
Abstract
Mesenchymal stem cells (MSCs) are powerful immunomodulators that regulate the diverse functions of immune cells involved in allogeneic reactions, such as T cells and natural killer (NK) cells, through cell–cell contact or secreted factors. Exosomes secreted by MSCs may be involved in their regulatory functions, providing new therapeutic tools. Here, we showed that fetal liver (FL) MSC-derived exosomes inhibit proliferation, activation, and cytotoxicity of NK cells. Exosomes bearing latency associated peptide (LAP), TGFβ, and thrombospondin 1 (TSP1), a regulatory molecule for TGFβ, induced downstream TGFβ/Smad2/3 signaling in NK cells. The inhibition of TGFβ, using a neutralizing anti-TGFβ antibody, restored NK proliferation, differentiation, and cytotoxicity, demonstrating that FL-MSC-derived exosomes exert their inhibition on NK cell function via TGFβ. These results suggest that FL-MSC-derived exosomes regulate NK cell functions through exosome-associated TGFβ.
Introduction
Mesenchymal stem cells (MSCs) are nonhematopoietic multipotent stem cells that can be isolated from various adult and fetal tissues, such as bone marrow (BM), fat, liver, and umbilical cord [1]. MSCs have been widely tested in clinical trials due to their regenerative and immunomodulatory abilities. They can downregulate functions of immune effector cells (IEC) of both innate and adaptive immunity, including T cells, natural killer (NK) cells, B cells, and macrophages [2]. MSCs act on T cells by inhibiting their proliferation and cytotoxicity and by committing them to become regulatory cells [3]. They also regulate NK-cell proliferation and cytotoxicity independently of the KIR haplotypes presented by NK cells [4,5]. Thus, MSCs are attractive candidates for cellular immunotherapeutics to induced tolerance in allogeneic transplantation and/or autoimmunity. The mechanism of the immunomodulatory activity of MSCs is unclear, but accumulating evidence suggests that MSCs exert their therapeutic effects mainly through the delivery of paracrine factors, including TGFβ, IDO, PGE2, and others [6 –8]. Some of these factors are directly secreted, whereas others are released associated with secreted membranes, such as exosomes, which are important mediators of the paracrine effect of MSCs [9,10].
Exosomes are 30–150-nm bilipid membrane-bound extracellular cargo vesicles, containing protein and RNA, which are actively secreted by diverse cell types. They are involved in intercellular communication through the transfer of proteins and RNA. Exosomes bear common surface markers (eg, tetraspanins such as CD9, CD63, and CD81 and lipid raft-associated proteins including flotillin-1) as well as internal markers such as Alix and Tsg101. They originate from cellular multi-vesicular endosomes (MVE) and are secreted by the cell after MVE fusion with the plasma membrane. Exosomes have a common phenotype as the original cells and, at least in part, exert some of the same functions [11]. Several published studies have reported that MSC-derived exosomes have similar functions as those of MSCs, such as repairing injured tissues, modulating immune responses, and anti-inflammatory effects [12 –14]. Recently, extracellular vesicles released by MSC have been demonstrated to regulate the activation of macrophages through microRNAs (miRNAs) [15].
MSCs are used for therapeutic purposes to regulate the immune system in transplantation, autoimmunity, and regenerative medicine [16 –19]. They can be locally delivered, but are generally injected intravenously, which can be hampered by the intravascular trapping in the lungs due to their size [20]. Therefore, administration of derived products of MSC such as exosomes, which are smaller, should reduce lung accumulation and favor their delivery to the targeted organ. However, exosomes, which are limited membrane fragments derived from multivesicular bodies and released by MSC, might have different functions from parental cells.
Here, we studied the immunomodulatory effect of MSC-derived exosomes, relative to that of MSCs, on the function of T cells and NK cells, which are considered to play a crucial role in allograft rejection. BM-MSCs have a limited effect when maintained in culture [21 –24]. Thus, we used MSCs derived from human fetal liver (FL-MSC), which have been shown to have a much longer in vitro lifespan than BM-MSCs, while exhibiting a similar immunomodulatory potential [24,25]. We show that similar to their parent cells, FL-MSC exosomes (FL-exo) inhibit NK in a TGFβ-dependent manner.
Materials and Methods
Cell culture
Human FLs (6–9 weeks of gestation) were obtained from women after voluntary abortion. Written consent was obtained from the patients in accordance with the Declaration of Helsinki for the use of abortion products for research. Tissue collection and use were performed according to the guidelines and with the approval of the French Biomedicine Agency. FL-MSCs were prepared as previously described [21,25]. Differentiation studies were carried out as previously described [26]. All experiments were performed between passages 5 and 20. Human peripheral blood mononuclear cells (PBMCs) were isolated from the blood of healthy volunteers by density Ficoll-Paque gradient centrifugation. NK cells were isolated from PBMCs by negative selection using the NK Cell Isolation Kit (Miltenyi Biotec), according to the manufacturer's instructions. Freshly purified CD56+CD3− NK cells of at least 95% cell purity, evaluated by flow cytometry using monoclonal antibodies (mAbs) anti-CD3-PEVIO770 and anti-CD56-APC, were cultured in RMPI 1640 medium supplemented with 10% heat-inactivated human AB serum (Sigma), 2 mM
GW4869 (10 μM; Sigma) was used to inhibit exosome production by FL-MSC.
Purification and characterization of FL-exo
FL-MSCs, at 80% confluence, were washed twice with phosphate-buffered saline (PBS) and then incubated with exosome-depleted FBS α-MEM complete medium. Exosome-depleted FBS α-MEM complete medium was obtained by 2 h of centrifugation at 100,000g at 4°C of FBS. The number of exosomes calculated with nanosight was reduced by 99%. FL-exo were isolated from the supernatant of FL-MSC cultures by differential ultracentrifugation and characterized as previously described [27]. FL-exo were quantified by using the micro BCA™ Protein Assay Kit (Thermo Fisher Scientific). Purified exosomes were analyzed by transmission electron microscopy to confirm particle form and size. Their size was also determined by Nanoparticle Tracking Analysis with Nanosight. FL-exo purity was confirmed by western blotting to assess the presence of the membrane-marker tetraspanins (anti-CD9, anti-CD63, and anti-CD81, kindly provided by Dr. Rubinstein; Ascites) and TSG101 (R&D Systems), as previously described [27], and the absence of intracellular markers such as cytochrome C for mitochondria, calnexin for the endoplasmic reticulum, and syntaxin 6 for Golgi apparatus.
Immunophenotyping
Flow cytometry analyses of FL-MSC surface markers were performed as previously described [25,28]. The surface phenotype of FL-MSC was assessed by using specific FITC, PE, or APC-conjugated mAbs, including CD73, CD14, CD45, HLA-ABC (BD Pharmigen), CD29, CD90, HLA-DR (ImMiexmunotools), CD105, CD34, and CD31 (Miltenyi Biotec). For flow cytometry analysis of FL-exo, purified exosomes were adsorbed to 4-μm Aldehyde/Sulfate latex beads (Life Technologies). Briefly, 5 μg FL-exo were mixed with 10 μL latex beads for 15 min at room temperature (RT). Then, 1 mL PBS was added to each sample and they were incubated on a rotating wheel for 2 h. Then, 110 μL 1 M glycine was added to the samples and they were mixed at RT for 30 min. Bead-bound exosomes were collected by centrifugation for 3 min at 5,000g. The beads were washed three times with PBS/0.5% bovine serum albumin (BSA) and resuspended in 500 μL PBS/0.5% BSA. Finally, 10 μL of bead-bound exosomes was stained with specific mAbs against tetraspanins, TGFβ, thrombospondin 1 (TSP1; R&D Systems), and other MSC markers.
Proliferation assays
To assess the immunomodulatory capabilities of FL-MSCs and FL-exo on NK cells, CD3− CD56+ cells were cultured for 7 days with mitomycin- (Sigma-Aldrich) treated FL-MSCs (25 μg/mL, 37°C, 30 min) at IEC/MSC ratios from 1:1 to 16:1 and their proliferation was analyzed by flow cytometry. Cells were stained with 5,6-carboxyfluorecein diacetate succinimidyl ester (CFSE; Molecular Probes, Eugene, OR), as previously described [25,28]. In some cases, NK cells at a concentration of 0.5 × 106 mL were stimulated with 200 U/mL Il-2 (R&D Systems) for 7 days. Activated IECs were also cultured with FL-exo (at various concentrations from 50 to 400 μg/mL). Activated NK cells alone were used as controls. Surface markers and proliferation were evaluated by flow cytometry. The following mAbs were used to evaluate IEC surface markers: anti-CD3-PEVIO770, anti-CD16-PE, and anti-CD56-APC.
Cytotoxicity assays of NK cells
Cytotoxicity assays were performed by using a flow cytometry-based method [29]. IL-2 stimulated NK cells from different donors were cultured with/without FL-MSC/FL-exo, for 4 days in 96-well flat-bottom plates. The K562 cell line was used as the target. Target cells were cultured in complete RPMI medium with human AB serum and labeled with 1 μM VPD450 (BD Biosciences) for 10 min, at 37°C on day 4 to discriminate them from NK cells. NK cells were incubated with VPD450-labeled K562 target cells at a ratio of 1:2 IEC/T in 96-well flat-bottom plates. K562 cells were used alone as a negative control. After 4 h of coculture at 37°C in 5% CO2, the cell mixtures were washed and stained with 5 μL 7-AAD (BD Biosciences) for 15 min in the dark before analysis; CD107a degranulation and various activation receptors were also analyzed by flow cytometry using the following mAbs: anti-CD107a-FITC (BD Biosciences), anti-NKG2D-PE, and anti-NKp30-PEVIO770 (Miltenyi Biotec). The involvement of TGFβ in the immunomodulation of NK cells by FL-MSCs and FL-exo was analyzed by culturing the cells with 50 μg/mL anti-TGFβ neutralizing antibody (R&D Systems) under the same conditions.
Flow cytometry
Flow cytometric analysis of cell surface markers was performed as previously described [25,28]. Immunophenotyping was performed on a BD Accuri™ C6 flow cytometer. The in vitro functional analysis of IEC was performed on an LSR Fortessa instrument by using FACSDiva software (BD Biosciences).
Quantitative real-time reverse transcriptase polymerase chain reaction for NK-activating receptors
Messenger RNA (mRNA) was extracted from NK cells after 2 days in culture, with or without FL-exo, by using the mRNA Catcher Plus Purification Kit (Thermo Fisher Scientific), and complementary DNA (cDNA) synthesis was performed directly in the mRNA Catcher PLUS plate by using the RevertAid H Minus First-Strand cDNA Synthesis Kit (Thermo Fisher Scientific), according to the manufacturer's instructions. The cDNA was then stored at −20°C until it was used for polymerase chain reaction (PCR) analysis. Amplification of cDNA was monitored by using a QuantiNova SYBR Green PCR kit (Qiagen) on an Mx3005P quantitative real-time RT-PCR (qPCR) System (Agilent Technologies). Pre-designed specific primers for NKG2D (FW: 5′-ACA-GCA-AAG-AGG-ACC-AGG-ATT-3′; RV: 5′-GGT-TGG-GTG-AGA-GAA-TGG-AG-3′), NKp30 (FW: 5′-CAC-TTG-CTT-CTT-CCC-GTT-TC-3′; RV: 5′-GAT-GTT-CTT-TCT-CCA-CCA-CCA-3′), and GAPDH (FW: 5′-AATCCCATCACCATCTTCCA-3′; 5′-TGGACTCCACGACGTACTCA-3′) were used for mRNA quantification for each sample (Eurogentec). Reactions were performed in a total volume of 20 μL, which included 5 μL cDNA sample, 2 μL primers (10 μM), and 10 μL 2 × SYBR Green PCR Master Mix. The PCR was performed as follows: 2 min at 95°C and 45 cycles of 5 s at 95°C, 10 s at 60°C, and one final cycle of 1 min at 95°C, and 30 s at 55°C. The amount of each receptor mRNA relative to GAPDH mRNA was calculated by using the comparative CT method.
Immunoprecipitation
FL-exo were lysed with 1 × TNT buffer at pH 7.4 containing 1 mM Orthovanadate, 0.01 mM NaPO4, 2 mM NAF, 0.03 mM NEM, 12.4 nM β-glycerophosphate, and 1 × protease inhibitor cocktail. After 30 min at 4°C, protein lysates were collected by centrifugation at 2,300g for 30 s. The supernatant was incubated with beads (Protein G sepharose fast flow; Sigma; 20 μL beads/100 μL protein lysate) for 1 h at 4°C, on a rotating wheel, to eliminate non-specific binding, followed by centrifugation for 5 min at 2,300g. Supernatants were incubated with 3 μg anti-TSP1 or 2 μg anti-latency associated peptide (LAP) overnight at 4°C on a rotating wheel. Then, 20 μL of beads was added to the supernatants and the mixture was incubated for 2 h at 4°C. After centrifugation, immune-complexed beads were collected and the supernatant was analyzed by western blot (lysate after immunoprecipitation). Beads were washed three times with TNT buffer and resuspended in 20 μL Laemmli buffer, with or without β-mercaptoethanol, and boiled for 5 min. Supernatants containing the precipitated proteins were subjected to gel electrophoresis and transferred onto PVC membranes for western blot analyses. All antibodies were obtained from R&D Systems.
Intracellular Phospho-Smad2/3 assay
Stimulated NK cells were treated with or without 400 μg/mL FL-exo or 10 ng/mL human TGFβ (Miltenyi Biotec) for 30 min in 96-well flat-bottom plates. Intracellular phosphoprotein staining with anti-Phospho-Smad2/3 (BD Pharmigen) was performed as previously described [30]. The samples were then analyzed by flow cytometry. Untreated samples were used as negative controls, and samples were treated with TGFβ as positive controls.
Immunofluorescence
Stimulated NK cells were treated with or without 400 μg/mL FL-exo or 10 ng/mL human TGFβ for 30 min in 96-well flat-bottom plates. Cells were collected, washed with PBS, and fixed with 4% PFA for 15 min at RT. Then, the cells were washed and permeabilized with 0.05% Triton X-100, and they were incubated for 30 min at RT with 10% PBS/FBS/100 mM NH4CL to block non-specific binding. Cells were stained with anti-Smad2/3 (Abcam) for 1 h, followed by Alexa Fluor 488-conjugated anti-rabbit IgG for another hour, and Hoechst for nuclear staining. Cells were then resuspended in 100 μL 10% PBS/FBS, deposited on poly-
Statistical analysis
GraphPad Prism software (Graph-Pad) was used for statistical analysis with the Wilcoxon test to compare treated and untreated cells. Data are expressed as the mean ± standard error of the mean. All experiments were performed at least three times. Differences were considered to be statistically significant for *P < 0.05, **P < 0.01, and ***P < 0.0001.
Results
Characterization of human FL-MSC and FL-exo
After 14–18 days in culture in standard medium, isolated FL-MSCs were positive for the specific MSC markers CD73, CD105, CD90, CD29, and HLA class I (HLA-ABC). They were negative for the hematopoietic markers CD45, CD34, CD31, CD14, and HLA class II (HLA-DR) (Fig. 1A). Moreover, we demonstrated the multipotent ability of FL-MSC by differentiating them into osteogenic, adipogenic, and chondrogenic lineages, resulting in high extracellular deposition of calcium, accumulation of intracellular lipid droplets, and active synthesis of proteoglycans, respectively (Fig. 1B).
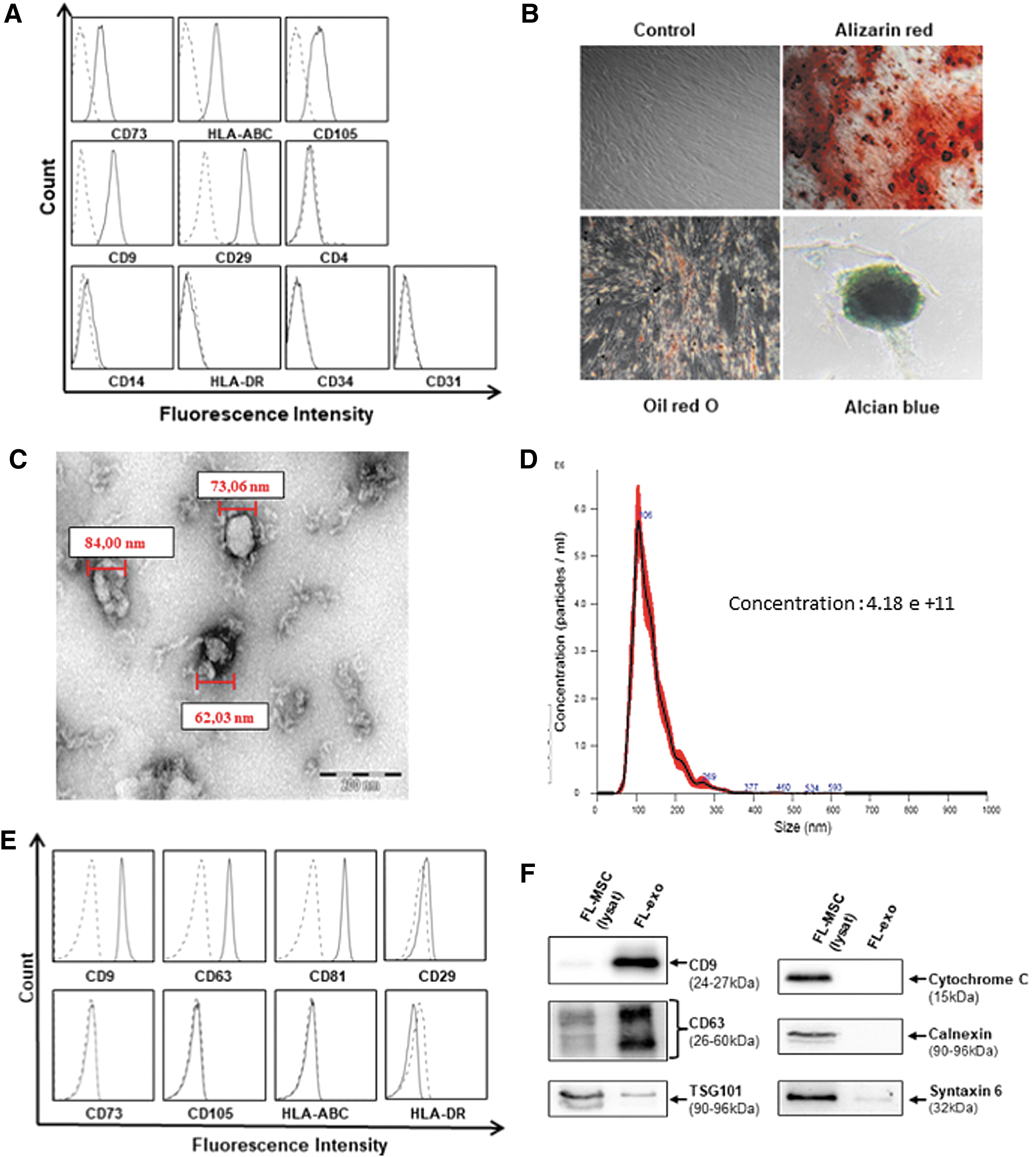
Characterization of FL-MSC and FL-exo.
FL-exo were isolated by sequential centrifugation. Electron microscopy analysis of vesicles showed them to be between 60 and 150 nm in diameter (Fig. 1C), corresponding to the normal size of exosomes. When determined by Nanosight, their mean size was 101.5 ± 11 nm (four independent preparations of FL-exo) (Fig. 1D). Immunophenotyping of exosomes showed the expression of tetraspanins (CD9, CD63, CD81), which are enriched on exosomal membranes. The exosomes also expressed the mesenchymal marker CD29, but not other markers, such as CD73 and CD105. They also did not express HLA class I or II (Fig. 1E) or markers specific to intracellular compartments, such as cytochrome C for mitochondria, calnexin for the endoplasmic reticulum, and syntaxin 6 for Golgi apparatus (Fig. 1F).
The immunosuppressive effect of FL-exo on IEC proliferation
We tested the effect of FL-MSCs and FL-exo on NK-cell proliferation. IL-2 activated NK cells were labeled with CFSE and cocultured for 7 days, with or without FL-MSCs or FL-exo, at various MSC/NK ratios (from 1:1 to 1:16) or various concentrations of FL-exo (from 50 to 400 μg/mL). Both FL-MSC and FL-exo prevented the proliferation of activated NK cells, as assessed by the CFSE dilution assay (Fig. 2A), in a ratio- and dose-dependent manner, respectively (Fig. 2B).

FL-exo have immunosuppressive effects on NK cells.
To evaluate the role played by exosomes in the regulation of NK-cell proliferation, MSC have been incubated with GW4869 (10 μM), an inhibitor of exosome formation. The use of CW4869 partially restores the proliferation of IL2 activated NK cells in the presence of FL-MSC, suggesting that part of the regulatory effect of MSC is dependent on the release of exosomes (Fig. 2C).
FL-exo prevent IL-2-induced NK-cell activation and inhibit NK-cell cytotoxicity
We investigated the role of FL-exo in NK-cell activation and cytotoxicity relative to that of FL-MSCs. The cytotoxicity of activated NK cells against K562 cells strongly depends on the expression of the activator receptors NKG2D and NKp30. NKG2D and NKp30 were strongly downmodulated when NK cells were treated with FL-MSCs or FL-exo (Fig. 3A, B). This was also observed at the mRNA level when activated NK cells were treated with exosomes, as assessed by RT-qPCR (Fig. 3C).
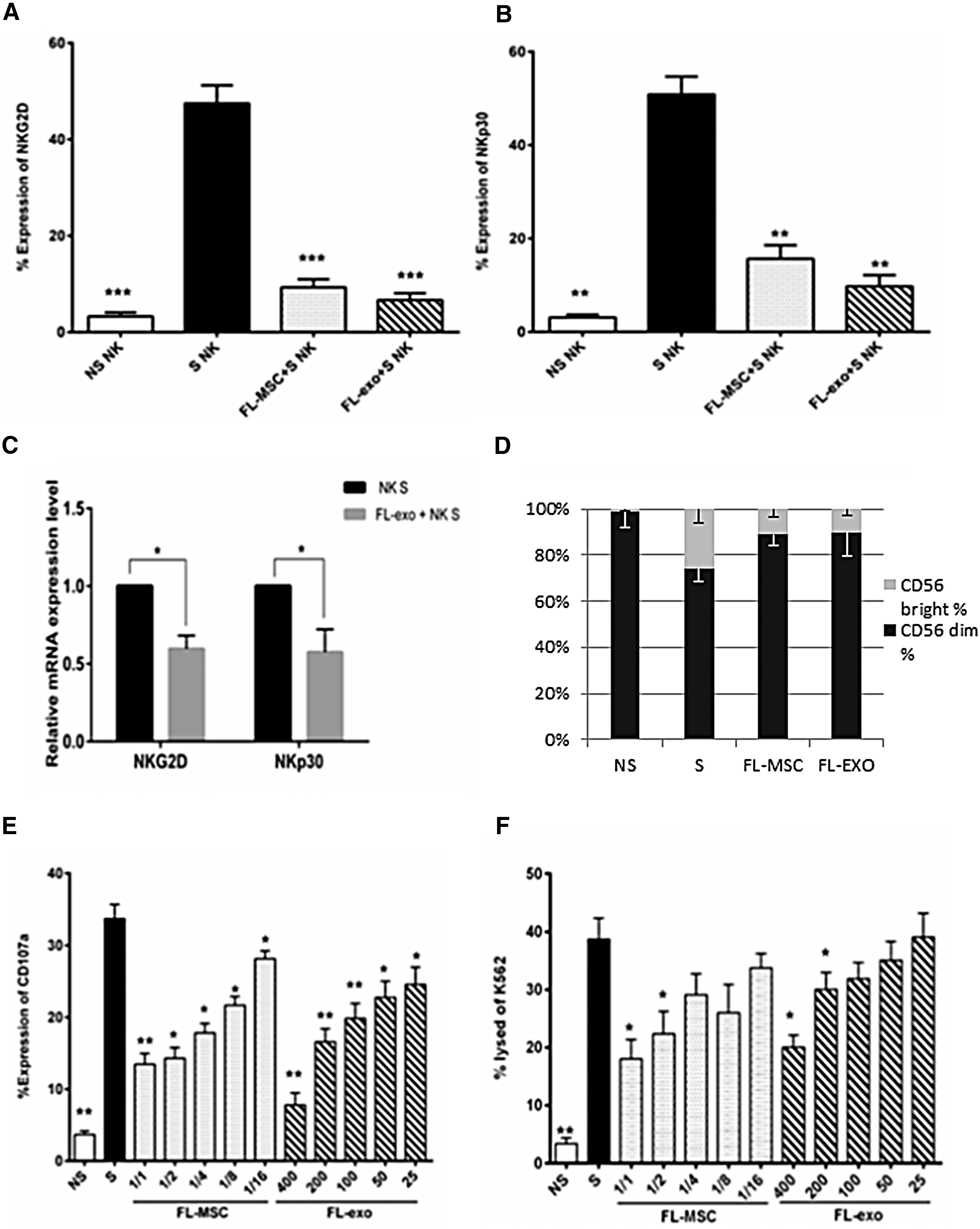
FL-exo alter NK-cells phenotype and cytotoxic function. Freshly purified CD3−/CD56+ NK cells (5 × 104/well) cultured under basal conditions, or stimulated with IL-2 (10 ng/mL), were incubated with FL-MSCs or FL-exo. NK cells (n = 8) were cultured with FL-MSCs (NK:FL-MSC ratio 1:2) or FL-exo (400 μg/mL) for 4 days. NK cells were collected, and surface expression of the activating receptors NKG2D
We also analyzed the phenotype of NK cells, especially their expression of CD56, since CD56-dim are associated with cytotoxic activities and CD56-bright NK cells are associated with the secretion of cytokines. The proliferation of both NK cells (CD56-dim and CD56-bright) is reduced in the presence of Fl-MSC or FL-exo. When we analyzed the repartition of these 2 populations, the stimulation by IL2 increased the CD56-bright fraction which is significantly reduced in the presence of FL-MSC or FL-exo (Fig. 3D). Altogether, these results suggested that both MSC and their exosomes impaired the activation, proliferation, and differentiation of NK cells.
We evaluated the cytotoxic degranulation capacity of NK cells by flow cytometry through the expression of CD107a (lysosomal-associated membrane protein-1, LAMP-1) in the presence of target cells. We did not detect CD107a expression in resting NK cells after incubation for 4 h with VPD450-labeled K562 target cells. However, CD107a was strongly expressed on the cell surface of activated NK cells, but it was lower when activated NK cells were cocultured with FL-MSCs or FL-exo, in a ratio- and concentration-dependent manner, respectively (Fig. 3E). We analyzed the cytotoxic effect of NK cells on K562 target cells by flow cytometry, by determining the incorporation of 7AAD into VPD450-labeled targeted cells. Resting NK cells exhibited minimal cytotoxicity against K562 cells, whereas IL-2-activated NK cells showed increased 7AAD staining of target cells, which was also efficiently impaired by FL-MSCs or FL-exo, in a ratio- and dose-dependent manner, respectively. (Fig. 3F).
These data indicate that FL-exo are able to inhibit NK-cell activation and cytotoxic functions in vitro.
Exosomes express TGFβ on their surface and induce the activation of the TGFβ/Smad pathway in NK cells
Several soluble factors have been reported to be involved in MSC-mediated inhibition of NK cells, such as PGE2, IDO, and TGFβ. We performed a proteome analysis by mass spectrometry to identify factors that could be associated with the inhibitory effects of exosomes. This analysis showed the presence of TSP1 (Supplementary Data and Supplementary Figure S1; Supplementary Data are available online at
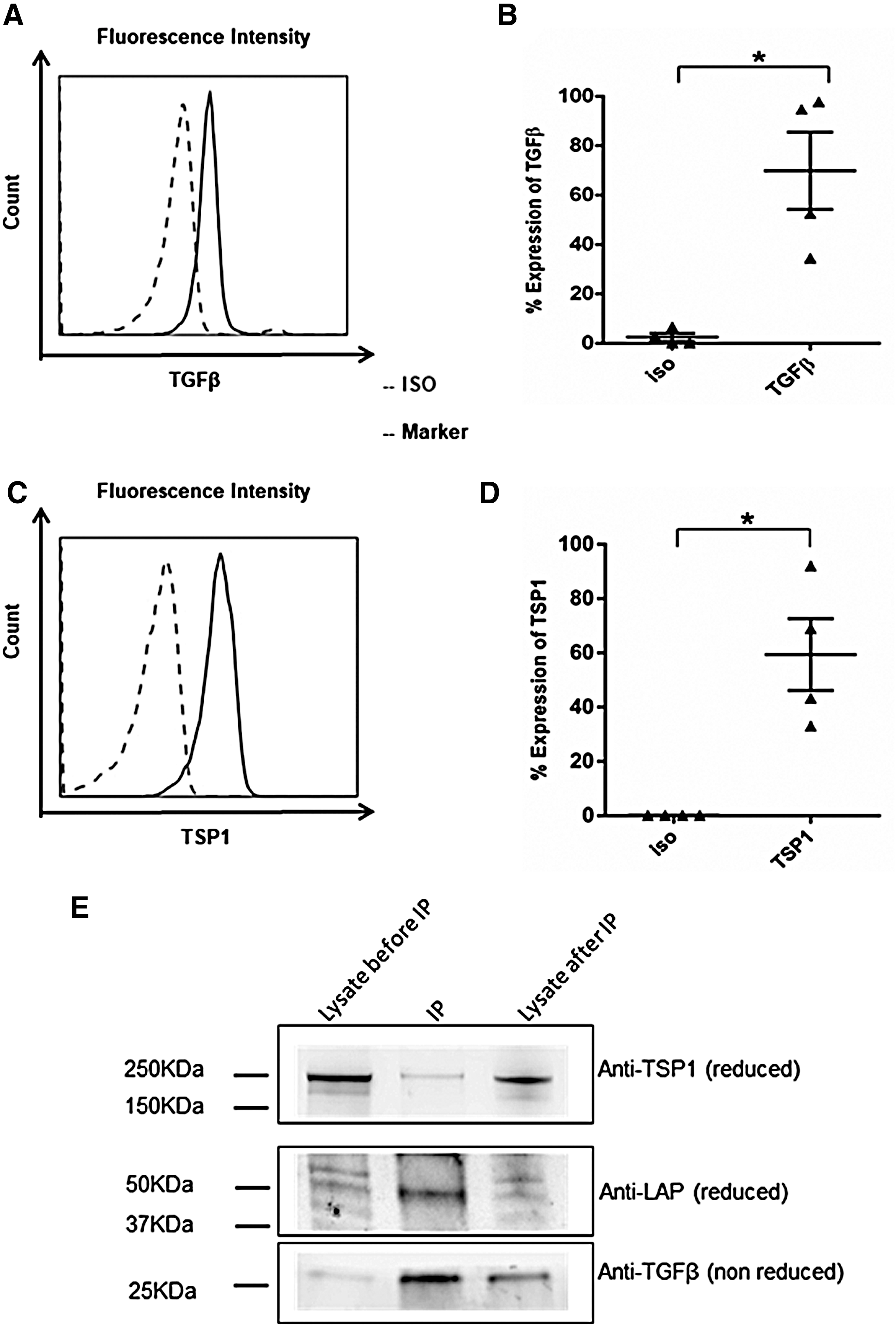
FL-exo associated TGFβ and TSP1 as molecular regulators. Surface expression of TGFβ1
We next determined whether the inhibitory effect of exosomes on NK-cell function is dependent on the surface expression of TGFβ. We investigated whether exosomes induce the TGFβ/Smad pathways on NK cells. IL-2-activated NK cells do not exhibit phosphorylation of Smad2/3, as assessed by flow cytometry. We observed Smad2/3 phosphorylation after 30 min of TGFβ treatment. Treatment with FL-exo also induced Smad2/3 phosphorylation in NK cells (Fig. 5A). We investigated the sub-cellular localization of Smad2/3 in IL-2-activated NK cells, in the presence or absence of TGFβ or FL-exo, by immunofluorescence. Almost all Smad2/3 staining remained localized in the cytoplasm in untreated NK cells. TGFβ treatment of IL-2-activated NK cells for 30 min induced partial translocation of Smad2/3 into the nucleus. Similarly, we observed complete nuclear translocation of Smad2/3 in activated NK cells after treatment with FL-exo for 30 min (Fig. 5B). We obtained similar results in four independent experiments.
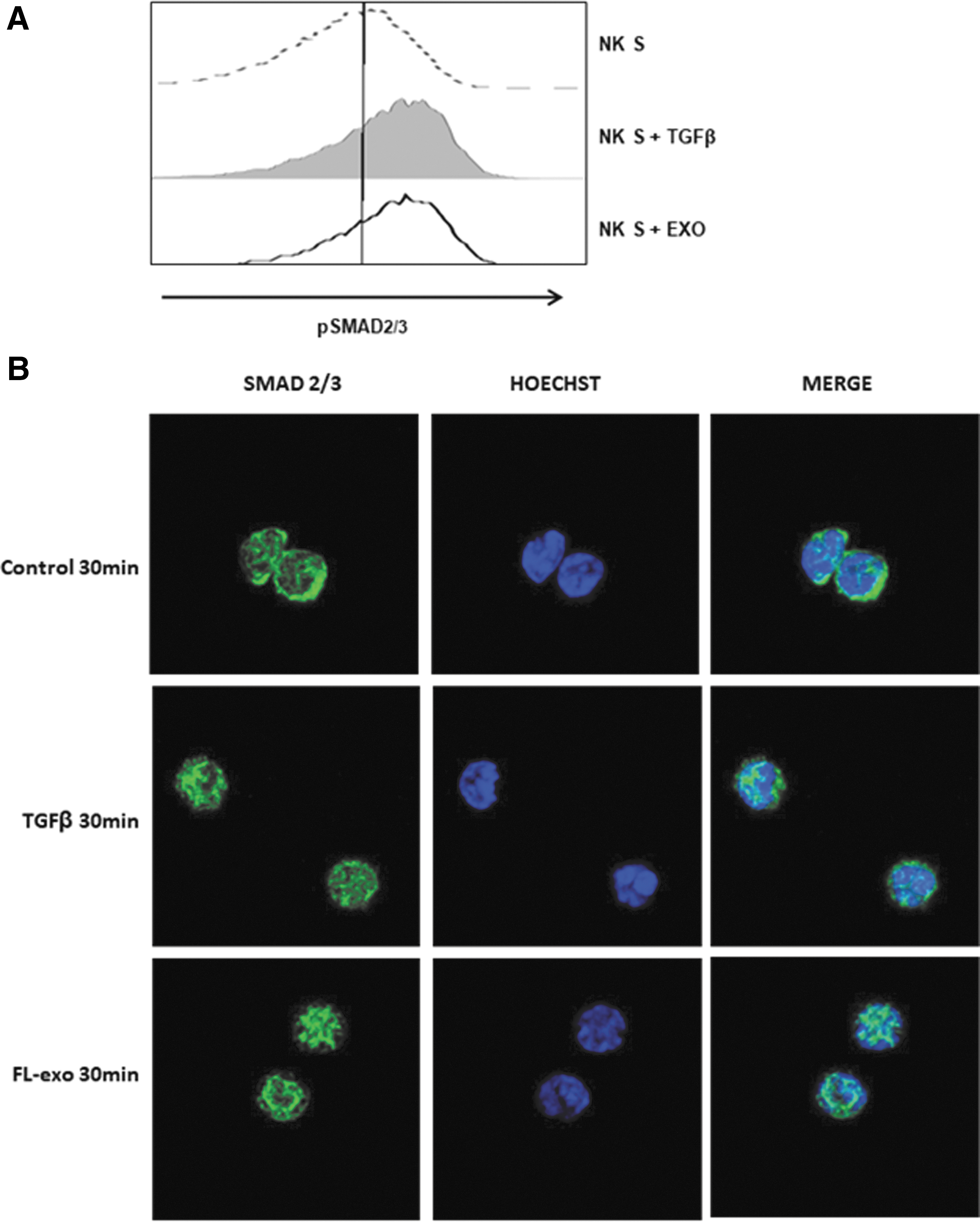
The TGFβ1/Smad pathways suppress NK-cell effectors functions. Freshly purified CD3−/CD56+ NK cells (5 × 104/well, n = 3) were stimulated with IL-2 (10 ng/mL) and treated with FL-exo (400 μg/mL) or TGFβ1 (10 ng/mL) for 30 min.
Reduced NK cell function is at least, in part, induced by exosomal TGFβ
We further investigated whether the inhibitory effect of exosomes is mediated by TGFβ by performing a functional assay using an anti-TGFβ neutralizing antibody (50 μg/mL). IL-2-activated NK cells were cultured for 4 days, in the presence or absence of FL-exo, and a TGFβ1-neutralizing antibody. We then assessed NK activation and cytotoxicity by flow cytometry. The expression of the NK-activating receptors NKG2D and NKp30 by activated NK cells was significantly restored by TGFβ1-neutralizing antibodies (Fig. 6A, B). We next analyzed the TGFβ1-neutralizing antibody-mediated inhibition of the effect of FL-exo by CD107a expression on NK cells and 7-AAD uptake by K562 target cells. TGFβ1-neutralizing antibodies restored the degranulation and cytolytic ability of activated NK cells inhibited by FL-exo (Fig. 6C, D).
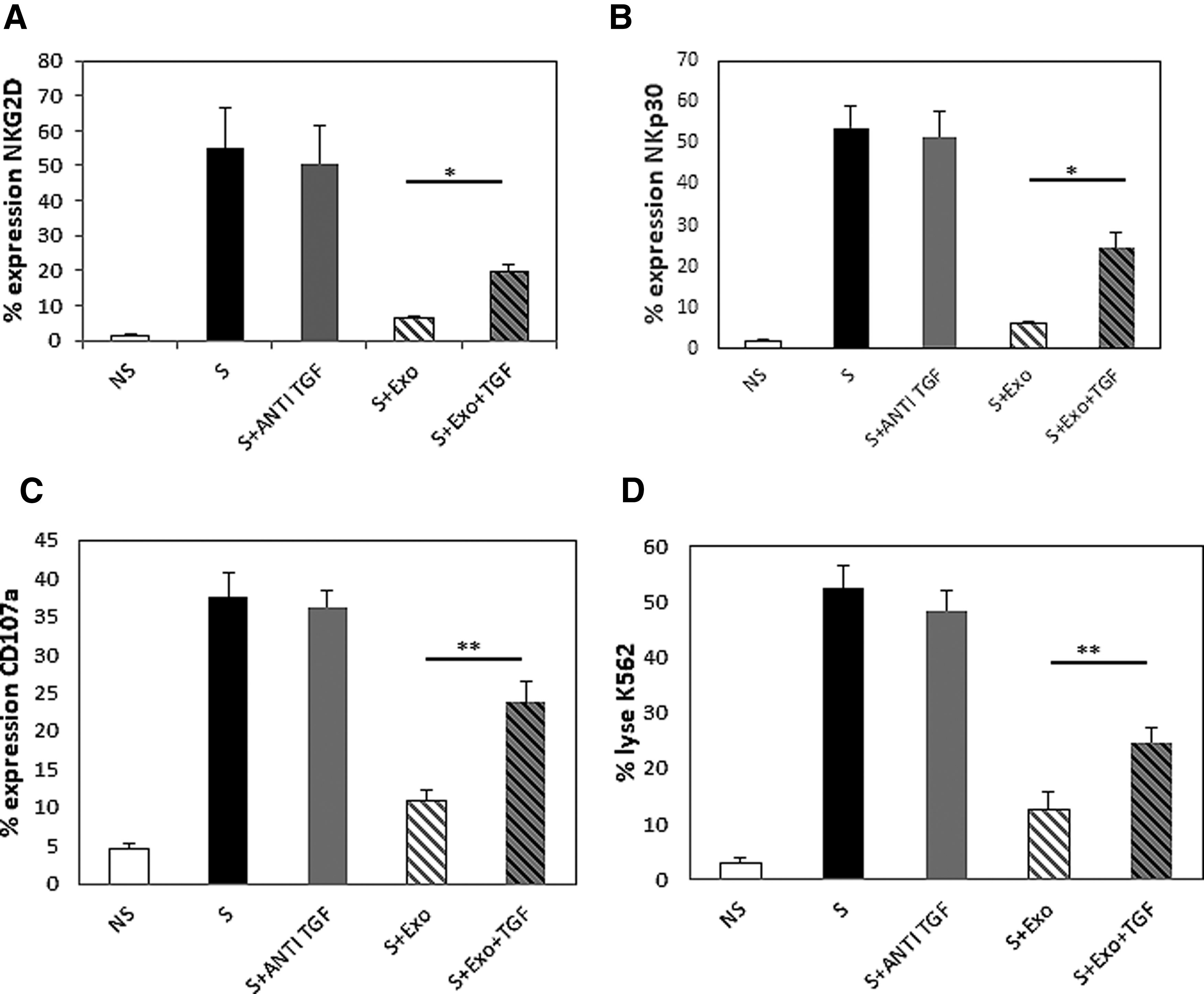
Down-regulation of NK activation is driven by FL-exo associated TGFβ1. Freshly purified CD3−/CD56+ NK cells (n = 4; 5 × 104/well) were cultured under basal condition or after stimulation with IL-2 (10 ng/mL) and treated with FL-exo (400 μg/mL) or TGFβ1, with or without anti-TGFβ1 (50 μg/mL) for 4 days. Surface expression of NKG2D
Our results suggest that the inhibitory effect of FL-exo and FL-MSC on NK cells is, at least, partially dependent on their TGFβ expression.
Discussion
We previously reported that FL-MSCs support long-term in vitro expansion better than BM-MSCs, while maintaining long-lasting immunoregulatory properties. These features make FL-MSCs an attractive therapeutic tool. We also reported that MSCs exert their immunoregulatory function through various secreted factors [28].
Here, we demonstrated that coculture with FL-exo and FL-MSCs inhibits purified IL-2-activated NK-cell functions. NK cells are responsive to IL-2-induced differentiation and activation. In vitro stimulation of peripheral blood NK cells by IL-2 alone induces their proliferation, in particular the CD56 bright subset [32]. Here, we demonstrated that exosomes inhibit NK proliferation. They also inhibited the proliferation and/or differentiation of the CD56 bright and dim NK subset. NK cells act in the first line of defense in immunity, and their ability to lyse target cells does not depend on prior sensitization. We show that the transcription of NK-activating receptors is inhibited by MSC-derived exosomes. NKG2D is an NK-cell-activating receptor that plays a major role in NK-mediated cytotoxicity by recognizing various antigens on stressed or infected cells and tumor cells [33]. The importance of NKG2D has been demonstrated in organ transplantation in animal models, as well as in human transplantation, magnifying innate and adaptive immune responses [34]. Depletion or inhibition of the expression of NK-activating receptors could account for the FL-exo-mediated inhibition of NK cell function [35]. Here, we showed that decreased expression of two NK-activating receptors, indeed, correlates with decreased cytotoxicity of NK cells against target cells.
The modification of IL-2-activated NK-cell cytotoxicity by MSCs has been previously reported by our group [25]. In our coculture system, FL-exo have a similar inhibitory profile as FL-MSCs, despite the fact that MSCs are also able to inhibit the cells and associated functions of both innate and adaptive immunity [2]. Our results are in accordance with other studies reporting variable effects of MSC-derived extracellular vesicles on various IECs in vitro [36]. Discrepancies may be due to the source of MSCs and different experimental protocols, suggesting that the origin of the cells, the type of soluble particles produced, and the isolation method could lead to varying effects on target cells [37 –39]. In our study, the analysis of the preparation of exosomes by electron microscopy and immunoblot indicated that the exosomes were pure with negligible contamination by microvesicles.
NK-cell function is controlled by various mechanisms. TGFβ is an important regulatory molecule that impairs NK-cell expansion, cytotoxicity, and the expression of activating receptors [40,41]. It is an important secreted factor involved in MSC immunomodulation [6]. TGFβ has been broadly reported in tumor invasion and metastases because of their immunosuppressive effect [42]. We showed that inhibitory functions of FL-exo on NK cells are, at least, partially mediated by TGFβ, using an anti-TGFβ blocking antibody. In addition, inhibition of NK cells by TGFβ occurs via the Smad-dependent pathway [43]. In our system, exosome/NK-cell coculture showed activation of the Smad signaling pathway, which mimicked the results of TGFβ treatment of IL-2-stimulated NK cells. Moreover, the addition of anti-TGFβ restored the effector functions of NK cells, as well as NKG2D and NKp30 expression. These data suggest that FL-exo contain biologically active molecules, notably TGFβ, which impair NK-cell activation and function.
We observed the inhibitory functions of exosomes on NK cells despite extensive washing of the membranes during exosome preparation, suggesting that TGFβ is carried on the surface of exosomes. TGFβ is secreted by various cell types as an inactive precursor, called latent TGFβ, which can be activated by TSP1 through a proteolytic process [31]. Mokarizadeh et al. used flow cytometry to demonstrate that murine BM-MSC-derived microvesicles carry membrane-bound TGFβ [44]. We show that FL-exo carry TSP1, LAP, and TGFβ on their surface. Moreover, LAP and TSP1 can be coprecipitated, suggesting release of the active form of TGFβ, as previously demonstrated [31]. Other molecules have also been associated with the inhibitory function of MSC. MSC-secreted IDO, which exerts a major inhibitory effect on T cell and NK-cell proliferation, is strongly induced by inflammatory cytokines, such as INFγ. In our study, IDO was entirely absent from exosomes isolated from the media of resting or primed MSCs (data not show). However, other molecules such as miRNA can participate in the inhibitory effects of MSC [45]. These different molecules could be involved depending on the micro-environment during the inflammatory phase or immune response in vivo.
It has been proposed that the immunomodulatory activity of MSCs is mediated by the synergism of several secreted factors or through contact-dependent mechanisms, corresponding to membrane-bound factors [46]. We showed that the exosomes in our system did not regulate T cell proliferation, whereas parental MSCs did. This result suggests that exosomal expression of TGFβ in our system is not sufficient for T cell inhibition, suggesting that other mechanisms involved in T cell regulation are not worn by FL-exo.
In conclusion, our studies provide evidence for an immunoregulatory function of FL-exo. The control of the cytotoxic activity of NK cells may be explained by biologically active tolerogenic factors carried by FL-MSC-derived exosomes. FL-exo show great potential to become a new immunosuppressive tool.
Footnotes
Acknowledgments
This work was supported by the Groupe Collaboratif en Transplantation d'Ile de France (GCIF) and the Institut Francilien de Recherche en Néphrologie et Transplantation (IFRNT).
Author Disclosure Statement
The authors have nothing to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.