
Abstract
Pluripotential reprogramming has been examined using various technologies, including nuclear transfer, cell fusion, and direct reprogramming. Many studies have used differentiated cells for reprogramming experiments, and nearly all type of somatic cells can acquire pluripotency. However, within the embryo, other cells types are present in addition to somatic cells. The blastocyst stage embryo consists of two main types of cells, inner cell mass and trophectoderm (TE). TE cells are the first differentiated form of the totipotent zygote and differ from epiblast cells. Thus, we examined whether extraembryonic cells can be reprogrammed using a cell–cell fusion method. Trophoblast stem cells (TSCs), which can be obtained from the TE, are known to acquire pluripotency by transcription factor Oct4 overexpression or somatic cell nuclear transfer. In this study, we demonstrated that TSCs can acquire pluripotent properties by cell fusion with embryonic stem cells (ESCs). TSC-ESC hybrids reactivated Oct4-GFP and displayed self-renewal properties. They expressed the pluripotency markers Oct4 and Nanog, whereas the expression of Cdx2 and Tead4, trophoblast lineage markers, was diminished. Moreover, these cells developed into three germ layers similarly to other pluripotent stem cells. RNA-seq analysis showed that global gene expression patterns of TSC-ESC hybrids are more similar to ESCs than TSCs. Thus, we demonstrated that TSCs successfully complete reprogramming and acquire pluripotency by cell fusion-induced reprogramming.
Introduction
T
Somatic cells originated from the epiblast can be converted into the pluripotent state by several reprogramming technologies, such as nuclear transfer, transduction of transcription factors, and cell fusion with pluripotent stem cells [7 –10]. Diverse somatic cell types originating from the epiblast have been subjected to reprogramming to successfully obtain pluripotency or totipotency through various reprogramming methods [11 –13]. Recently, extraembryonic cells, TSCs, were used as donor cells for reprogramming into pluripotent cells by overexpression of reprogramming factors [14,15]. As TE and ICM are irreversibly determined after blastocyst formation [16], reprogramming of extraembryonic cells into the pluripotent state may require extensive force for the cell fate transition, which is not required for embryonic somatic cells. Induced pluripotent stem cells (iPSCs) can be generated using extraembryonic cells, such as TSCs, amnion, and yolk sac [14,15,17]. Interestingly, TSCs can be reprogrammed into the pluripotent state by overexpressing only one factor, Oct4 [14]. In addition, iPSCs can be derived from human and mouse extraembryonic amnion and yolk sac cells by retroviral transduction of Oct4, Sox2, Klf4, and c-Myc [17]. Recently, Ogawa et al. reported that cloned embryos using TSCs developed into a blastocyst, indicating that TSCs were reprogrammed into ICM, which is the origin of the embryonic lineage, following nuclear transfer [18]. However, reprogramming of TSCs by cell fusion has not been attempted.
Cell fusion with pluripotent cells is also an efficient reprogramming method for inducing pluripotency from differentiated cells; pluripotency from somatic cells was obtained within 2 days [19,20]. Somatic cells can acquire pluripotency by cell fusion with various types of pluripotent fusion partners, including ESCs, embryonic germ cells, embryonic carcinoma cells, and iPSCs [21 –23]. Cell fusion reprograms somatic cells very efficiently, regardless of the somatic cell type [24]. Although many approaches have been used to reprogram somatic cells by cell fusion, it remains unclear whether extraembryonic cells can be efficiently reprogrammed into pluripotent cells following cell fusion with pluripotent fusion partners. Thus, we attempted to reprogram extraembryonic cells, TSCs, into a pluripotent state through cell fusion-induced reprogramming. We established pluripotent fusion hybrid cells using ESCs and TSCs. These TSC-ESC fusion hybrid cells were further characterized to determine whether TSCs can acquire pluripotency and lose their own properties by cell fusion-induced reprogramming.
Materials and Methods
The methods were carried out in accordance with the approved guidelines, and all experimental protocols were approved by the Institutional Animal Care and Use Committee of Konkuk University.
Mice and cell culture
All mouse strains were either bred and housed at the mouse facility of Konkuk University, Korea or bought from Jackson Laboratory (Bar Harbor, ME). Animal handling was conducted in accordance with the Konkuk University animal protection guidelines and Korean animal protection laws.
Derivation of TSCs
TSCs were derived from OG2+/−/ROSA26+/− transgenic blastocysts as previously described [25]. To obtain OG2+/−/ROSA26+/− transgenic blastocysts, homozygous OG2 males were crossed with ROSA26 females. At 3.5 days postcoitum (dpc), female mice were sacrificed, and blastocysts were harvested from the uterus. Blastocysts were then transferred onto dishes layered with mitomycin C-treated mouse embryonic fibroblast (MEF) feeders with TS medium (RPMI 1640; Gibco BRL, Grand Island, NY) supplemented with 20% fetal bovine serum (FBS), 1 × penicillin/streptomycin/glutamine, sodium pyruvate, 0.1 mM β-mercaptoethanol (Gibco BRL), and 20 ng/mL FGF4, 1 μg/mL heparin). Upon attachment of the blastocyst on a feeder-layered dish, Oct4-GFP-positive ICM aggregates were removed, and remaining cells were further cultured for outgrowth of the TE cells. After 5–7 days of culture, the cells were passaged into TS medium containing 70% feeder-conditioned RPMI.
Cell culture
ESCs or TSC-ESC hybrids were grown on mitomycin C-treated MEF feeders with standard mouse ESC culture medium: Dulbecco's modified Eagle's medium (DMEM) supplemented with 15% FBS, 1 × penicillin/streptomycin/glutamine, 1 mM nonessential amino acids (Gibco BRL), 0.1 mM β-mercaptoethanol (Gibco BRL), and 1,000 U/mL leukemia inhibitory factor (LIF; ESGRO, Chemicon, Temecula, CA). TSCs were grown on mitomycin C-treated MEF feeders with TSC culture medium: RPMI 1640 supplemented with 20% FBS, 1 × penicillin/streptomycin/glutamine, sodium pyruvate, 0.1 mM β-mercaptoethanol (Gibco BRL), and 20 ng/mL FGF4, and 1 μg/mL heparin.
Cell fusion and subsequent culture
ESCs were mixed with TSCs in a 1:1 ratio (105:105) and washed in phosphate-buffered saline (PBS). The mixture was centrifuged for 5 min at 130 g in conical tubes (Falcon; BD Biosciences, San Jose, CA). The supernatant was thoroughly removed, and 1 mL of a prewarmed solution of 50% polyethylene glycol 1500 (PEG1500; Roche, Basel, Switzerland) was added to the cell pellet over 1 min. An additional 20 mL of DMEM was added to the cell suspension over 5 min with constant stirring. The cells were centrifuged at 130 g for 5 min to remove the polyethylene glycol (PEG) and further washed gently with DMEM and cultured in ESC medium. After 4–5 days, Oct4-GFP+ colonies were picked and plated on feeder layer in ESC medium. Expanded colonies were dissociated into single cells by 0.25% trypsin treatment and passaged every 2–3 days. To prevent contamination of nonfused cells, we picked Oct4-GFP+ single cell and established clonal cell line.
Karyotype analysis
The 50% confluent cells in a 6-cm dish were treated with 0.3 mg/mL nocodazole for 3 h. Cells were recovered by trypsinization and replated on gelatin-coated (0.1% in PBS) dishes for 30 min to induce attachment of feeder cells. Nonattached cells were recovered and treated with hypotonic (0.56%, w/v) KCl solution for 15 min. The cells were spun down at 500 rpm, fixed by washing three times in fresh fixative (3:1 methanol:acetic acid), and dropped onto clean glass slides. The slides were air-dried, stained with 3% Giemsa (Sigma), and observed under a microscope.
In vitro differentiation
Cells were recovered by trypsinization and replated onto a bacteriological dish in DMEM (15% FBS) in the absence of LIF for 4 days. After embryoid bodies were formed, they were cultured for 4 days in a bacteriological dish and plated onto gelatin-coated (0.1% in PBS) tissue culture dishes for 8–14 days. These differentiated cells were used for immunocytochemistry analysis.
Chimera generation
TSC-ESC hybrids were aggregated and cultured with denuded postcompacted 8-cell-stage mouse embryos. Morula embryos were flushed from 2.5-dpc B6C3F1 female mice. Clumps of TSC-ESC hybrids (10–20 cells per clump) were selected after brief trypsinization and transferred into microdrops of G2 medium under OVOIL-containing zona-free morula embryos. Aggregates were cultured overnight at 37°C and 5% CO2. After 24 h of culture, the aggregated blastocysts were transferred into one uterine horn of a 2.5-dpc pseudopregnant recipient.
Trophoblast lineage differentiation
Cells were plated on gelatin-coated (0.1% in PBS) tissue culture dishes with TSC basal medium; RPMI 1640 supplemented with 20% FBS, 1 × penicillin/streptomycin/glutamine, sodium pyruvate, and 0.1 mM β-mercaptoethanol (Gibco BRL) without any factors. Medium was replaced every day for 7 days. These differentiated cells were used for RNA isolation and real-time reverse transcription–polymerase chain reaction (RT-PCR) analysis.
Immunocytochemistry
Cells were stained for Oct4 and Nanog, which are markers of pluripotent stem cells. For immunocytochemistry, the cells were fixed with 4% paraformaldehyde for 20 min at 4°C. After washing with PBS, the cells were treated with PBS containing 3% bovine albumin serum and 0.03% Triton X-100 for 45 min at room temperature. Primary antibodies used were anti-Oct4 (Oct4; 1:500) and anti-Nanog (Nanog; 1:200, Abcam). To detect primary antibodies, fluorescence-labeled (Alexa fluor 568; Molecular Probes, Eugene, OR) secondary antibodies were used according to the manufacturer's instructions.
RNA isolation and real-time quantitative reverse transcription-polymerase chain reaction analysis
Total RNA was isolated using the RNeasy Mini Kit
X-gal staining
OG2ROSA26 TSCs or TSC-ESC hybrids, which were derived from OG2/ROSA26 transgenic mice, were stained with X-gal. Cells were rinsed with PBS and fixed in 4% paraformaldehyde for 20 min at 4°C. Cells were rinsed three times at room temperature in PBS containing 5 mM EGTA, 0.01% deoxycholate, 0.02% NP40, and 2 mM MgCl2. Cells were washed with PBS and stained in X-gal staining solution: PBS supplemented with 1 mg/mL 5-bromo-r-chloro-3-indolyl-galactosidase (X-gal; Promega, Madison, WI, USA), 5 mM K2Fe(CN)6, 5 mM K4Fe(CN)6, and 1 mM MgCl2. Blue staining was visualized by light microscopy.
RNA sequencing
Total RNA samples were converted into cDNA libraries using the TruSeq Stranded mRNA Sample Prep Kit (Illumina). Starting with 100 ng of total RNA, polyadenylated RNA (mainly mRNA) was selected and purified using oligo-dT-conjugated magnetic beads. This mRNA was physically fragmented and converted into single-stranded cDNA using reverse transcriptase and random hexamer primers, with the addition of Actinomycin D to suppress DNA-dependent synthesis of the second strand. Double-stranded cDNA was created by removing the RNA template and synthesizing the second strand in the presence of dUTP (deoxy-ribo-uridine triphosphate) in place of dTTP (deoxythymidine triphosphate). A single A base was added to the 3′ end to facilitate ligation of sequencing adapters, which contain a single T base overhang. Adapter-ligated cDNA was amplified by polymerase chain reaction to increase the amount of sequence-ready library. During this amplification, the polymerase stalls when it encounters a U base, rendering the second strand a poor template. Accordingly, the amplified material used the first strand as a template, thereby preserving the strand information. Final cDNA libraries were analyzed for size distribution and using an Agilent Bioanalyzer (DNA 1000 kit; Agilent), quantitated by qPCR (Kapa Library Quant Kit; Kapa Biosystems, Wilmington, MA), then normalized to 2 nM in preparation for sequencing.
Bisulfite genomic sequencing
To differentiate between methylated and unmethylated CG dinucleotides, genomic DNA was treated with sodium bisulfite to convert all unmethylated cytosine residues into uracil residues using the EpiTect Bisulfite Kit (Qiagen) according to the manufacturer's protocol. In brief, purified genomic DNA (0.5–1 mg) was denatured at 99°C and then incubated at 60°C. After desulfonation, neutralization, and desalting, the modified DNA was diluted in 20 mL of distilled water. Subsequently, bisulfite PCR (BS-PCR) amplification was carried out using 1–2-mL aliquots of modified DNA for each PCR. The primers used for BS-PCR were as follows: Oct4 first sense 5′-TTT GTT TTT TTA TTT ATT TAG GGG G-3′, Oct4 first antisense 5′-ATC CCC AAT ACC TCT AAA CCT AAT C-3′; Oct4 second sense 5′-GGG TTG GAG GTT AAG GTT AGA GGG-3′, Oct4 second antisense 5′-CCC CCA CCT AAT AAA AAT AAA AAA A-3′; Elf5 first sense 5′-GAA GTA TTG GGT TGA GGA AAA TG-3′; Elf5 first antisense 5′-CAA CCA CTT TAA CCA AAC AAT C-3′; Elf5 second sense 5′-ATA GTA GTT GGG GTT ATT GTT TT-3′; and Elf5 second antisense 5′-CAA TTA TAA AAA TCT ACT TAC CTT C-3′. In brief, the amplified products were verified by electrophoresis on a 1% agarose gel. The desired PCR products were used for subcloning using the TA cloning vector (pGEM-T Easy Vector; Promega). Reconstructed plasmids were purified, and individual clones were sequenced (Solgent Corporation, Daejeon, Korea).
Results
Derivation of TSCs and their characteristics
As an extraembryonic fusion partner with ESCs, we derived TSCs from OG2+/−/ROSA26+/− double transgenic blastocysts, in which Oct4-GFP (OG2) is activated when TSCs are reprogrammed into the pluripotent state, and Lac Z (ROSA26) is ubiquitously expressed. Following disaggregation of the outgrowth and subculture in TSC medium containing FGF4 and heparin, TSCs displaying GFP-negative flat colonies with distinct borders were established (Fig. 1A). TSCs expressed Cdx2 and Sox2 as typical TSC markers, but did not express Oct4 (Fig. 1B). RT-PCR analysis confirmed that other TSC markers, Tead4 and Eomes, were expressed in TSCs without expression of pluripotency markers (Fig. 1C).

Generation and characterization of OG2ROSA TSCs.
TSCs can be reprogrammed into a pluripotent state by cell–cell fusion
To investigate whether TSCs can be reprogrammed into the pluripotent state by cell fusion, we fused OG2+/−/ROSA26+/− transgenic TSCs with wild-type E14 ESCs by PEG-mediated cell fusion (Fig. 2A). On day 4 after cell fusion, Oct4-GFP+ colonies emerged (Fig. 2B). In terms of Oct4-GFP activation, reprogramming at day 4 was much slower case than that in somatic cell-ESC fusion, in which Oct4-GFP activation required at least 2 days [19]. Moreover, reprogramming efficiency was about 0.002% (two Oct4-GFP+ cells from 1 × 105 TSCs).
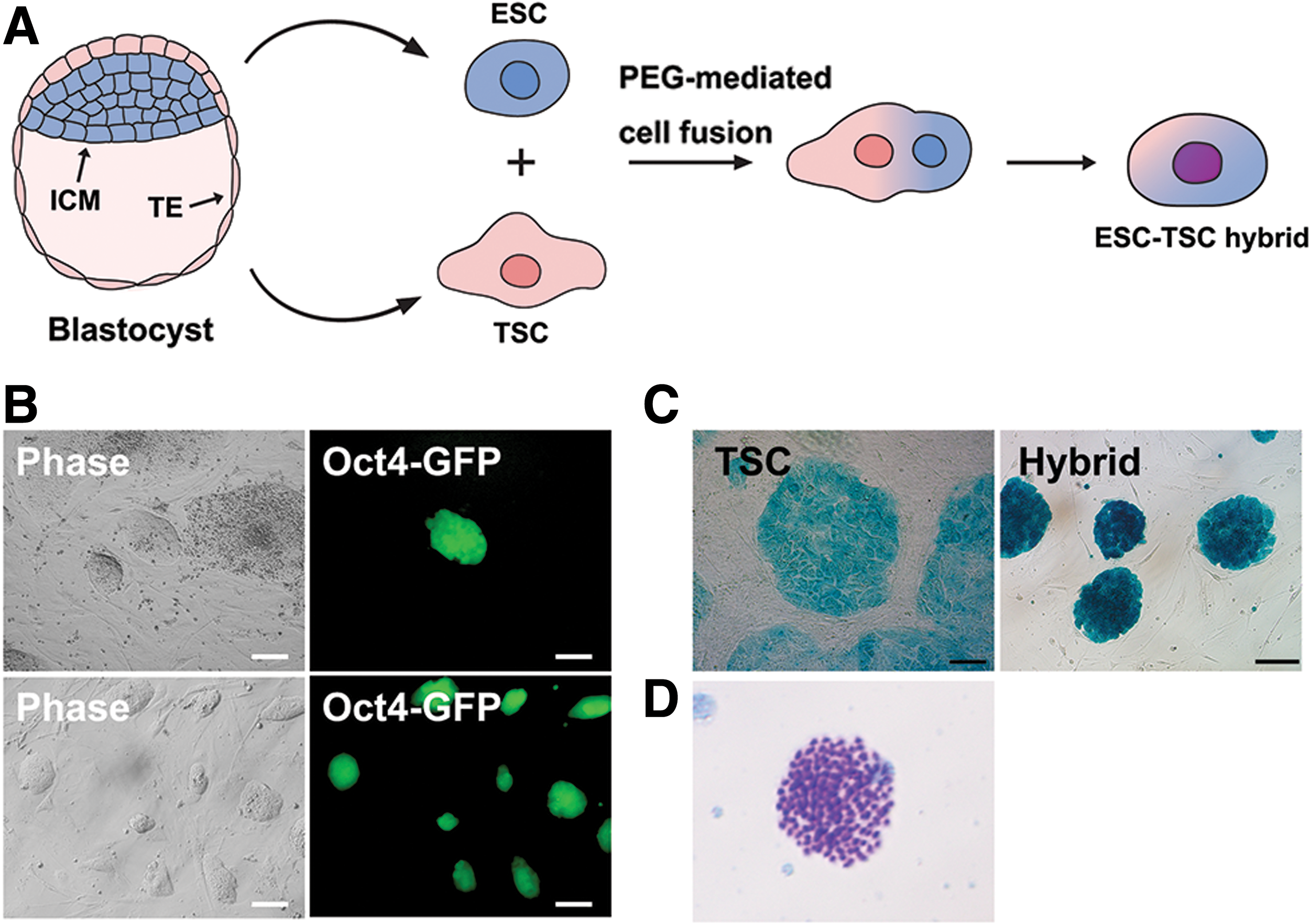
Reprogramming of TSCs by cell fusion with ESCs.
Oct4-GFP+ cells were X-gal positive, indicating that TSC-ESC hybrids express Lac Z from the TSC genome (Fig. 2C). Thus, Oct4-GFP+/X-gal-positive cells that formed ESC-like colonies were predicted to be reprogrammed fusion hybrid cells. To confirm whether these Oct4-GFP+ cells were fusion hybrid cells, we conducted karyotyping. Karyotype analysis demonstrated that TSC-ESC hybrids were near tetraploid (4n) (Fig. 2D). As negative controls, TSC-TSC and ESC-ESC fusions were conducted, which did not show any GFP+ cells (data not shown), indicating that only the ESC-TSC fusion can result in Oct4-GFP+ reprogrammed hybrid cells.
TSC-ESC hybrid cells exhibited pluripotent properties
Oct4-GFP+ TSC-ESC hybrids formed a dome-like morphology (Fig. 2B) and were alkaline phosphatase-positive (Fig. 3A). Immuonocytochemistry analysis revealed the expression of Oct4 and Nanog, two major pluripotency transcription factors (Fig. 3B), which were not detected in TSCs. Quantitative RT-PCR analysis also revealed overexpression of Oct4 and Nanog, but repression of the TSC-specific markers Cdx2 and Tead4 in TSC-ESC hybrids indicated that the expression pattern of pluripotency- and tissue-specific markers of TSCs were successfully reprogrammed to the pluripotent state by cell-fusion with ESCs (Fig. 3C). Next, we examined the DNA methylation patterns of pluripotency- and TSC-specific genes in ESCs, TSCs, and fusion hybrid cells. As DNA methylation is known to regulate gene expression levels, ESCs and TSCs display different DNA methylation patterns. Bisulfite analysis showed that the Oct4 promoter regions were nearly unmethylated in ESCs, but hypermethylated in TSCs (Fig. 3D). The DNA methylation pattern of Elf5, a major transcription factor of TSCs, showed the opposite pattern as Oct4. The Elf5 promoter region was highly methylated in ESCs but hypomethylated in TSCs. TSC-ESC hybrids showed similar DNA methylation patterns in the Oct4 and Elf5 promoter regions as ESCs: erasure of DNA methylation in Oct4 regions but gain of DNA methylation in the Elf5 region indicated that TSCs were reprogrammed at the epigenetic level to a pluripotent state. Collectively, along with Oct4-GFP activation and inactivation of TSC markers (Cdx2 and Tead4), the change in DNA methylation suggests that the TSC genome is reprogrammed in fusion hybrids.

Acquisition of pluripotency in TSC-ESC hybrid cells.
Differentiation potential of reprogrammed TSC-ESC hybrid cells
Pluripotent cells have differentiation potential and contribute all three germ layer-derived cells. TSCs can only differentiate into the extraembryonic trophoblast lineage and lack differentiation potential into embryonic lineages. Therefore, if TSC-ESC hybrids obtained pluripotency after reprogramming, they can differentiate into cells of all three germ layers as ESCs [26]. Thus, we investigated whether TSC-ESC hybrids had pluripotent differentiation potential as standard pluripotent stem cells. Through embryoid body formation in vitro, TSC-ESC hybrids were successfully differentiated into the ectodermal (Tuj-1+), mesodermal (SMA+), and endodermal (HNF3β+) lineages (Fig. 4A). To confirm the differentiation potential, we examined the chimera formation ability of hybrid cells. TSC-ESC hybrid cells successfully integrated into the ICM of the blastocyst and contributed to the chimeric embryo after aggregation with morula and transplantation into the uterus of the surrogate mother. However, the hybrid cells did not contribute to the germline, as determined by the absence of Oct4-GFP+ populations in the gonad of 13.5 dpc chimeric embryos. Notably, these TSC-ESC hybrid cells contributed to the placenta (Fig. 4). These results agree with those of our previous study showing that tetraploid hybrid cells contribute to both embryonic and extraembryonic tissue, but not to germ cells [26] (Fig. 4B).
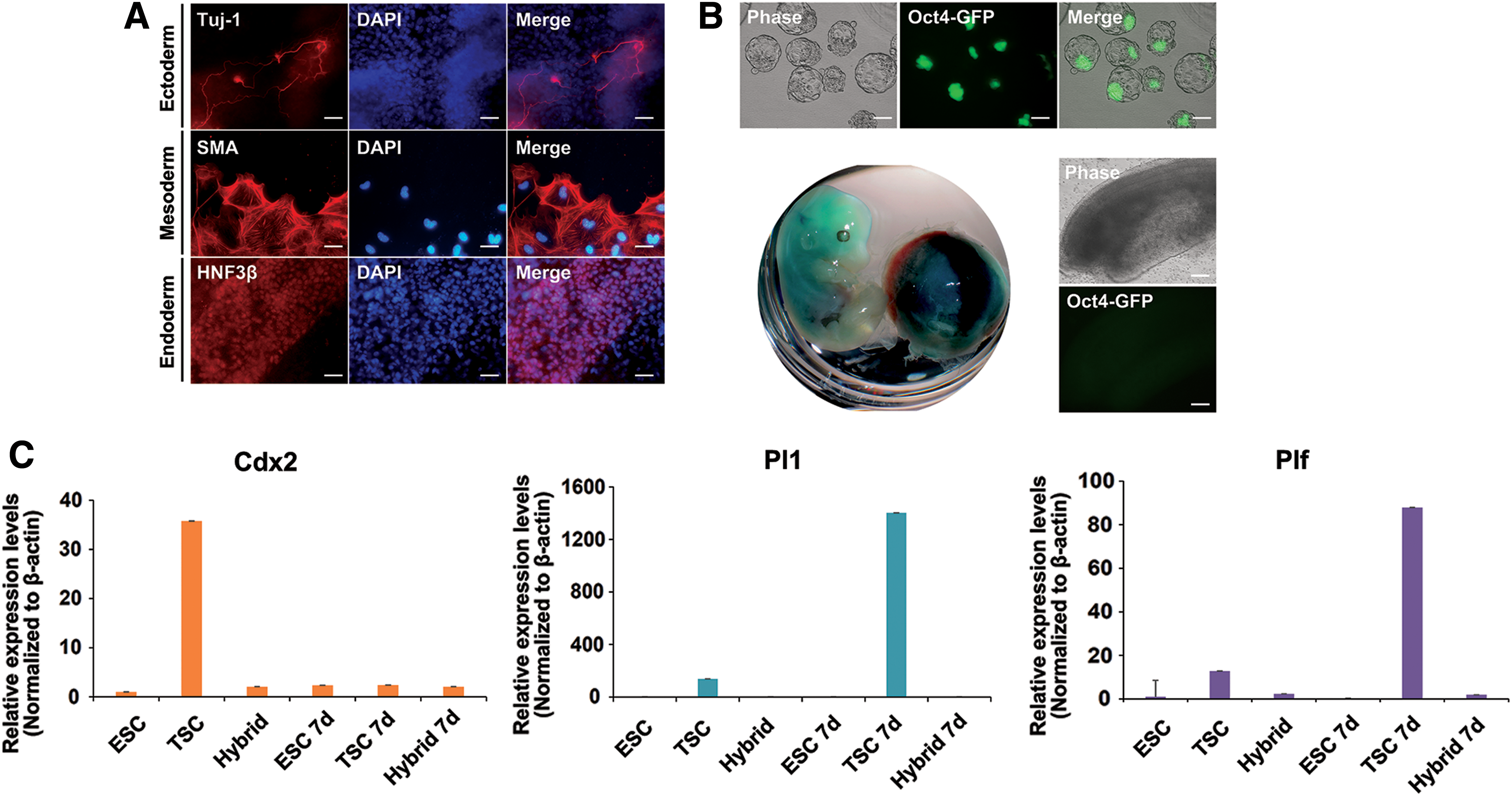
Differentiation potential of TSC-ESC hybrid cells.
Because TSC-ESC hybrids originate from TSCs, we tested their ability to contribute to TE when they were induced to differentiate. We performed trophoblast lineage differentiation of ESCs, TSCs, and TSC-ESC hybrids in vitro. Cells were induced to differentiate in TSC basal medium without any factors for 7 days (Fig. 4C). TSCs are multipotent stem cells that can differentiate into the several types of cells that make up the placenta. After 7 days, TSCs expressed the trophoblast giant cell marker placental lactogen 1 (Pl1) and proliferin (Plf) simultaneously with the loss of Cdx2. In contrast, in ESCs and TSC-ESC hybrids, there was no sign of trophoblast lineage differentiation and no expression of Pl1 and Plf, indicating that TSCs lost their memory to differentiation into the trophoblast lineage in vitro after reprogramming by cell fusion (Fig. 4D). This result supports that the contribution of TSC-ESC hybrids to the placenta in the chimeric embryo may be because of the tetraploidy of the hybrid cells.
Global gene expression patterns of TSCs, ESCs, and TSC-ESC hybrids
Next, we performed RNA-seq analysis to compare the transcriptomes of TSC-ESC hybrids with TSCs or ESCs. Heat map and hierarchical clustering analyses showed that gene expression pattern of TSC-ESC hybrids was different from that of the originating TSCs (Fig. 5A, B). TSC-ESC hybrids were more closely clustered together with ESCs, but were far different from TSCs (Fig. 5B). Pairwise scatter plot analysis also supported that the TSC-ESC hybrids obtained pluripotency to become ESC-like state through reprogramming process (Fig. 5C). We found that 1,613 genes were upregulated and 1,154 genes were downregulated in TSC-ESC hybrids versus ESCs. There was 2,743 upregulated genes and 2,593 downregulated genes when we compared TSC-ESC hybrids with TSCs. Also, to a similar degree, 2,835 upregulated genes and 2,737 downregulated genes were observed in TSCs versus ESCs (Fig. 5D). In more detail, we examined the expression of trophoblast- and pluripotency-related genes in TSCs, ESCs, and TSC-ESC hybrids. TSC-ESC hybrids expressed pluripotency-related genes (Tbx3, Nr5a2, Nanog, and Oct4) with the similar degree to ESCs. However, trophoblast-related genes (Cdx2, Gata3, Krt7, Hand1, Eomes, Tead4, and Ets2) were downregulated in TSC-ESC hybrids (Fig. 5E). By using RNA-seq analysis, we verified that global gene expression pattern of TSCs was successfully converted into ESC-like pluripotent state by cell–cell fusion with ESCs.
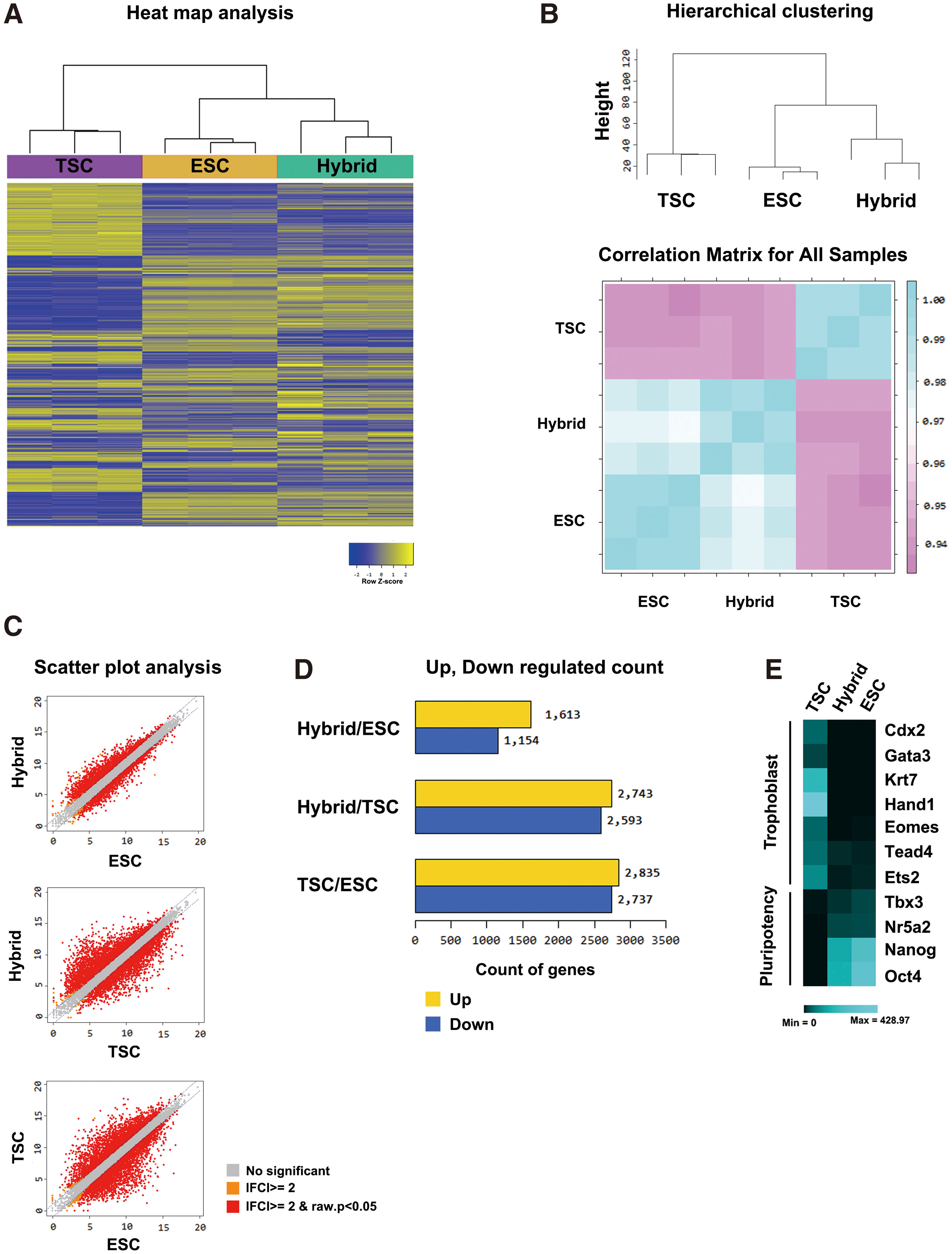
RNA-sequencing analysis of ESCs, TSCs, and TSC-ESC hybrid cells.
Discussion
In the present study, we established pluripotent fusion hybrid cells from two different stem cell lines, pluripotent ESCs and extraembryonic multipotent TSCs, which were irreversibly segregated after blastocyst formation [16]. Similar to the somatic cell-ESC hybrids, TSC-ESC hybrid cells successfully reprogrammed and exhibited pluripotent properties. They expressed pluripotency markers, while TSC markers were downregulated. Moreover, Oct4 promoter regions, which were hypermethylated in TSCs, were demethylated after reprogramming through cell fusion. In contrast, the TSC-specific Elf5 promoter region, which was hypomethylated in TSCs, became methylated after reprogramming. These results suggest that the TSC genome itself was successfully reprogrammed into a pluripotent state by cell fusion-induced reprogramming. Furthermore, TSC-ESC hybrids showed similar in vitro differentiation potential to ESCs; they were differentiated into all three germ layers through embryoid body formation and chimeric embryo formation. Global gene expression comparison using RNA sequencing analysis also showed that gene expression pattern of TSC-ESC hybrid cells was more similar to ESCs than that of TSCs.
Cell fusion-induced reprogramming is the fastest method for acquiring pluripotency compared to nuclear transfer and direct reprogramming through transduction of reprogramming factors. Yamanaka factors (Oct4, Sox2, Klf4, and c-Myc) induced reprogramming at approximately more than 7 days posttransduction. When somatic cells were fused with pluripotent cells, the Oct4-GFP reporter was activated within 2 days postfusion. Once somatic cells were successfully fused with ESCs, reprogramming efficiency was ∼95% [24]. In this study, however, we found that the reprogramming efficiency of TSCs, in which Oct4-GFP was activated at day 4 postfusion, was remarkably lower than that of somatic cells in fusion-induced reprogramming.
Reprogramming of extraembryonic cells has been attempted by using nuclear transfer and direct induction of pluripotency (iPSCs). Nuclear-transferred embryos using TSC donors may develop into the blastocyst and form the ICM, the origin of the embryonic lineage [18]. However, the pluripotency of the TSC-derived ICM has not been well characterized because a pluripotent stem cell line has not been established from cloned blastocysts. The pluripotency of TSC-derived cells was well characterized in iPSCs from TSCs generated by lentiviral infection of Oct4, Sox2, Klf4, and c-Myc [14]. Primary ESC-like colonies formed after 2–3 weeks of lentiviral infection of Yamanaka factors, which was much slower than reprogramming of somatic cells, which required ∼1 week [27]. This was also observed in fusion-induced reprogramming [24] in this study. Initial Oct4-GFP+ colonies were formed at day 4 after fusion of TSCs with ESCs, which was about 2 days slower than the fusion of neural stem cells (NSCs) with ESCs [24]. We could not clearly explain the slower activation of Oct4-GFP in TSC-ESC hybrids. One possible explanation is that the transcription factor Cdx2 expressed in TSCs suppresses Oct4-GFP and endogenous Oct4 as Cdx2 is known to repress Oct4 expression and vice versa [28]. Other trophoblast lineage-specific transcription factors, such as Gata3, Tcfap2c, and Elf5, may also repress pluripotency-related genes [29 –31]. This slow reprogramming was also observed in iPSC derivation from TSCs [15]. The efficiency of TSC reprogramming to iPSCs was 0.0055% at day 28, which was comparable to the value of 0.2% in reprogramming using fibroblast cells. Moreover, in nuclear transfer using TSCs as somatic cells, nuclear-transferred embryos showed low efficiency of the development to blastocysts [18]. Collectively, although TSCs can be reprogrammed to the embryonic lineage or pluripotent stem cells, genomic reprogrammability was relatively low compared with that of embryonic lineage somatic cells. Interestingly, Wu et al. suggested that TSCs could be reprogrammed to iPSCs by only one factor, Oct4, as TSCs express Sox2, c-Myc, and Esrrb, which substitute for Klf4 [14]. Oct4 lentiviral infection required 16 days to form colonies as shown in four factor-driven reprogramming. NSCs also can be reprogrammed by only Oct4, but reprogramming of NSCs (judged by Oct4-GFP activation) by one factor (Oct4) required 4–5 weeks [32,33]. Thus, Wu et al. suggested that for reprogramming by one factor, TSCs are relatively easier to reprogram than NSCs. However, this result requires confirmation; TSCs and NSCs must be compared in the same laboratory and using the same methods.
We showed that TSC-ESC hybrid cells contribute to the placenta as well as somatic tissues. Our previous reports also revealed the contribution of hybrid cells to the placenta [26]. In contrast, TSC-derived iPSCs did not contribute to the trophoblast lineages, suggesting that erasure of the epigenetic memory during reprogramming blocked the preferential differentiation to trophoblast tissues [15]. In the present study, we also showed that TSC-ESC hybrids did not show trophoblast lineage differentiation in vitro as was observed for ESCs, suggesting that TSCs lost their memory to differentiate into the trophoblast lineage after reprogramming by cell fusion (Fig. 4D). Thus, the contribution of the TSC-ESC hybrid to the placenta may not be because of the TSC origin in hybrids, but may be related to the tetraploidy of the hybrids [26].
In conclusion, our results and those of previous studies suggest that the extraembryonic lineage cannot block pluripotential reprogramming.
Footnotes
Acknowledgment
This work was supported by Konkuk University in 2017.
Author Disclosure Statement
No competing financial interests exist.