
Abstract
The crucial trace element zinc stimulates osteogenesis in vitro and in vivo. However, the pathways mediating these effects remain poorly understood. This study aimed to investigate the effects of zinc on osteoblast differentiation in human bone marrow-derived mesenchymal stem cells (hBMSCs) and to identify the molecular mechanisms of these effects. In hBMSCs, zinc exposure resulted in a dose-dependent increase in osteogenesis and increased mRNA and protein levels of the master transcriptional factor RUNX2. Analyzing the upstream signaling pathways of RUNX2, we found that protein kinase A (PKA) signaling inhibition blocked zinc-induced osteogenic effects. Zinc exposure increased transcriptional activity and protein levels of phospho-CREB and enhanced translocation of phospho-CREB into the nucleus. These effects were reversed by H-89, a potent inhibitor of PKA. Moreover, zinc exposure led to dose-dependent increases in levels of intracellular cyclic adenosine monophosphate (cAMP). These findings indicate that zinc activates the PKA signaling pathway by triggering an increase in intracellular cAMP, leading to enhanced osteogenic differentiation in hBMSCs. Our results suggest that zinc exerts osteogenic effects in hBMSCs by activation of RUNX2 via the cAMP-PKA-CREB signaling pathway. Zinc supplementation may offer a promise as a potential pharmaceutical therapy for osteoporosis and other bone loss conditions.
Introduction
O
Although zinc is a trace element, it is a constituent of numerous enzymes and proteins that perform multiple biological functions necessary for maintaining health. With respect to skeletal biology, zinc plays crucial roles in bone metabolism [8] and normal growth processes [9]. Several studies have reported that dietary deficiency of zinc disrupts normal bone growth and maturation in humans, as well as in animal models [10,11]. In addition, zinc has been found to increase alkaline phosphatase (ALP) activity [12] and collagen synthesis during bone formation [13]. In clinical studies, zinc intake has been found to positively correlate with bone mineral density in premenopausal women [14] and to negatively correlate with bone loss in postmenopausal women [15]. Taken together, these findings suggest that zinc likely plays an important role in the signaling pathways of osteogenesis.
In another recent study, the zinc transporter protein ZIP13 was shown to be involved in the BMP/TGF-β signaling pathway during connective tissue development, a process which includes osteoblast maturation [16]. Another zinc transporter, ZIP14, has been proposed to control G-protein-coupled receptor-mediated signaling, which is required for mouse systemic growth [17]. Although previous studies have reported the existence of a relationship between zinc and osteogenesis, the molecular mechanisms accounting for this relationship have remained to be elucidated.
In this study, we aimed to characterize the effects of zinc exposure on osteoblast differentiation in human bone marrow-derived mesenchymal stem cells (hBMSCs). We focused on investigating the mechanistic role of zinc with respect to RUNX2, a master transcriptional factor in osteogenesis. Our hypothesis was that zinc exposure might increase RUNX2 levels via the cAMP-PKA-CREB signaling pathway, thereby inducing enhanced osteoblast differentiation and stimulating osteogenesis.
Materials and Methods
Cell culture and reagents
Human bone marrow aspirates from the posterior iliac crest were obtained from seven adult donors (four males and three females; age range: 50–60 years old). From these bone marrow samples, hBMSCs were isolated based on cell ability to adhere to a plastic culture flask. Following selection, cells were maintained in low-glucose Dulbecco's modified Eagle medium (DMEM-LG) (Gibco; Grand Island, NY) supplemented with 10% fetal bovine serum (FBS) (Gibco) and 1% antibiotic–antimycotic solution (Carlsbad, CA) at 37°C in a 5% CO2 atmosphere. hBMSCs were grown to 70%–80% confluency and then harvested by incubation with 0.05% trypsin/EDTA (Invitrogen, Carlsbad, CA) followed by centrifugation at 1,300 revolutions per minute (rpm) for 3 min. Cells obtained through this process were then replated at a density of 5,000 cells/cm2 and subcultured when they were 80%–90% confluent. hBMSC characteristics were confirmed for this protocol in our previous studies [18 –20]. To determine the optimal concentration of ZnSO4 (Sigma; St. Louis, MO) for enhancing osteogenic differentiation in hBMSCs in vitro, cultured cells were assigned to one of the following study groups: control group or one of five experimental groups (ZnSO4 treatment groups: 10, 20, 50, 100, and 200 μM). AKT1/2 kinase inhibitor (A6730; Sigma), H-89 (Sigma), was used at a concentration of 0.1 μM, and DKK1 (R&D Systems, Minneapolis, MN) was used at a concentration of 0.1 μg/mL in hBMSCs. This study protocol was approved by the Institutional Review Board (IRB) of the Yonsei University College of Medicine (IRB No. 4-2017-0232) and informed consent was obtained from all patients. All methods were performed according to the relevant guidelines and regulations of the institution.
In vitro osteogenesis and adipogenesis
The medium used for stimulating in vitro osteogenesis by hBMSCs has been described previously [21]. In brief, hBMSCs were seeded at 4 × 104 cells/well in 24-well plates or 1 × 104 cells/well in 96-well plates. To induce osteogenic differentiation, hBMSCs were maintained for 5–10 days in OM [DMEM-LG containing 10% FBS, 1% antibiotic–antimycotic solution, 10 mM β-glycerophosphate (Sigma), 100 nM dexamethasone (Sigma), and 50 μg/mL ascorbic acid (Gibco)]. To adipogenic differentiation, hBMSCs were maintained for 7–21 days in adipogenic medium (AM) (DMEM-LG containing 10% FBS, 1% antibiotic–antimycotic solution, 0.5 mM isobutyl-methylxanthine [Sigma], 1 μM dexamethasone [Sigma], 200 μM indomethacin [Sigma], and 5 μM insulin [Sigma]). For each experimental group, ZnSO4-supplemented medium was replaced every 2 days during this differentiation period.
Cell viability assay
Cell viability was evaluated using an EZ-Cytox Cell Viability Assay Kit (Daeil Lab Service, Seoul, Korea) according to the manufacturer's instructions. For this assay, hBMSCs were seeded in 12-well culture plates at a density of 1 × 104 cells/well. Cells were then maintained in DMEM-LG for 5 days, and medium was replaced once per day during the assay period. As detailed in the kit instructions, at the end of the assay period, after cells were washed with phosphate-buffered saline (PBS), 10 μL of EZ-Cytox (tetrazolium salts) solution was added to each well, and these were incubated at 37°C for 3 h (h). After incubation, conditioned media were transferred to 96-well plates. Absorbance was then measured at 410 nm. All samples were tested in triplicate.
Colony-forming units-fibroblast assay
Human bone marrow mononuclear cells (hBMMs) were seeded at 5 × 105 in 100-mm culture dishes and maintained in DMEM-LG supplemented with 20% FBS for 12 days. ZnSO4-treated media was replaced every 2 days during fibroblast colony formation for ZnSO4-treated hBMMs. Subsequently, cells were fixed in a 1:1 acetone: methanol fixative, stained with 20% crystal violet solution (Merck; Darmstadt, Germany) for 30 min in darkness, and washed in distilled water (DW). Human bone marrow fibroblast colony-forming ability of the stained cells was then evaluated and counted in triplicate.
Flow cytometry
hBMSCs were cultured with 100 μM ZnSO4 for 3 days and harvested with 0.02% EDTA and washed twice in fluorescence-activated cell sorting (FACS) buffer (PBS containing 1% FBS and 0.05% sodium azide). The hBMSCs (2 × 105) were stained with CD73-PE (Miltenyi Biotec, Auburn CA), PE-mouse-IgG2a isotype control (Miltenyi Biotec), CD90-FITC (Miltenyi Biotec), FITC-mouse IgG2a isotype control (Miltenyi Biotec), CD105-APC (Miltenyi Biotec), CD146-APC (Biolegend, San Diego, CA), and APC-mouse IgG1 isotype control (Miltenyi Biotec) for 1 h at 4°C. Thereafter, the stained cells were washed with PBS, centrifuged at 300g for 5 min, and then resuspended in 500 μL of ice-cold FACS buffer. The samples were subjected to FACS analysis using a FACS Verse™ flow cytometer (BD Biosciences). With an unstained sample (3 × 105) as the negative control, the stained cells were analyzed using FlowJo software (FlowJo LLC, Ashland, OR).
ALP/Alizarin red S staining and quantification
For ALP staining, after fixation in a 2:3 citrate buffer:acetone fixative, hBMSCs treated with test concentrations of ZnSO4 were stained for ALP using an alkaline staining solution mixed with fast blue RR salt (Sigma) in naphthol AS-MX phosphate alkaline solution (Sigma) for 30 min in darkness. After washing in DW, cells were stained with Mayer's hematoxylin solution (Sigma) for 5 min and then rinsed in tap water. ALP activity was normalized to Alamar blue. For alizarin red S staining, after cells were fixed in ice-cold 70% ethanol, freshly prepared 3% alizarin red S solution (weight/volume [wt/vol]) (Sigma) was added. Then samples were incubated for 30 min. For quantification of alizarin red S, absorbance was detected at 595 nm, following destaining with 10% cetylpyridinium chloride monohydrate (Sigma) for 10–20 min.
Oil red O staining for adipogenic differentiation
hBMSCs were rinsed with PBS, fixed in 10% neutral-buffered formalin buffer (Sigma) for 30 min, washed by DW. Then, cells were stained with and 0.18% oil red O solution (sigma) for 1 h, followed by repeated washing with DW. For quantitative analysis of stained cells, absorbance was detected at 500 nm after destaining with 100% isopropanol for 30 min.
Quantitative real-time polymerase chain reaction
Quantitative real-time polymerase chain reaction (qPCR) analysis was performed as previously described [19]. In brief, total RNA was isolated using a RNeasy kit (Qiagen; Valencia, CA) according to the manufacturer's instructions. One microgram of total RNA was reverse-transcribed using an Omniscript Kit (Qiagen). The primer sets (Bioneer; Seoul, Korea) were as follows: GAPDH (P267613), PPAR-γ (P102359), RUNX2 (P229954), ALPL (P324388), COL1A1 (P157768), Osteonectin (P279949), Bone sialoprotein (P188040), and PKA (P312739). There were no validated primers for Osteopontin and Osteocalcin. Thus, the primers for these were designed as follows: Osteopontin, 5′-CCGTTGCCCAGGACCTGAA-3′ (sense) and 5′-TG TGGCTGTGGGTTTCAGCA-3′ (antisense); Osteocalcin, 5′-AGAGCCCCAGTCCCCTACC C-3′ (sense) and 5′-AGGCCTCCTGAAAGCCGATG-3′ (antisense). Mean cycle threshold values from triplicate (n = 3) measurements were used to calculate gene expression, with normalization to GAPDH as an internal control.
Western blot
For total protein extraction, hBMSCs were lysed in passive lysis buffer (Promega; Madison, WI). Protein concentrations were determined with a Bio-Rad protein assay (Bio-Rad Laboratories, Inc.; Hercules, CA), and 10–30 μg of protein was analyzed by 10% sodium-dodecyl sulfate-polyacrylamide gel electrophoresis (Sigma). Transferred membranes were blocked with 5% skim milk (BD; Sparks, MD) or 5% bovine serum albumin (BSA) (Sigma), and incubated for 12 h with antibodies against RUNX2 (Millipore; San Diego, CA), PKA (Santa Cruz Biotechnology; Santa Cruz, CA), p-CREB (Cell Signaling; Danvers, MA), CREB (Cell Signaling), active β-CATENIN (Cell Signaling), β-CATENIN (Abcam; Cambridge, United Kingdom), p-ERK1/2 (Abcam), ERK1/2 (Abcam), p-SMAD2 (Cell Signaling), SMAD2 (Abcam), p-SMAD 1/5/8 (Abcam), SMAD 1/5/8 (Abcam), p-AKT (Cell Signaling), AKT (Cell Signaling), p-GSK-3β (Santa Cruz Biotechnology), GSK-3β (Santa Cruz Biotechnology), PPAR-γ (Santa Cruz Biotechnology), FABP4 (Santa Cruz Biotechnology), Lamin B (Santa Cruz Biotechnology), and lactate dehydrogenase (Santa Cruz Biotechnology). Membranes were further probed with an antibody against HSP90 (Santa Cruz Biotechnology) and β-ACTIN (Santa Cruz Biotechnology), which served as a loading control.
Nuclear and cytosolic fractionation
hBMSCs were collected by trypsinization and washed with PBS three times before nuclear and cytosolic fractionation. Nuclear and cytoplasmic fractionation was performed using the NE-PER Nuclear and Cytoplasmic Extraction Reagents kit (Thermo Fisher Scientific; Rockford, IL) according to the manufacturer's instructions. Each separated protein was analyzed by western blot analysis.
Lentiviral small hairpin RNA transduction
Methods for lentiviral small hairpin RNA (shRNA) transduction and infection were previously described [22]. In brief, 3 × 106 HEK-293FT cells per 100-mm dish were transfected with lentiviral shPKA vectors (TRCN0000356094 or TRCN0000233527, Sigma) using Lipofectamine 2000 (Invitrogen). After 6 h, HEK-293FT cells were sustained in DMEM-LG supplemented with 10% FBS for 2 days, and then the supernatant fractions were collected. For lentivirus infection, 5 × 104 cells per well were seeded on six-well plates and then exposed to virus-containing supernatants. Lentivirus-infected cells were selected using puromycin (10 mg/mL) (Sigma) 72 h later.
Immunocytochemistry
hBMSCs were seeded at 1,000 cells/cm2 onto four-well glass chamber slides (Nalge Nunc International; Rochester, NY), and the cells were incubated in a 5% CO2 incubator at 37°C. After an overnight incubation, the cells were washed with PBS, followed by fixation with 4% paraformaldehyde (Sigma) for 30 min. Permeabilization was performed with 0.25% Triton X-100 in PBS for 10 min, followed by blocking for 1 h with 3% BSA in PBS. The cells were incubated with 1:100 dilution of primary antibodies against CREB (Cell Signaling) overnight at 4°C. After washing three times with PBS, the cells were incubated with fluorescein isothiocyanate (FITC)-conjugated secondary antibody in a 1:5,000 dilution in 3% BSA-containing PBS for 1 h at room temperature in darkness. The nuclei were stained with 4,6-diamidino-phenyindole (Sigma) and then examined using a confocal microscope (LSM700, Carl Zeiss MicroImaging GHBH; Jena, Germany).
Luciferase reporter assay
hBMSCs were seeded at 1 × 105 cells per well in six-well plates, and cells were transfected with 1 mg of either CREB (SwitchGear Genomics; Menlo Park, CA), AKT (SwitchGear Genomics), TOP-flash, or FOP-flash reporter vectors, with 1 ng of pRL-SV40 as control using Lipofectamine 2000. After 24 h, the cells were maintained for 3 days in OM with ZnSO4 or H-89. The cells were analyzed using a simple dual-luciferase assay (Promega).
Measurement of intracellular cyclic adenosine monophosphate
Intracellular cyclic adenosine monophosphate (cAMP) was measured with the direct cAMP ELISA kit (Enzo Life Sciences; Plymouth Meeting, PA). In brief, cell culture supernatants were loaded into plates, treated with antibody, and incubated at room temperature for 2 h on a microplate shaker. Then, plates were read with a spectrophotometric microplate reader at 405 nm, and cAMP levels were analyzed according to the manufacturer's protocol.
Phosphodiesterase activity assay
Phosphodiesterase (PDE) activity was measured by PDE activity assay kit (Colorimetric) (Abcam; ab139460). In brief, 1 × 106 cells were homogenized in DW and disrupted by sonication. The supernatant was desalted by gel filtration, and then PDE activity was measured according to manufacturer's protocols. IBMX (10 μM), which is a PDE inhibitor, was treated as negative control.
Statistical analyses
Data are presented as mean ± standard deviation. We performed the Shapiro–Wilk normality test for checking normal distributions of the groups. If normality tests passed, two-tailed, unpaired Student's t-test, and if normality tests failed, Mann–Whitney tests were used for the comparisons between two groups. For more than two groups, we used one-way ANOVA if normality tests passed, followed by Tukey's multiple comparison test for all pairs of groups. The GraphPad PRISM version 6.0 was used for data management and statistical analyses. Values of *P < 0.05 or **P < 0.01 were considered statistically significant.
Results
Zinc promotes osteoblast differentiation in hBMSCs
To determine whether zinc is cytotoxic to hBMSCs, we examined the viability of the cells. hBMSCs treated with up to 100 μM ZnSO4 were viable for 5 days (Fig. 1A). We then examined the fibroblast colony-forming abilities exposed to a range of ZnSO4 concentrations using hBMMs for 12 days. The results of these colony-forming unit-fibroblast assays revealed that ZnSO4 had no effect on the number of colony-forming cells, compared with cells not treated with ZnSO4 (Fig. 1B). Also, we analyzed the expression of MSC-associated markers using flow cytometry in the absence or presence of ZnSO4. As a result, ZnSO4 treatment did not change expression pattern of MSC-associated markers (Supplementary Fig. S1; Supplementary Data are available online at
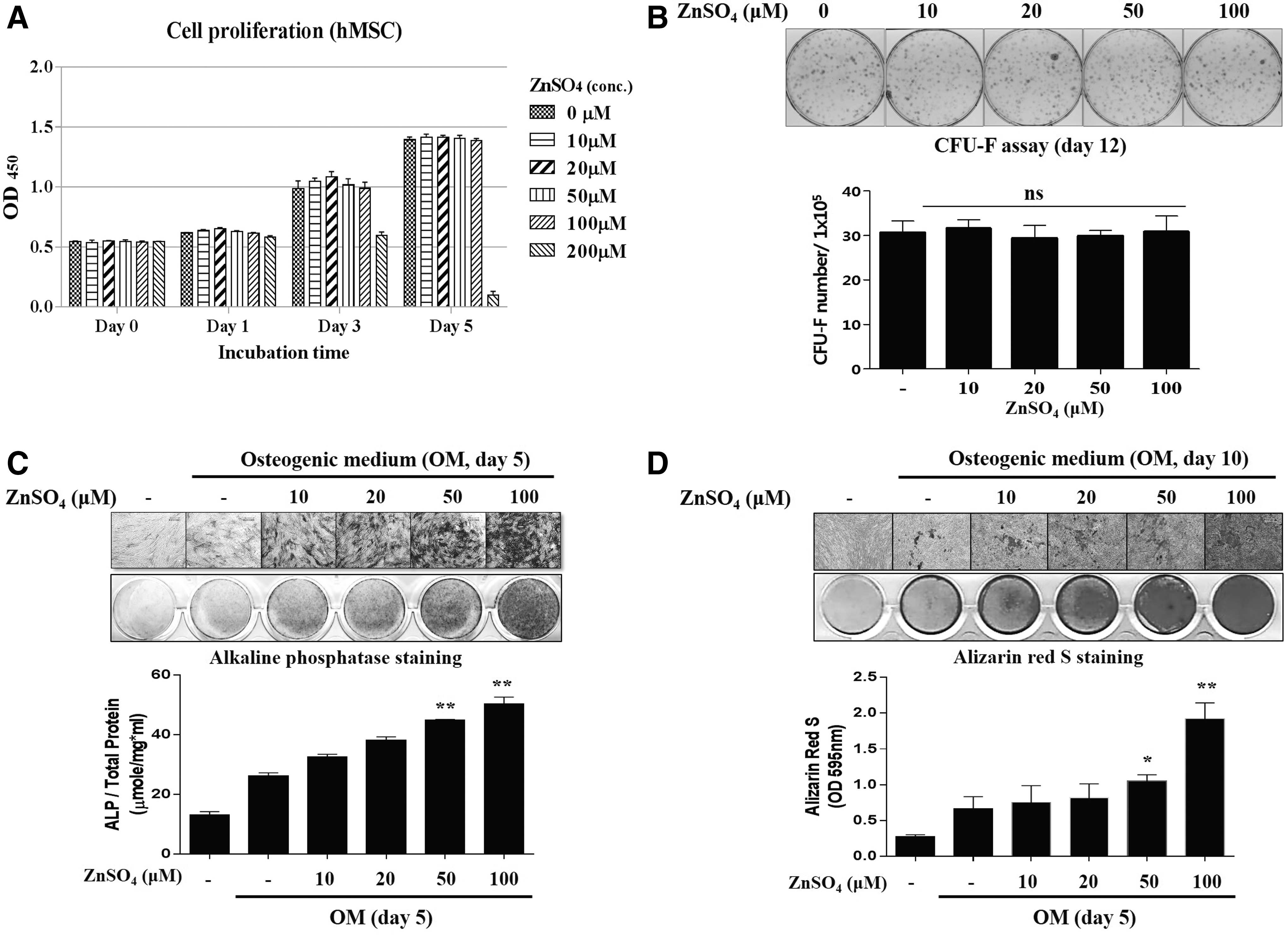
The effects of zinc on proliferation, fibroblast colony-forming ability, and osteogenic differentiation.
Zinc enhances expression of RUNX2 and phosphorylation of PKA during osteoblast differentiation in hBMSCs
In the first set of experiments, we confirmed that 100 μM ZnSO4 enhanced osteogenic differentiation, while ZnSO4 had no effects on proliferation and fibroblast colony-forming ability. Thus, we designated 100 μM ZnSO4 as the maximal concentration that was nontoxic to hBMSCs. To gain insight into the molecular mechanisms mediating the osteogenic effects of zinc, we then analyzed mRNA levels of the master transcriptional factor RUNX2 during osteoblast differentiation in the presence of ZnSO4. During periods of osteogenesis in hBMSCs, mRNA levels of RUNX2 gradually increased until day 3, followed by a decrease thereafter, and expression of RUNX2 was higher in the presence of ZnSO4 than OM without supplemental zinc (Fig. 2A). As expected, the overall protein levels of RUNX2 were enhanced by treatment with ZnSO4 during osteogenesis (Fig. 2B). Among the many genes related to osteoblast differentiation, mRNA levels of RUNX2 and its target genes, such as ALP, COL1a1, OPN, ON, BSP, and OCN, were increased in the presence of zinc (Fig. 2C). It has been reported that RUNX2 expression is regulated by several signaling pathways such as BMP, WNT, TGF-β, PTHrP, IGF, and FGF [23 –27]. To analyze how zinc enhances RUNX2 expression, we screened the downstream targets of each signaling pathway involved in osteogenic differentiation in hBMSCs. ZnSO4 was not found to impact downstream targets of the BMP, TGF-β, or FGF signaling pathways, while zinc exposure did increase phosphorylation of PKA and CREB, well-known to be downstream of the PTHrP pathway during osteogenic differentiation (Supplementary Fig. S3A and Fig. 2B). ZnSO4 also activated AKT and β-CATENIN, downstream targets of the IGF pathway and WNT pathway (Supplementary Fig. S3B). These results indicate that ZnSO4 promotes osteogenic differentiation via RUNX2 upregulation.
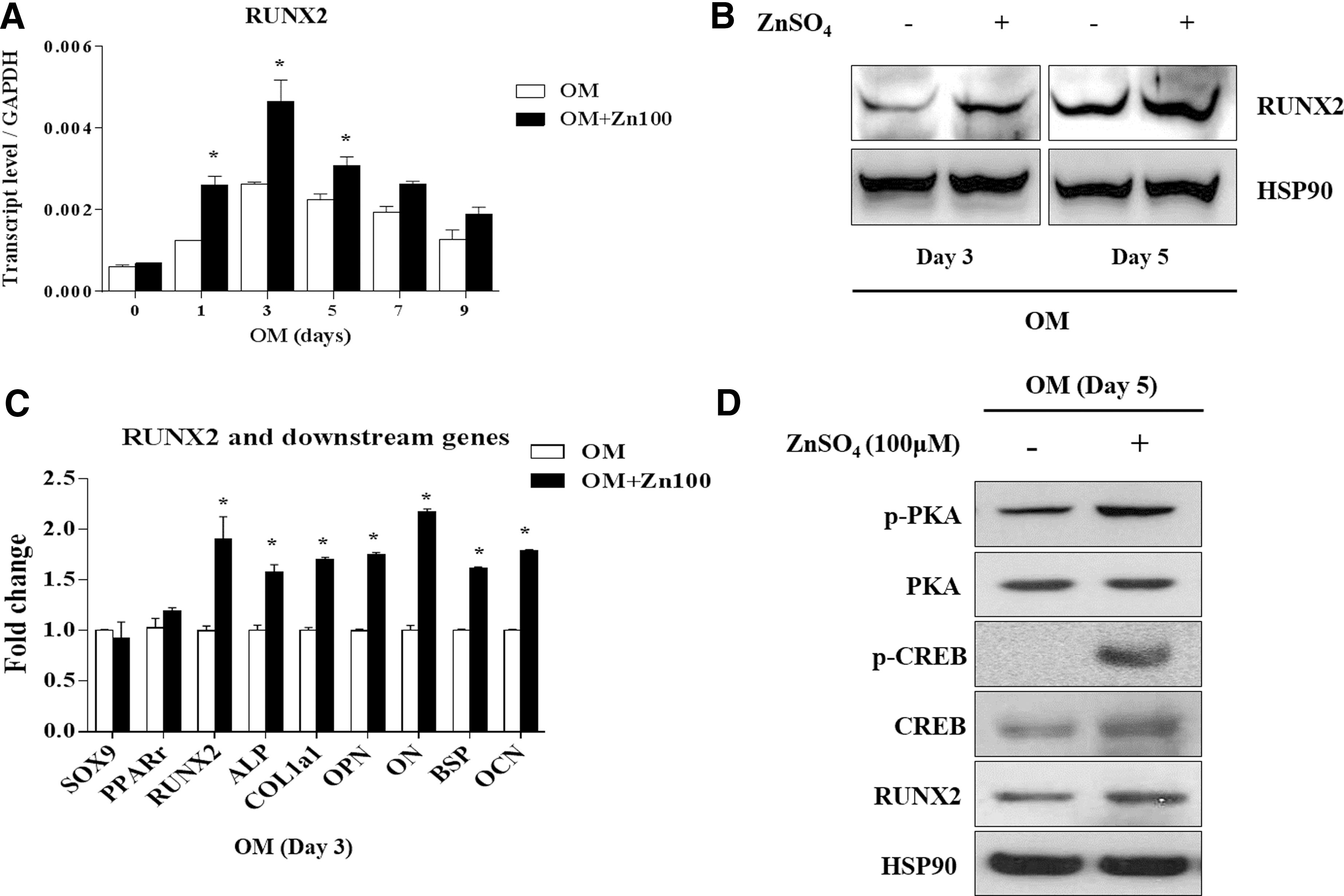
Zinc increases expression of RUNX2 and its target genes through phosphorylation of PKA during osteogenic differentiation of hBMSCs.
PKA inhibition abolishes the osteogenic effect of zinc in hBMSCs
To investigate which signaling pathway could regulate RUNX2 expression induced by ZnSO4, we targeted specific signaling pathways using inhibitors during treatment with varying doses of ZnSO4. First, we selected inhibitor concentrations for each signaling pathway to efficiently suppress osteogenic differentiation. Interestingly, zinc exposure did not reverse the inhibition of early and late osteogenic effects by H-89, an inhibitor of PKA (Fig. 3A, B). However, WNT- and AKT-inhibited osteogenic effects were reversed by zinc treatment in a dose-dependent manner (Supplementary Fig. S4A, B). Thus, we hypothesized that PKA signaling might be the main signaling pathway for zinc-induced osteogenic differentiation in hBMSCs. To test this hypothesis, we infected hBMSCs with shRNA targeting PKA. The efficiency of the shPKA was then confirmed by qPCR and western blot analysis (Fig. 3C). Then, we treated PKA-knockdown hBMSCs with ZnSO4 during osteogenic differentiation for 5–10 days. During osteogenic differentiation of hBMSCs in the presence of zinc, shPKA significantly suppressed ALP activity and mineralization (Fig. 3D, E), suggesting that PKA inhibition abolishes zinc-induced pro-osteogenic effects in hBMSCs.
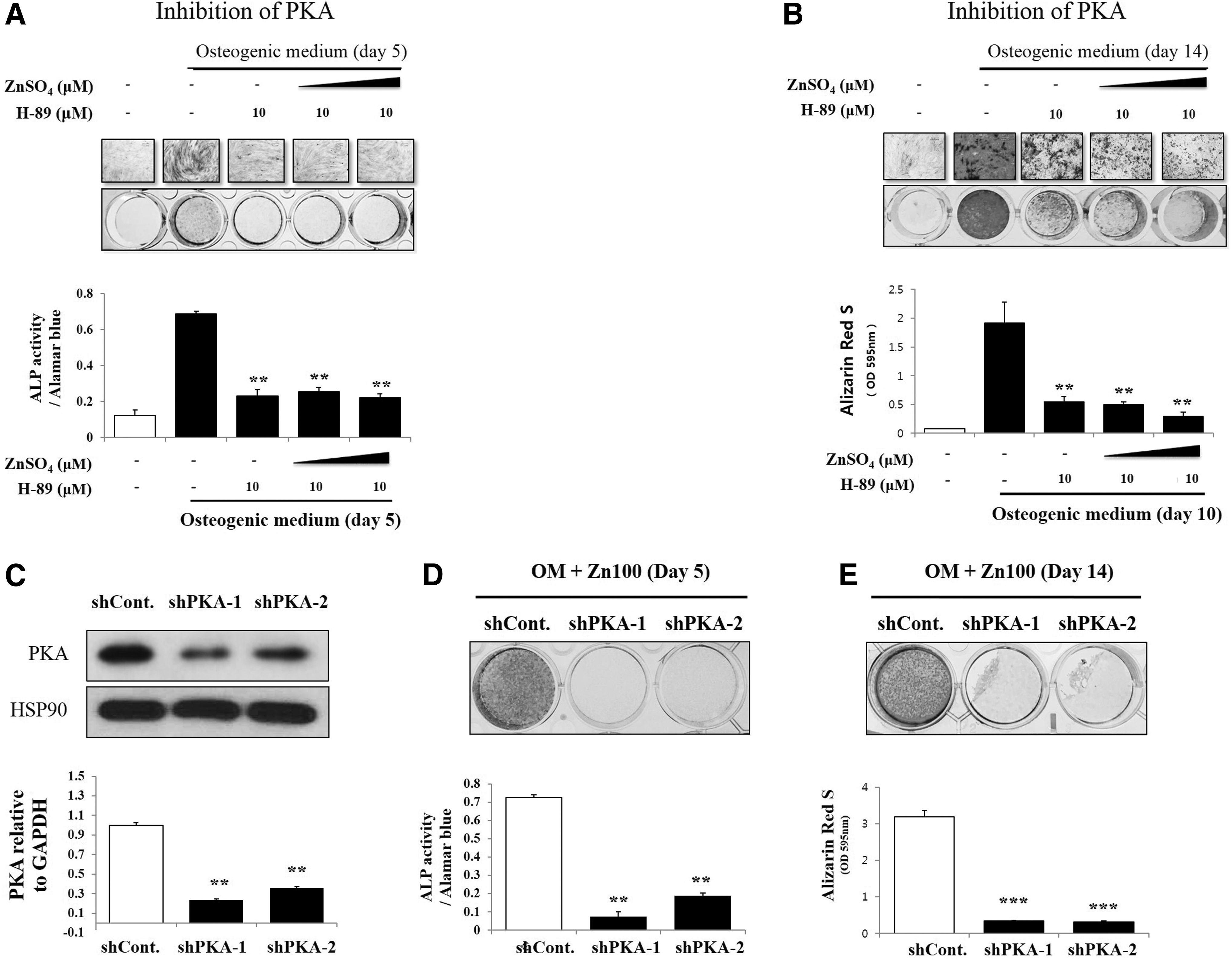
Zinc exposure did not reverse suppression of osteogenesis by PKA inhibition in hBMSCs. hBMSCs (4 × 104 cells/well in 24-well plates) were cultured with OM, and 10–20 μM of ZnSO4, followed by the addition of H-89 (0.1 μM).
PKA inhibitor reverses zinc-mediated activation of PKA downstream genes during the osteogenic differentiation
To confirm whether transcriptional activity and protein level of phospho-CREB are regulated by PKA signaling in the presence of zinc, we treated ZnSO4 and then inhibited PKA signaling using H-89 during osteogenic differentiation for 5 days. As a result, zinc-induced RUNX2 expression and phosphorylation of CREB were suppressed by inhibition of PKA (Fig. 4A). As phosphorylation of CREB is known to lead to nuclear import [28,29], we investigated the cellular localization of phospho-CREB. We observed that PKA inhibition blocked the nuclear translocation of phospho-CREB induced by ZnSO4 during osteogenic differentiation (Fig. 4B, C). Next, we examined whether zinc-mediated cellular localization of CREB could regulate CREB transcription levels. A luciferase reporter assay showed that ZnSO4 increased transcription activity of CREB. In contrast, PKA inhibition significantly repressed its activity (Fig. 4D). Previously, ZnSO4 was also found to activate AKT and β-CATENIN during osteogenic differentiation of hBMSCs (Supplementary Fig. S3B). It has also been reported that PKA can induce phosphorylation of AKT and activate β-CATENIN by phosphorylating GSK-3β [30,31]. PKA inhibition likewise suppressed phosphorylation of AKT and GSK-3β induced by ZnSO4 (Supplementary Fig. S5A). Moreover, while zinc increased the transcription activities of AKT and β-CATENIN, their activities were repressed by PKA inhibition (Supplementary Fig. S5B, C). These results are consistent with previous reports that PKA signaling activates AKT and β-CATENIN. Overall, these results suggested that zinc-induced osteogenesis is mediated by the PKA signaling pathway.
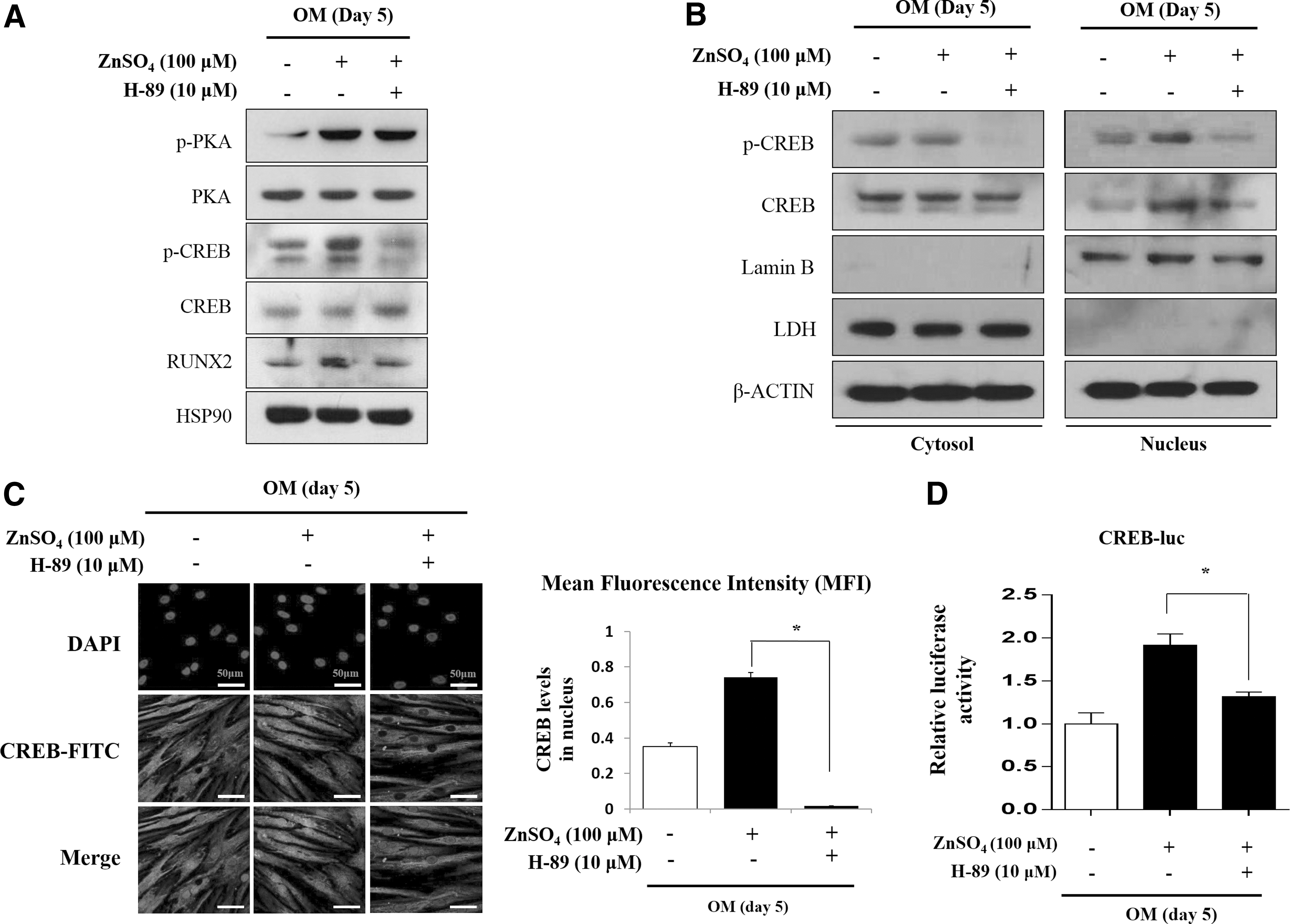
PKA inhibition suppresses zinc-induced activation of PKA target genes during osteogenesis. hBMSCs were incubated with OM with ZnSO4 (100 μM) for 5 days.
Zinc activates PKA signaling pathway via increasing intracellular cAMP
It has been well-established that the activity of PKA depends on cellular level of cAMP [32,33]. Therefore, we hypothesized that zinc regulates the PKA signaling pathway by increasing cellular cAMP levels during osteogenic differentiation in hBMSCs. To test this hypothesis, we measured the levels of intracellular cAMP activity in hBMSCs treated with ZnSO4. Zinc increased intracellular cAMP levels in a dose-dependent manner (Fig. 5A). However, the activities of PDEs, which is known to hydrolyze cAMP into inactive 5′-nucleotide monophosphates [34], showed no significant change during treatments with varying doses of ZnSO4 (Fig. 5B). These findings indicated that zinc activates the PKA signaling pathway by increasing intracellular cAMP levels with PDE-independent manner (Fig. 5C).
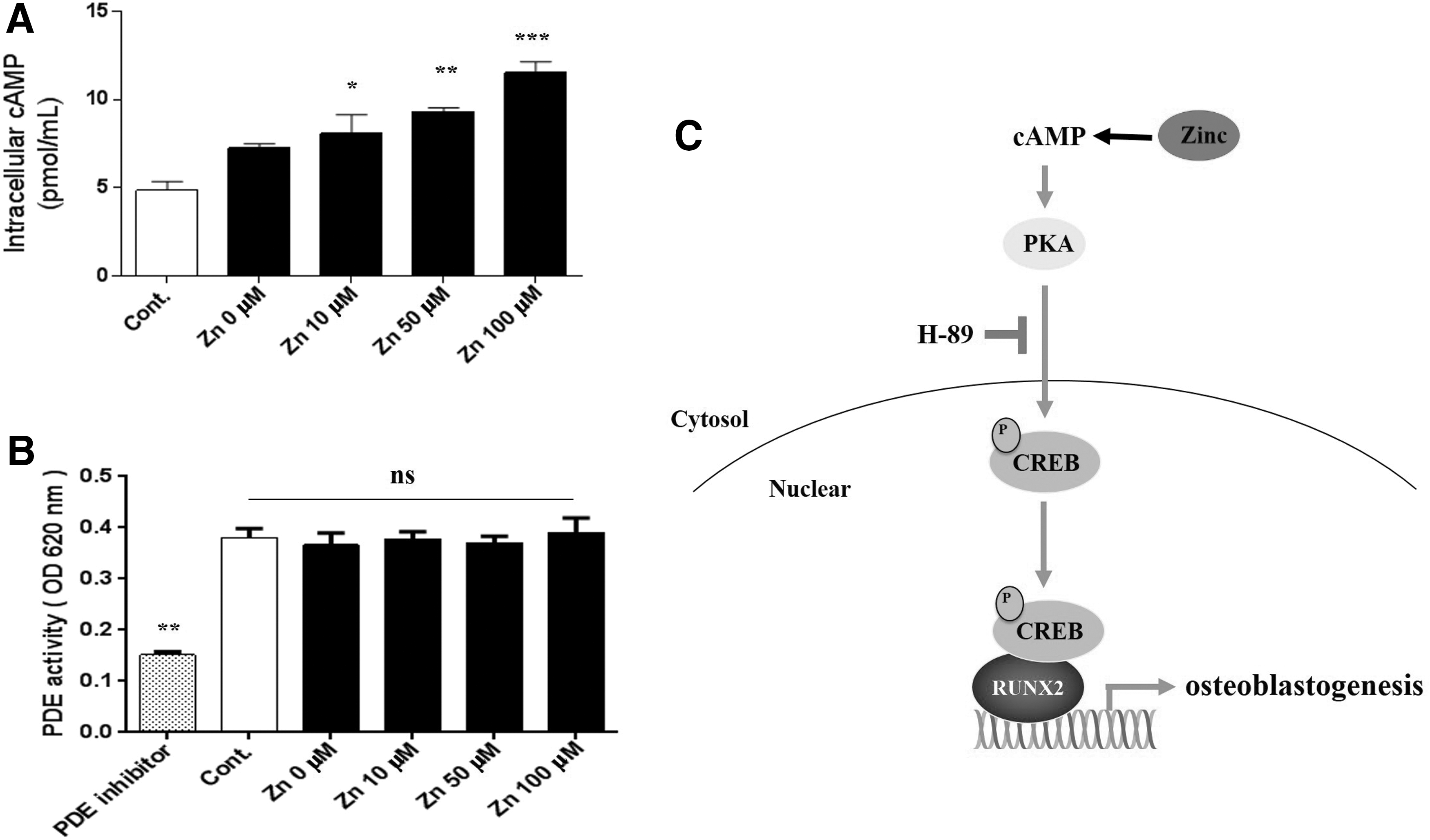
Zinc increases intracellular cAMP in a dose-dependent manner during osteogenesis.
Discussion
In this series of experiments, we have demonstrated that zinc promotes phospho-CREB translocation into the nucleus by increasing PKA activity during osteogenesis in hBMSCs. This effect ultimately enhances expression of RUNX2 and activates osteoblast differentiation. Interestingly, in this study, zinc exposure was found to result in a dose-dependent increase in intracellular cAMP in OM, indicating that enhanced PKA activity may be caused by increased intracellular cAMP. Based on the results of this investigation, we propose that zinc promotes osteogenesis via its effects on the downstream signaling pathways of the cAMP-PKA-CREB axis.
Guo et al. have previously suggested that zinc deficiency may induce apoptosis in osteoblastic MC3T3-E1 cells and cause loss of bone mass [35]. However, there have been no reports demonstrating effects of high-dose zinc on osteoblast apoptosis. In this investigation, we began by establishing a concentration of zinc (100 μM) that was nontoxic to hBMSCs. Although a 100 μM concentration of zinc was found to significantly activate ALP activity in hBMSCs, it was not observed to affect the viability of these cells (Fig. 1A, C). As shown in Figure 1B, the number of hBMSC colonies did not decrease following zinc treatment, indicating that the tested concentrations of zinc did not impact the capacity of hBMSCs for self-renewal. These findings likewise suggest that the osteogenic effects of zinc exposure do not result from enhanced self-renewal capacity.
Many previous studies have established that RUNX2 is a master regulator of osteoblast differentiation [36]. The functions of RUNX2 during osteoblast differentiation have been reported from both in vitro and in vivo studies. RUNX2 induces osteoblastic differentiation of MSCs by driving the expression of many bone matrix protein genes at the early stages of differentiation, while later RUNX2 inhibits osteoblast maturation [37]. Supporting the important role of RUNX2 in bone biology, RUNX2-deficient mice completely lack mature osteoblasts and mineralized bone formation [38]. Regulation of RUNX2 activity is therefore among the most important temporal events that occur during osteogenic differentiation of hMSCs. In mouse osteoblast cell lines, zinc supplementation increases RUNX2 expression at the mRNA and protein levels, while zinc deficiency reduces RUNX2 expression and nuclear RUNX2 protein levels during osteogenic differentiation [39]. Similarly, rats fed a zinc-free diet have been reported to have significantly decreased expression of RUNX2 mRNA measured from the distal femur [40]. Given the central role of RUNX2 in osteogenic differentiation and the evidence that zinc likewise may strongly influence osteogenesis, we hypothesized that zinc might contribute to regulation of RUNX2 during osteogenic differentiation of human mesenchymal stem cells, In this investigation, we aimed to further explore this hypothesis and clarify the molecular mechanisms responsible for the effects of zinc on osteogenic differentiation.
Consistent with previous reports, in this study, mRNA and protein levels of RUNX2 were found to increase upon zinc treatment as osteoblast differentiation progressed (Fig. 2A, B). These findings suggested that zinc accelerates osteoblast differentiation in human mesenchymal stem cells. However, as shown in Figure 4D, the transcriptional activity of CREB was not fully inhibited by PKA inhibitor during zinc treatment. Some residual activity was found to persist after treatment with PKA inhibitor. We propose that other signaling pathways, apart from PKA, may also partly contribute to CREB activation in response to zinc treatment, although these other pathway effects might not be sufficient to enhance osteoblast differentiation. Previous study reported that zinc deficiency leads to decreased osteoblastogenesis due to reduced Runx2 expression through the inhibition of Wnt/β-catenin signaling via the suppression of GSK3β inhibition and Akt activation preceded by the reduced level of SP-1 protein [40]. However, in this study, neither AKT inhibitor nor Wnt inhibitor (DKK1) completely reversed the osteogenic effects of zinc (Supplementary Fig. 4), although protein levels of p-AKT and active β-catenin were increased during osteoblast differentiation (Supplementary Fig. S3B). In addition, the protein levels and transcriptional activities of p-AKT and β-catenin were partially suppressed by PKA inhibitor (Supplementary Fig. S5). These results indicated that zinc-induced AKT and β-catenin activities are mediated by the PKA signaling pathway, not by IGF or WNT signaling and PKA signaling may probably the main mechanism in the zinc effect.
Levels of intracellular cAMP are controlled by a balance between its synthesis by adenylate cyclase (AC) [41] and its hydrolysis into inactive ATP by PDE [34]. So far, controversy exists as to whether zinc influences intracellular cAMP level. It has been reported that zinc could prevent degradation of cAMP through inhibition of PDE activity in Sf9 cells [42], whereas zinc also inhibits forskolin-stimulated AC activity in a mouse neuroblastoma cell line [43]. However, our results demonstrate that zinc dose-dependently increases intracellular cAMP levels in hBMSC. We checked whether zinc could inhibit PDE activity in hBMSC, thus promoting intracellular cAMP level. However, zinc did not affect PDE activity in hBMSCs (Fig. 5A, B), suggesting that the increase of intracellular cAMP after zinc treatment was independent with PDE and may be related with AC activity.
Zinc is one of the nutrients essential for maintenance of bone health. In one animal model, zinc deficiency was reported to cause multiple bone abnormalities, including disturbances of bone formation, growth, and mineralization, thereby resulting in osteoporosis [44]. However, oral administration of zinc sulfate induced calcium contents in newborn rats [45]. In addition, when synthetic bone grafts incorporating zinc were implanted in the femoral condyles of ovariectomized rats, bone mass and bone mechanical properties were found to significantly improve in these animals [46]. Despite these previous studies, the mechanisms accounting for zinc-induced osteogenesis in hBMSCs had previously remained largely unclear. In this study, we present the results of a series of experiments which reveal the effects of zinc on regulation of the cAMP-PKA-CREB axis during osteoblast differentiation.
Osteoporosis is caused by an imbalance between osteoblast and osteoclast activity, which ultimately results in bone loss [47]. Among the drugs currently available for treating osteoporosis, most have only one effect domain, such as inhibition of bone resorption. A drug that only reduces resorption cannot restore bone loss previously caused by osteoclast activity. Therefore, a new strategy for enhanced treatment of osteoporosis is the development of drugs with dual effects, of not only blocking bone resorption but also promoting bone formation. Such drugs should also be cost-effective in order for these therapies to be available to all people who could benefit from using them. With respect to considerations of cost-effectiveness, zinc is notably inexpensive. In addition, several reports have indicated that zinc reduces osteoclast resorption activity [48]. In a previous study conducted by our group, we found that zinc inhibited osteoclast differentiation by suppression of the NFATc1 signaling pathway [49]. Subsequently, in this study, we explored the possibility that zinc also acts to effectively promote bone formation, and we established its molecular mechanism. Remarkably, zinc has now been shown both to increase bone mass and to decrease bone loss.
In conclusion, in this study, we report that zinc exerts osteogenic effects in hBMSCs by activation of RUNX2 via the cAMP-PKA-CREB signaling pathway. These findings provide mechanistic insights into the crucial role of zinc in bone formation. Zinc supplementation offers promise as a potential pharmaceutical therapy for osteoporosis and other bone loss conditions, particularly given the evidence from this study and others of its multimodal effects on bone resorption as well as bone formation.
Footnotes
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (no. NRF-2016R1D1A1B03933182) and from the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2017M3A9E8029722).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.