemotherapy and surgery (months; M or years; Y), Johnsen score (type of cells present in the seminiferous tubules) and Tanner stage of the patients.
ALL, acute lymphoblastic leukemia; THA, Beta-thalassemia major.
During the surgery, a single biopsy from one testis was taken in accordance with the approval of the Ethics Committee. Due to the small size of the biopsies used in the present study (around 3 mm3), most of the biopsies were used (as first priority) for in vitro culture in MCS. In three biopsies, which were larger than 3 mm3, an additional part was used for histological evaluation (fixation with Bouin's solution and embedding in paraffin) and/or for RNA extraction (stored at −70°C) (see study design in Fig. 1A).

In vitro culture system and markers of spermatogenesis development stages. A scheme of design of the study, which includes all the steps of using the biopsies of the patients, is presented
Testicular cell isolation and culture
Seven of the biopsies were immediately transferred in Dulbecco's phosphate buffer saline in ice from the operating theater to our laboratory for cell isolation. One biopsy from a cancer patient (Pt#3) was cryopreserved [29] and used after 11 months. In brief, after washing with phosphate buffer solution (PBS) to remove residual blood, the biopsy was divided to around 3 mm3 parts and cryopreserved in 1.8 cryovials that contained 1.5 mL cryoprotectant media composed of 5% DMSO, 10% human serum albumin, and 3.5% sucrose diluted in HBSS. Cryopreservation was performed similarly to program 1 [29] with minor modifications. The cooling rate was 0.5°C/min, with holding at 0°C for 9 min, followed by a cooling rate of 0.5°C/min, until −8°C with a holding of 5 min at this temperature. Thereafter, seeding was manually performed. After 15 min holding at −8°C, the vials were frozen to −40°C at a rate of 0.5°C/min. The vials were then frozen to −80°C at a rate of 0.7°C/min and then transferred to liquid nitrogen. The cryopreserved biopsy was thawed at room temperature and centrifuged (for washing) in the presence of Minimum Essential Media (Biological Industries). Part of the biopsy from three patients (Pt#2, Pt#3, Pt#6) were Bouin-fixed and paraffin-embedded to be used for histological analysis by hematoxylin and eosin (H&E) staining or immunostaining for different markers of spermatogenesis. The fixation was performed immediately following the surgery of patients #2 and #6, and after thawing the cryopreserved biopsy of patient #3. Biopsies were cut into small pieces of ∼1 mm and subjected to collagenase type V (2 mg/mL) (Sigma, St. Louis, MO), DNAse (8 μg/mL) (Sigma) and hyaluronidase (2 mg/mL) (Sigma) in a total volume of 4 mL for 20 min in a 32°C water bath shaker. Cells were precipitated by centrifugation (300 g, 10 min) and suspended with 4 mL of TrypLE Select (Gibco, Denmark) for 10 min in a water bath shaker at 32°C. Cell suspension was centrifuged (300 g, 10 min), and the precipitated cells were suspended in 200 μL Roswell Park Memorial Institute (Biological Industries, Beit Haemek, Israel) and counted.
Cells were cultured (4–5 × 104 cells/well/500 μL) in methylcellulose (MC) (R&D, Minneapolis) (42%) as a 3D culture system. In brief, the cells were diluted in 58% of media composed of 33% StemPro-34 medium and 25% KSR (knockout serum replacement) (Gibco) enriched with different factors and reagents such as human rEGF (recombinant epidermal growth factor) (20 ng/mL) (Biolegend, CA), human rGDNF (glial cell line-derived nerve growth factor) (10 ng/mL) (Biolegend), human rLIF (leukemia inhibitory factor) (10 ng/mL) (Biolegend), and human r-bFGF (basic fibroblast growth factor) (10 ng/mL) (Biolegend) [22,24,30; with modification]. Media containing isolated cells (58% final dilution in the well) were mixed with MC (42% final dilution in the well) and culture in the wells. Cells were cultured for 5–15 weeks in CO2 incubator at 37°C. We applied similar conditions (temperature; 37°C) as in our previous study using mouse system [25], which showed similar results of development of spermatogenesis compared to our other cultured systems under 35°C [26,27]. Every 10 days to 2 weeks, we evaluated the development of the cells in MCS under the microscope [according to the growth quality/viability (around 10% of the cells with apoptotic vacuoles in their cytoplasm) and morphology of the developed colonies] and added 50 μL/well of fresh concentrated ( × 10) enriched StemPro-34 medium (containing all the growth factors used in the primary culture) to the cell cultures to be followed up after additional 1–2 weeks (see design of the study in Fig. 1A). We tried to grow the cells in the culture as much as possible to be closer to the physiological timing of development of human spermatogenesis (around 3 months). At the end of the incubation period in MCS, we added 0.5 mL PBS to the culture wells that contained 0.5 mL MC mix by pipetting and collected the suspension to 15 mL tubes. The tubes were centrifuged in 1,600 rpm for 10 min. We removed most of the volume and collected the remainder around 100 μL from the bottom of the tube. This volume which contained the cells was smeared on a slide and/or collected and kept at −70°C to be used for RNA analyses (according to the number of colonies developed in the culture. If we had more than 10–15 small colonies, or 5 medium or large colonies in the well, we utilized the cells for both immunofluorescence (IF) and RNA analyses. Otherwise, the priority was for IF analysis).
Immunofluorescence staining
To determine the presence of a specific germ cell population distinctive to each spermatogenic phase in the testicular sections or in the isolated testicular cells before and after in vitro culture in MCS, IF staining was performed for markers which are known to be specific to each spermatogenic stage (Fig. 1C) [28,31].
Testicular tissues immunostaining
Testicular biopsies were fixed in Bouin's solution (Kaltek, Italy) and paraffin embedded. Sections of 5 μm were placed on Superfrost Plus slides (Thermo, Braunschweig, Germany) for IF staining of spermatogenic markers. Deparaffinated section slides were treated with xylene and ethanol (BIO-LAB, Jerusalem, Israel) for 20 min. After washing with PBS (Biological Industries), antigen retrieval of the sections was performed in heated 36% urea solution (Millpore) at a warm microwave degree (900 W) for 5 min (twice).
Following washing, nonspecific adhesion sites in the tissues and cells were blocked by 5% fetal calf serum (Biological Industries) for 30 min at room temperature. Following the removing of the blocking buffer, the first antibodies were added: polyclonal rabbit anti-human VASA (1:1,000; Santa Cruz, CA), polyclonal rabbit anti-human CD9 (1:200; Abcam, Cambridge, United Kingdom), polyclonal goat anti-human OCT4 (1:200; Santa Cruz), polyclonal rabbit anti-human α-6-INTEGRIN (1:200; Santa Cruz), polyclonal goat anti-human GFR-α (1:50; R&D, MN), monoclonal mouse anti-human PLZF (1:100; Santa Cruz), polyclonal rabbit anti-human c-KIT (1:200; Dako, CA), polyclonal goat anti-mouse vimentin (1:50; Santa Cruz), and polyclonal rabbit anti-mouse GDNF (1:100; Santa Cruz). After overnight incubation at 4°C, the slides were washed and the specific secondary antibodies were added compatibly to the first antibodies [donkey anti-rabbit IgG (Cy3), donkey anti-goat IgG (Cy3), and goat anti-mouse IgG (Rhodamine red) Jackson Immuno Research] for 40 min at room temperature. Following washing, the slides were dried and DAPI, which stains the nucleus in blue, was added to the tissues, and they were stuck with cover slides. The negative control was incubated in a blocking buffer instead of the first antibody. The specificity of the staining was also examined in testicular tissue using the relevant IgG isotype as negative control. The positive staining for the examined markers was performed on Bouin-fixed adult human testis tissue embedded in paraffin. Slides were examined for staining, using a fluorescence microscope (Nikon eclipse 50 I, Tokyo, Japan).
Immunostaining of testicular cells
Isolated cells were fixed in cold methanol for 20 min. The immunostaining process was similar to that mentioned above for testicular tissue immunostaining after the stage of antigen retrieval. Following the removal of the blocking buffer, the first antibodies were added: polyclonal rabbit anti-mouse SALL4 (1:400; Abcam), polyclonal rabbit anti-human VASA (1:100; Santa Cruz), polyclonal rabbit anti-human CD9 (1:10; Santa Cruz), polyclonal goat anti-human OCT4 (1:50; Santa Cruz), polyclonal rabbit anti-human α-6-INTEGRIN (1:100; Santa Cruz), polyclonal rabbit anti-human GFR-α (1:50; Santa Cruz), monoclonal mouse anti-human PLZF (1:100; Santa Cruz), polyclonal rabbit anti-human c-KIT (1:50; Santa Cruz), polyclonal rabbit anti-human BOULE (1:25; Santa Cruz), polyclonal rabbit anti-mouse CREM-1 (1:30; Santa Cruz), polyclonal goat anti-human LDH (1:50; Santa Cruz), polyclonal goat anti-mouse PROTAMINE (1:20; Santa Cruz), polyclonal rabbit anti-human ACROSIN (1:10; Santa Cruz), monoclonal mouse anti-human Ki67 (1:200; Dianova GmbH, Germany), and monoclonal rabbit anti-human Ki67 (1:200; Cell Marque). After overnight incubation at 4°C, the slides were washed and treated as mentioned above for testicular tissue immunostaining. Since our antibodies are cross-reactive with mouse, the positive staining for the examined markers was performed using isolated testicular cells from azoospermic patients with sperm in their biopsies and/or mouse testicular cells.
Counting of stained cells
Slides were divided by PAP pen to be used for staining of a few markers. We counted around 30 cells for each marker when the number of positive stained cells was dependent on the examined marker or patient and if the sample was from before or after culture. For example, in one case, we counted 5/32 VASA-positive stained cells, 6/23 PLZF-positive stained cells, and 5/31 LDH-positive stained cells.
MitoTracker staining
MitoTracker Green FM probes (Molecular Probes, Cat# 7514; Invitrogen), which stain mitochondria, were used to identify developed sperm in MCS, according to the supplier's protocol. In brief, 200 nM MitoTracker was added to collected cells from MCS (in an Eppendorf tube) and incubated at 37°C in CO2 incubator for 30 min. Thereafter, the tubes were centrifuged at 1,600 rpm for 5 min. At the end of the centrifugation, the supernatant was removed and the pellet was diluted with 50 μL PBS. DAPI was added to the tube (5 μL), and the suspension was smeared on a slide. Identification of sperm-like cells was performed immediately using a fluorescence microscope (Nikon eclipse 50 I, Tokyo, Japan).
Gene expression: polymerase chain reaction amplification
Isolated cells
Enzymatically isolated testicular cells and developed cells from in vitro cultures were mixed with 200 μL of lysis buffer (Dynabeads Kit; Dynal Biotech, Oslo, Norway). The lysates were frozen at −80°C for later RNA extraction using a Dynabeads kit (Invitrogen, Lithuania). The cDNA synthesis was performed according to M-MLV Reverse Transcriptase protocol (Invitrogen) using random hexamers, and polymerase chain reaction (PCR) was performed using specific primers for each examined spermatogenesis marker: OCT4 (forward: AATTTGCCAAGCTCCTGAAG; reverse: CGTTTGGCTGAATACCTTCC; product size, 337 bp), SALL4 (forward: TCCCAAACACCAGTTTCCTC; reverse: TGTGTCTGCATTGCTCCTTC; product size, 90 bp), VASA (forward: TGG AAA CAG AGA TGC TGG TG; reverse: CCT CTG TTC CGT GTT GGA TT; product size, 183 bp), PLZF (forward: AAGGCTGCAGTGGACA; reverse: CTGCATCATCATCTCCGTCTT; product size, 144 bp), α-6-INTEGRIN (forward: TTGTTTCGTAACACAGCATTG; reverse: GGCACTAGTATCTTTGGCTGA; product size, 146 bp), CD9 (forward: CCTACAACAAGCTGAAAACCA; reverse: GGATAGCACAGCACAAGATCA; product size, 282 bp), GFR-α1 (forward: AGCAGGGTCTGAGAATGAAAT; reverse: GCCATTGATTTTGTGGTTATG; product size, 171 bp), c-KIT (forward: TTCTACAAGATGATCAAGGAAGG; reverse: AGAATTGATCCGCACAGAAT; product size, 243 bp), the meiotic gene CREM (forward: ACGAGGTCCGCTACGTAAAT; reverse: GGCTCTCCAGACATTTTACATATT; product size, 249 bp), BOULE (forward: CCATTTATCAGCAACCTGCAT; reverse: GTGCAATTTCCACTGGTTGAT; product size, 153 bp) and LDH (forward: GGAACGGATTCAGATAAGGAA; reverse: TTCACAACATCTGAGACACCA; product size, 260 bp) and the postmeiotic gene PROTAMINE (forward: AAAGAAGTCGCAGACGAAGGA; reverse: TATTGGATGGTGGCATTTTCA; product size, 193 bp). The calibrator gene PPIA (forward: TATCCTAGAGGTGGCGGATTT; reverse: GAATGGTATCACCCAGGGAAT; product size, 150 bp). The PCR amplification reaction occurred as: 3 min at 95°C, 30 cycles of 15 s at 95°C, 15 s at 60°C (all the primers were designed with this specific annealing temperature), and 40 s at 72°C. The final elongation step lasted 5 min at 72°C.
Results
Histology and IF staining of spermatogenic cells in testicular tissue of biopsies from PCPBs
Histological sections of testicular biopsies from three prepubertal ALL patient boys (Fig. 2A: #2, B: #3) and one medulloblastoma (MD) patient (Fig. 2C: #6) showed similar diameters (μm ± SD; 133 ± 11, 124 ± 10, and 141 ± 10, respectively) of their STs. The presence of A dark (Ad) and A pale (Ap) SPGCs was distinguished according to the intensity of staining of the nucleus by H&E staining and their closeness to the basal membrane of the ST. Primary spermatocytes (SPC) were also distinguished in one biopsy according to their location inside the STs and the morphology of the nucleus (Fig. 2C). In the periphery of the ST (outside the tubules), we were able to identify peritubular cells. The interstitial compartment contained clusters of cells similar histologically to those characterized as Leydig cells (LCs) (Fig. 2B–D). However, the histology of the STs in patient #2 showed a wider lumen and were impaired histologically with a thin single layer of cells (Fig. 2A) compared with patients #3 and #6 (Fig. 2B, C, respectively).
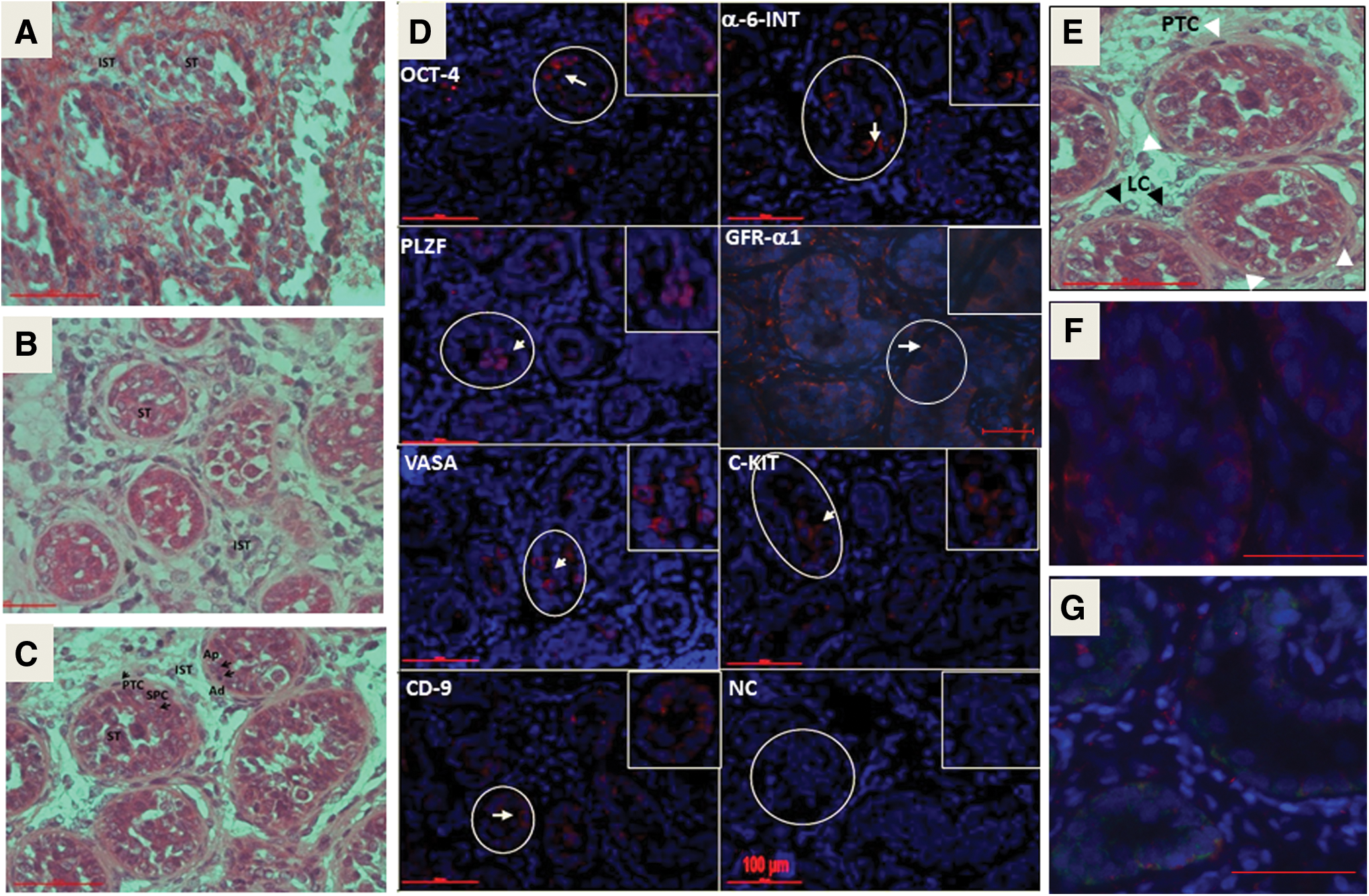
Histological and IF staining of testicular sections from a prepubertal cancer patient boys. The histological staining by H&E of testicular biopsy from prepubertal cancer patients (
IF staining of testicular biopsies from prepubertal cancer patients (n = 3; #2, #3, #6) showed positive staining for the premeiotic markers; VASA, PLZF, CD9, α-6-INTEGRIN, and c-KIT in all three examined biopsies, whereas OCT4 was stained in two of them (Pts#2 and #3), and GFR-α1 was stained only in one (Pt#6) (Fig. 2D and Table 2). We examined sections by IF and/or RNA expression for meiotic and postmeiotic markers, but those markers were undetectable. Immunostaining for Sertoli cells using specific antibodies to vimentin showed the presence of Sertoli cells in the STs of fixed tissue before enzymatic digestion (F). These Sertoli cells were active to produce GDNF, as examined by a double staining for Sertoli cells (vimentin; red) and GDNF (green) (G). This is important to confirm that Sertoli cells (and not germ cells that also present in the tubules and known to produce GDNF) are functional/active and produce GDNF. These supporting cells are present in the culture and support the development of germ cells. For the four other cancerous patients and control (the patient with β-thalassemia major), histological sections were unavailable due to small amounts of testicular tissue, which were used for other investigations in this current study. Therefore, the IF staining was performed on isolated cells from the biopsies of chemotherapy-treated patients and the control patient (untreated with chemotherapy before testicular biopsy; beta thalassemia major patient) (Fig. 3F). The results are summarized in Table 2.
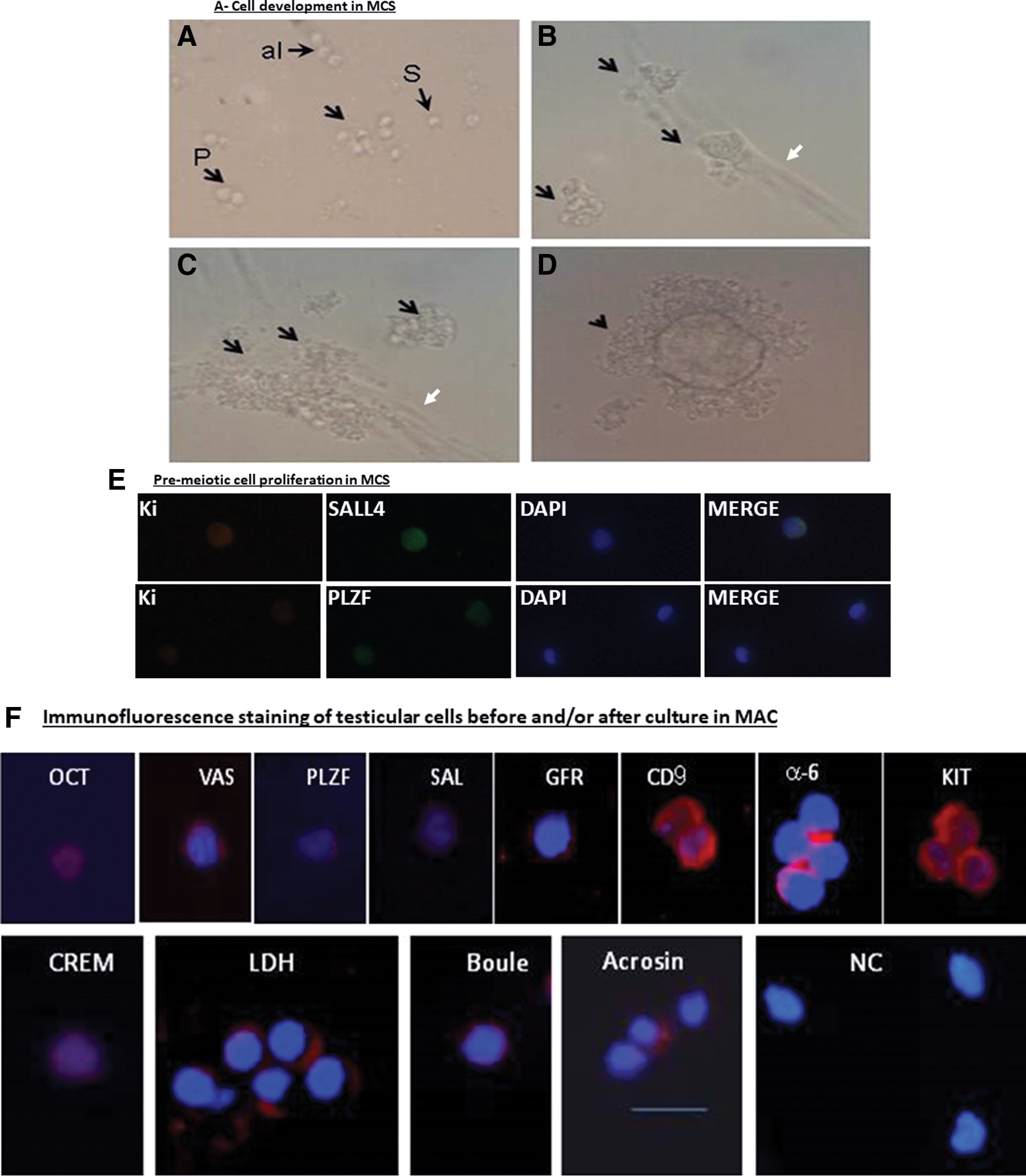
Proliferation, colony formation and development of spermatogenic cells in vitro from SPGCs isolated from testicular biopsies of prepubertal cancer patients. Isolated germ cells from testicular biopsies of prepubertal cancer patients developed after 5–15 weeks of culture to small (around 10/well), medium (4–5/well) and large (1–3/well) colonies in MCS culture.

Cells isolated from testicular biopsies of prepubertal cancer patients (n = 6) were cultured for number of weeks in vitro in MCS. Cells before and after culture in MCS were examined for the presence of spermatogenic cells by IF staining or for expression markers (RNA; R) by RT-PCR analysis using specific antibodies or primers (respectively) for each spermatogenic stage: pre-meiotic (PLZF, GFR-α1, SALL4, OCT4, CD9, α-6-Int, VASA, and c-KIT), meiotic (CREM, LDH, and BOULE) and post-meiotic (PRO and ACR), ES. Empty rectangles indicate not examined. Gray color indicates markers that were not expressed before in vitro culture and positively expressed after in vitro culture in MCS. (−) indicates negative expression. (+) indicates positive expression. Size indicates size of the developed colonies (S–small, M–medium, L–large) as described in Fig. 3A–D.
Ac, after culture and the number of weeks in the culture (Ws); ACR, Acrosin; Af, after culture; Be, before culture; Col, developed colonies in the culture; ES, elongated spermatid; IF, immunofluorescence; MCS, methylcellulose culture system; MD, medulloblastoma; PRO, protamine; Ws, weeks.
IF staining and expression of germ cell markers in isolated cells from testicular biopsies of prepubertal cancer patients
Cells isolated from testicular biopsies of the prepubertal patients (n = 8) before culture in MCS showed positive IF staining for cells belonging to the premeiotic stages of spermatogenesis (positive cells were very few, less than 10 cells from a total of around 30 cells. The same scenario existed for meiotic and postmeiotic cells, with a different percentage. It depended on the examined marker and patient), or expression of those markers when examined by reverse transcription polymerase chain reaction (RT-PCR) analysis (positive expression of markers of the different stages of spermatogenesis is presented in Fig. 3G) (summary of results in Table 2). This is not a background IF staining because we only considered positive staining when it was clearly stained in comparison to the negative control.
Isolated cells from patient #4 expressed only PLZF, CREM-1, and BOULE (Table 2). Our results showed that testicular tissues from prepubertal patient boys previously exposed to chemotherapy (including cyclophosphamide) contained premeiotic cells that stained for some markers (ranging from 1 to 7 markers) specific for SPGCs. It should be noted that the two patients (#4 and #5) at ages 10 and 13 years old, respectively, showed staining for only one premeiotic marker (Table 2).
Induction of spermatogenesis, including the generation of sperm-like cells in MCS
In two patients (#1, #7), the number of cultured isolated cells from testicular biopsies was very low (around 2 × 104 cells) and, therefore, in vitro culture was unsuccessful. Isolated cells from the remaining six testicular biopsies (6 patients) were cultured in MCS for a period of 5–15 weeks (Fig. 1A). During this period, cells were developed to form only single cells in MCS (cells, not clusters/colonies) and/or small colonies/clusters (up to 30 cells) (S) and/or medium colonies/clusters (up to 100 cells) (M) and/or large colonies/clusters (>150 cells) (L) (Fig. 3A–D). In patient #2, colonies started to develop after 8 weeks. Patient #3 developed large colonies after 8 weeks of culture. Patient #4 developed colonies after 7 weeks of culture. In patient #5, the colonies developed after 1 week of culture. In patient #6, colonies developed after 2 weeks of culture. Patient #8 developed colonies after 1 week of culture. In addition, we showed that SALL4- and PLZF-positive SPGCs from the developed cultures were costained with Ki67 (a marker of proliferation) by double immunostaining (Fig. 3E), which indicates proliferation of SPGCs in the cultures. Cells/colonies were collected and examined for the different stages of spermatogenesis (Fig. 1C) by IF and/or RT-PCR analyses (Fig. 3F, G, respectively; representative results) (summary of results in Table 2). In general, the number of examined markers was dependent on the amount of RNA extracted from the samples. We tried to compare markers, which were positive before culture to after culture (eg, BOULE and LDH in patients #3, 4, 5, 8). In one biopsy (#4), the isolated cells only expressed PLZF, CREM-1, and BOULE before culture, and in another biopsy (#2), the isolated cells did not stain for any of the examined spermatogenic markers before culture but stained in the tissue (before cell isolation) for PLZF, OCT4, CD9, α6-Int., VASA, and c-KIT (Table 2). Those cells from both biopsies did not express spermatogenic markers after in vitro culture in MCS (Table 2). Another biopsy (patient #5) expressed some premeiotic markers before and after culture in MCS, but did not develop meiotic or postmeiotic markers after culture in MCS (Table 2). Our results show that one biopsy (#6) expressed 6 premeiotic markers before culture, and after in vitro culture in MCS, it developed meiotic (CREM-1, LDH) and postmeiotic (expression of protamine) cells/markers. Cells from the biopsy of patient #8 (beta-thalassemia major [THA]) expressed and developed premeiotic markers before and after culture in MCS and also developed the meiotic marker BOULE, but did not develop postmeiotic markers. Cells from biopsy of patient #3 expressed and developed premeiotic markers before and after culture in MCS and also developed the meiotic marker BOULE (IF) (Table 2) and the postmeiotic marker acrosin in round spermatid [Fig. 3H (a, b)] and elongated-like cells [Fig. 3H (c)] (Table 2). Acrosin-stained cells were characterized as round or elongated spermatid, according to their morphology [size and shape of the nucleus; Fig. 3H (a–c)]. In addition, cells with sperm-like morphology that were developed in vitro were identified according to staining with MitoTracker [Fig. 3H (e1–e3)]. In Fig.3H (e1), the cytoplasm of sperm-like cells was large (similar to the morphology of i8 and i9 that are characterized in stages 8–12 of spermatid development in the human seminiferous epithelium VIII–XII stages [32]) and stained with MitoTracker (mitochondria in the cytoplasm stained green), while the nucleus with morphology of sperm-like cell nucleus is stained blue. It should be noted that some sperm-like cells showed a nucleus similar in their size [Fig. 3H(c, e1)] to normal sperm found in some biopsies of nonobstructive azoospermic patients [Fig. 3H (f)], even though the final morphological structure was different. However, other cells with sperm-like morphology exhibited a relatively large nucleus [Fig. 3H (e2–e3)] compared to the control [Fig. 3H (f)] but presented a neck (N) (with concentrated green color, which indicates the presence of concentrated mitochondria) and tail. These morphologies of sperm-like cells that developed in MCS are similar to those morphologies characterized in stages 8–12 of spermatid development in the human seminiferous epithelium VIII–XII stages [32] (Fig. 3I).
In summary, following culture in MCS, premeiotic markers were developed in at least 3/6 biopsies, meiotic markers were developed in 3/6 biopsies, and postmeiotic markers were developed in 2/6 biopsies. Cells with sperm-like morphology were identified in 1/6 of the biopsies (Fig. 3H and Table 2).
Discussion
This study is the first, to the best of our knowledge, to publish a successful in vitro induction of meiotic and postmeiotic stages from SPGCs harvested from some chemotherapy-treated prepubertal cancer patients, as shown by the development in culture of meiotic and postmeiotic germ cell types, and furthermore, the generation of cells with sperm-like morphology. In addition, we clearly showed that biologically active SSCs (based on the presence/expression of several premeiotic markers before culture and their subsequent proliferation and development in the 3D culture to meiotic and postmeiotic stages) were present in the testes of prepubertal cancer patients even after chemotherapy treatments. It should be emphasized that even though we found bioactive SSCs in chemotherapy-treated patients, for some patients, there is a very high risk of no residual SSCs. This is dependent on type, dosages, and combinations of drugs/radiation and length of period of treatment [5,12,33]. The developed cells from fresh and cryopreserved testicular tissues behaved similarly in MCS. Therefore, it is recommended to seek the advice of an oncologist regarding the need for cryopreservation of testicular biopsy before starting the chemotherapy/radiotherapy treatments. In one biopsy (#4), the isolated cells only expressed PLZF, CREM-1, and BOULE before culture, and in another biopsy (#2), the isolated cells did not stain for any of the examined spermatogenic markers before culture, but stained in the tissue (before cell isolation) for PLZF, OCT4, CD9 and α6-Int., VASA, and c-KIT (Table 2). Also, patient #6 showed a Johnsen score of 5 by histological evaluation of his testicular biopsy (contained SPC), but did not express meiotic markers in his isolated testicular cells. This could be related to the loss of cells following isolation procedure (some of the remaining cells are undetectable for some markers). Our results show that most of the biopsies (6/8 patients) showed (before culture) the presence/expression of more than three premeiotic marker (3–7 markers), and only two patients who were older (10 and 13 years old) (#4 and #5) expressed the presence/expression of only one marker (Table 2). The expression of only one premeiotic marker in testicular biopsy of patients #4 and #5 could be related to the fact that these patients are close to puberty in age (10 and 13 years old, respectively), in which SPGCs are more active in proliferation, which make them more sensitive to chemotherapy treatments [34]. This could also be related to type, dosages, combinations of drugs/radiation, and length of treatment (31 and 29 months, respectively) [5,12,33]. Nurmio et al. demonstrated the presence of some markers of SPGCs (four markers were examined) in testicular biopsies of some chemotherapy-treated prepubertal cancer patients. However, these markers were not detected in testicular biopsies of those patients who were treated with cyclophosphamide [35]. In contrast, our results showed the presence of biologically active SPGCs in cyclophosphamide-ALL treated patients. Furthermore, Poganitsch-Korhonen et al. demonstrated that the quantity of SPGCs (according to histology/cross section) decreased with treatment with alkylating agents [36]. However, our results show that the SPGCs, which were found in biopsies of some patients, were biologically active as they could proliferate and differentiate to different stages of spermatogenesis in MCS in vitro. We grew human SPGCs in MCS, in similar conditions (temperature; 37°C) used in our previous study with mouse systems [25], which showed similar results of development of spermatogenesis compared to our other cultured systems under 35°C [26,27]. The similar effect of the different temperature on the development of spermatogenesis in vitro (which is in contrast to in vivo effects) could be related to a different microenvironment present in the in vitro conditions, including the type of cells, proteins, constant conditions (flow/diffusion), and MC (not the normal extracellular matrices and 3D of the tubule) compared to in vivo conditions. The inability of isolated SPGCs from two biopsies from ALL-chemotherapy-treated patients (#2, #4) to proliferate and differentiate in MCS could be related to the quality and/or quantity of these cells and/or to the activity of the supporting cells present in the culture and originate from the biopsies of the patients (Table 2) (see discussion later). On the contrary, isolated SPGCs from the other three patients who received chemotherapy (#3, #5, #6), two out of whom even received cyclophosphamide (#3, #5), could proliferate and/or differentiate to meiotic and/or postmeiotic cells with no association to the type of disease or the kind of chemotherapy protocol (Table 2). These results may support our suggestion that the development of SPGCs in MCS could be related to the quality of SPGCs and to the activity of the supporting cells after chemotherapy treatment. On the contrary, the differences in timing of maturation in vitro for cells isolated from testicular biopsies of patients #3 and #6, even though they are of similar age (6 and 7 years old), could be due to the type of the disease (ALL and MD, respectively) and treatments they received. They even expressed different cell markers before culture. Also, it should be noted that isolated SPGCs from patient #3 (ALL-chemotherapy treated, including cyclophosphamide) developed in MCS meiotic (boule- and acrosin round positive cells), postmeiotic cells (acrosin-positive elongated cells,) and even cells with sperm-like morphology, as detected by MitoTracker staining (Fig. 3H). Some of the generated sperm-like cells were positively stained to acrosin and showed nucleus similar to that of normal sperm. However, other sperm-like cells showed nucleus larger than that of normal sperm. These results may indicate that some of the generated sperm-like cells in MCS may be morphologically normal, while others are still premature sperm. These developed sperm-like cells in MCS are similar to those described in stages 8–12 of spermatid development in the human seminiferous epithelium VIII–XII stages [32]. The different morphology of sperm-like cells developed in the culture system compared to the morphology of the sperm from the “positive control,” could be related to either the degree of development (stage of development) of the sperm-like cells and/or to the culture conditions that may affect the morphology of the developed sperm. Thus, these results may suggest the ability of SPGCs from some ALL-cancer patients (even after chemotherapy treatment) to achieve almost complete spermatogenesis under certain in vitro conditions such as MCS. The expression of boule and protamine (RNA but not protein; IF staining was undetectable) in isolated cells of this patient before culture may indicate the presence of cells from the early stages of meiosis that express these markers. Indeed, recently it was shown that protamine could be expressed from the pachytene stage of meiosis; however, the protein is expressed only at the spermiogenesis stage [37].
In some cases, the expression (RNA) and the translation (protein–stained by IF) are not in parallel. When protein was detected but RNA did not, this could be related to regulation of RNA expression or its stability. However, when RNA was detected but protein was not, this could be related to translational regulation. In addition, either protein levels and/or RNA expression could be related to the stage of cell development [37].
Since we started our human experiments with a very small biopsy, which contained a very small number of SPGCs, we were unable to make additional examinations to confirm protein/expression results such as the ploidy of the cells.
The in vitro culture system we used in this study to induce spermatogenesis was composed of MCS (a 3D system that mimics the in vivo conditions of the STs). In addition to the 3D conditions, different growth factors (GDNF, LIF, FGF), StemPro media, and KSR were present in the MCS. These factors induced proliferation of mouse and human SPGCs [21,22,30]. In addition, Sertoli cells that produce functional factors, which are involved in induction proliferation and differentiation of SPGCs were present in the culture. Also, peritubular and LCs that may support the microenvironment of SPGC development in vivo were cultured in our system. Due to the fact that we are limited in the number cells developed in the human culture, we were unable to perform additional IF staining or additional analyses to confirm the presence of Sertoli cells and factors produced by these cells, and in addition, to confirm ploidy, DNA quality and epigenetics of the developed cells. We suggest that the 3D culture conditions (provided by MCS) and growth factors, in addition to the somatic cells present in the biopsies that remain in the culture, provide a microenvironment that supports and enables the development of spermatogenesis, including, in one case, the generation of sperm-like cells. The conditions of the in vitro culture are not yet optimized, and the type and quality of cells from the biopsies may also vary from one case to another. Furthermore, the reasons for not observing all the postmeiotic markers examined in the same culture could be related to different stability of these markers in vitro, or the possibility that different markers of the postmeiotic stage are not expressed in the same time point and, thus, the number of cells expressing one marker could possibly not be in another marker. Since the in vitro culture of human SPGCs from PCPBs is new in our system (MCS), we preferred to examine the development of these cells from fresh biopsies rather than from frozen/thawed material. Once we establish a fresh successful culture system, we will, of course, move on to frozen/thawed material. Using animal models, it was shown that cryopreserved SPGCs and/or isolated SPGCs from cryopreserved testicular tissue are still actively able to develop spermatogenesis in vivo [38]. Future studies should be performed to compare the efficiency of proliferation and differentiation of germ cells and the activity and viability of the supporting cells from cryopreserved and fresh biopsies in MCS. It is valuable to validate the efficiency of MCS for possible future use in the clinic.
Our results are preliminary and need to be optimized, but they could be considered as a proof of concept for the probability of induction of human SPGCs from prepubertal cancer patients to develop almost last postmeiotic stages under specific in vitro conditions.
These results may encourage future therapeutic strategies using novel technologies (such as in vitro maturation or others) that may induce SPGCs to generate round spermatids and/or sperm. Recently, it was shown that injection of human round spermatids to oocytes led to development of embryos and even to birth of newborns [39]. Our results still require further enhancement to induce the development of more meiotic and postmeiotic cells, including the generation of normal and fertile sperm. Our results show the presence of biologically active SSCs in testicular biopsies of chemotherapy-treated patients. Therefore, it is important to suggest that these survival cells may also develop in vivo and recover spermatogenesis in the cured patient after puberty, and the generated sperm could be used by assisted reproductive techniques to fertilize oocytes. It is important to emphasize the possible DNA damage and apoptotic triggered by the chemotherapy of SSCs as well as the supporting somatic cells in the testis. Therefore, it is recommended to cryopreserve testicular tissue from PCPBs before chemotherapy/radiotherapy and cryopreservation of testicular biopsy after chemotherapy/radiotherapy is an alternative approach for PCPBs who have undergone cancer treatment without collection of testicular biopsy for fertility preservation. Thus, it is crucial to examine the epigenetic and DNA content of the cells before and after in vitro culture. Should this system be further validated and improved for the production of fertilization competent gametes, then it will circumvent the problem of fertility preservation of prepubertal cancer male patients who are receiving aggressive chemotherapy and/or radiation and still have some SSCs in their testes. This therapeutic approach will prevent the risk of reintroducing cancer cells into survivors by autotransplanting testicular tissue/cell technologies. Furthermore, this technology may assist nonobstructive azoospermic patients in whom no sperm has been found in their testicular biopsies.
Our results encourage the approach of cryopreserving testicular biopsies from prepubertal cancer patients that still contain biologically active SPGCs to be used in future developed fertility therapeutic strategies (in vitro or in vivo).
Footnotes
Acknowledgments
Our thanks to the staffs of IVF and to the staff of Pediatric Oncology and Hematology Department of Pediatrics, Soroka Medical Center, Beer-Sheva, Israel and to the staffs of Pediatric Oncology Units at: Sheba Medical Center Ramat Gan, Tel Aviv Sourasky Medical Center, Tel Aviv; Rambam Health Care Campus—Hospital, Haifa; and Rabin Medical Center, Schneider Hospital, Petach Tikva, Israel for referring patients to the Soroka Medical Center; to Dr. Anat Stein, Male Infertility and Sperm Bank, Schneider Hospital for assisting in cryopreservation of a biopsy; and to Mrs. Caroline Simon for excellent editing.
This study was partially supported by The Kahn Foundation, The Natural Science of China (NSFC) - Israel Science Foundation (ISF) (NSFC-ISF) and The United States-Israel Binational Science Foundation (BSF).
Author Disclosure Statement
No competing financial interests exist.