
Abstract
Astronauts on missions beyond low-Earth orbit are exposed to a hostile environment in which they are continually bombarded with unique high-energy species of radiation, while in conditions of microgravity (μG), which can alter radiation response and immunity. In the present studies, we examined the impact exposing human hematopoietic stem/progenitor cells (HSC) to μG had upon their capacity to repair DNA damage and their ability to generate immune cells critical for mounting an effective antitumor response. To this end, we first treated a human HSC-like cell line with an acute dose of the radiomimetic drug bleomycin, cultured them in normal gravity (1G) or simulated μG, and quantitated double-strand breaks through γ-H2AX foci. Calculating the median fluorescence intensity ratio at 1-to-4 h post-bleomycin revealed a 26% decrease in 1G, but a 20% increase in μG, suggesting that μG compromised HSC DNA damage repair and thus has the potential to enhance the genotoxic effects of space radiation. We next examined whether μG negatively affected the development of dendritic cells (DC), critical regulators of both the innate and acquired arms of the immune system. Primary human HSC were cytokine induced in 1G or μG and analyzed for generation of plasmacytoid (CD123+) and myeloid (CD11c+) DC. HSC cultured in 1G gave rise to significantly higher numbers of both myeloid and plasmacytoid DC than those cultured in μG, suggesting μG impairs production of these critical antigen-presenting cells. Our studies thus indicate that conditions of μG present during spaceflight perturb multiple pathways that could potentially enhance astronaut risk from exposure to space radiation.
Introduction
O
In addition to being exposed to high energy radiation, astronauts are subjected to conditions of microgravity (μG) during spaceflight, which can also exert a wide range of untoward effects on the body [13 –23]. Considering μG-induced alterations of direct relevance to cancer risk, numerous studies have collectively demonstrated that conditions of μG present during spaceflight deleteriously affect the immune system [24 –40]. Specifically, alterations of leukocyte subset distribution [24,26,33,39,40], cytokine production [25,27,31,35], inhibition of leukocyte blastogenesis [40], and reduction of total NK activity [29,31,32] have all been reported. Data from manned spaceflights have also shown decreased resistance to infection in some astronauts during their missions, immediately after return, or during the first week of landing [41]. Impaired cellular immunity, as assessed by delayed hypersensitivity skin test responses to common recall antigens, has also been clearly demonstrated in astronauts during short- and long-term spaceflights [42 –47].
The immune system has developed over millennia as a combination of defenses not only against invading organisms and foreign cells but also as a surveillance system for cancerous cells. Its two major components, the innate and the adaptive, work together in an intricate net to preserve normal homeostasis. Innate immunity mediates the initial protection against infections and cancer [48 –51], and delivers regulatory signals to promote an adaptive immune response. Among the key cells involved in the innate immune response, dendritic cells (DC) are a unique leukocyte population that sits at the crossroads of the innate and adaptive immune systems, and they are the most effective antigen-presenting cells for initiating an immune response. DC, like all other immune cells, are generated from HSC through the process of hematopoiesis, which occurs in the bone marrow (BM) of adults in a unique microenvironment that is responsible for both maintaining primitive HSC and inducing their differentiation to specific lineages as the need arises [52 –57]. Once generated, DC exit the BM and circulate in immature form in the peripheral blood, as distinct subsets [58 –62]. From the blood, DC migrate to different tissues and organs and, depending on the tissue of residency, migration pattern, and/or activation status, specific DC phenotypes have also been identified. Once in the peripheral immune system, DC behave like quiescent sentinels, rapidly activating when a pathogen or a tumor cell is detected by Toll-like receptors, scavenger receptors, or other surface structures. Once activated, DC can direct the differentiation of T helper (Th) cells, thus launching the immune response to eliminate the offending entity. Importantly, activated DC also communicate with NK cells, inducing resting NK cells to begin proliferating, producing IFN-γ, expressing the activation marker CD69, and exhibit pronounced cytotoxicity, all of which combine to enhance the ability of NK cells to recognize and eliminate tumors [63]. As such, DC play a pivotal role in the body's ability to recognize and eliminate both microbial invaders and tumor cells [64 –66].
While numerous previous studies have demonstrated that alterations occur in the immune system as a result of space travel, the level at which these mechanisms exert their effect, which is at the level of the mature immune cell or earlier at the progenitor or stem cell stage, is not known. In the present study, we employed in vitro studies to determine whether exposure to μG impairs immune function by altering the ability of HSC to efficiently generate cells of the immune lineages. Given their crucial role in multiple facets of the immune response, we chose to focus our experiments on the effects μG exerts on the differentiation of human HSC into specific subsets of DC. With the exception of two fairly early studies [67,68], this is an area that has not been explored in a great deal of detail. Studies in recent years have also provided evidence that conditions of μG can adversely affect the DNA damage response (DDR) of cells, potentially altering their ability to respond appropriately and effectively to the damaging effects of ionizing radiation [69 –71]. Herein, we show that HSC differentiation into DC is markedly impaired in simulated μG, and that human HSC DDR is compromised in conditions of μG, which results in an accumulation of double-strand breaks that cannot be resolved over time.
Materials and Methods
Human HSC isolation
Healthy human BM mononuclear cells (BMMNC) were purchased from AllCells, LLC (Alameda, CA). CD34+ HSC were isolated from BMMNC using anti-human CD34 MicroBeads and the MiniMACS magnetic-based positive-selection system (Miltenyi Biotec, Inc., Auburn, CA), as we have previously described [72 –75].
Culture conditions
Isolated human CD34+ cells (HSC) were cultured in serum-free QBSF-60 media (Quality Biologicals, Inc., Gaithersburg, MD) supplemented with the following concentrations of cytokines: 10 ng/mL IL-4, 10 ng/mL GM-CSF, 4 ng/mL SCF, 10 ng/mL Flt-3 L, 2 ng/mL TNF-α (all cytokines were purchased from PeproTech, Rocky Hill, NJ). Alternatively, HSC were cultured in IMDM containing 15% fetal bovine serum (HyClone FBS, Thermo Fisher Scientific, Raleigh, NC) with 10 ng/mL IL-4, 10 ng/mL GM-CSF, 4 ng/mL SCF, 10 ng/mL Flt-3 L, and 2 ng/mL TNF-α in 25-cm2angled neck cell culture flasks (T25; Corning®, Sigma-Aldrich, St. Louis, MO). Cells were incubated for a period of 14–26 days. Cells were grown at a temperature of 37°C, in 98% humidity with 5% CO2 tension.
To model conditions of μG, isolated CD34+ cells were cultured in the Synthecon Rotary Cell Culture System (RCCS)-4 system using 10 mL HARV vessels. The RCCS is a fluid-filled rotating culture chamber that keeps the cells in a continuous state of free fall. Cells were incubated in the RCCS (or in standard T25 culture flasks) as a normal gravity (1G) control/reference for a period of 14–21 days, also at a temperature of 37°C, in 98% humidity with 5% CO2 tension. The normal gravity controls in T25 flasks were placed in the same incubator as the RCCS, on the same shelf as the device, to control for any microvibrations or temperature fluctuations/aberrations caused by the presence of the RCCS device in the incubator, as is routinely done by investigators in the field [76 –80]. As an additional control, we performed pilot studies, in which we placed the human CD34+ cells in the 10-mL HARV vessels, using the same DC-inductive media, but simply rested the HARVs on the shelf of the incubator, rather than attaching them to the RCCS and rotating them. We then performed flow cytometric analysis on these cells, and compared the outcome to those obtained with the same cells cultured in T25 flasks under the same conditions. No significant difference was observed in the phenotype/dendritic differentiative potential of these cells under these two conditions (data not shown), providing strong evidence that the materials/geometry of the HARV do not, in and of themselves, alter the ability of human HSC to undergo differentiation along the dendritic lineage.
Flow cytometry to assess differentiation
To perform flow cytometric analysis at each time point, 5 mL of cell suspension was collected from the RCCS (μG) chamber and 2.5 mL of cell suspension was collected from the T25 flask (1G reference). The cultures were refed with equivalent amounts of fresh media and put back into the incubator. Analysis occurred from day 3–21 at certain checkpoints. Cell count and viability was accomplished using 0.4% Trypan Blue (Gibco). Cell populations from each respective sample were spun at 400 × g for 5 min, and cell pellets were resuspended in phosphate buffered saline (PBS). The cells were then stained with a previously described three-color protocol [81] using 10 μl of each antibody directly conjugated to a specific fluorochrome (FITC, PE, or PerCP) to allow simultaneous visualization and assessment of expression of three antigens. Each sample was stained (in various combinations) with antibodies to “Lin 1,” CDw123, CD11c, and HLA-DR. Lin 1 is a “cocktail” that contains FITC-labeled antibodies to CD3, CD14, CD16, CD19, CD20, and CD56. Appropriately labeled mouse immunoglobulins (γ1-FITC, γ2-PE, or γ1-PerCP) were used as isotype controls to account for any nonspecific staining. All antibodies employed in these studies were purchased from Becton Dickinson Immunosystems (San Jose, CA).
Following addition of the respective antibodies, cells were incubated in the dark for 15 min at room temperature. Cells were then washed with PBS +0.1% sodium azide, and pelleted by 5 min of centrifugation at 400 × g. Cells were fixed using 1% formaldehyde and either analyzed immediately or stored at 4°C overnight. All flow cytometric acquisition was performed on a FACSCalibur cytometer, and all data were then analyzed using CellQuest software (both from Becton Dickinson Immunosystems, San Jose, CA).
Morphologic analysis by Cytospin
At various time points in the differentiative process, one million cells were collected from each sample of HSC grown under conditions of normal (1G) and simulated μG, and divided into four aliquots. Each aliquot was pipetted into a separate Shandon™ Cytospin™ Cytofunnel with slow absorption-pad chambers. Cell suspensions were centrifuged in a Shandon Cytospin at 500–1,000 rpm for 3 min onto noncoated glass slides (all cytospin products were purchased from Thermo Fisher Scientific, Grand Island, NY). Slides were immediately fixed and stained with PROTOCOL™ Hema-Quik™ II Stain Solution according to the manufacturer's instructions (Fisher HealthCare™, Thermo Fisher Scientific, Grand Island, NY), and evaluated on an Olympus BH-2 compound microscope (Olympus America, Inc., Melville, NY) at 10X and 100X magnifications. Images were obtained on an Olympus IX71 inverted fluorescence and phase contrast microscope (Olympus Americas, Inc., Melville, NY). Adobe Photoshop CS6 (Adobe Systems, San Jose, CA) was then used to assemble multipanel figures, and perform minimal global processing, such as brightness, contrast adjustment, and color balance.
Growth of human HSC-like cell line to study HSC DDR
The human HSC-like cell line, KG1a (ATCC® CCL246.1™), was grown in suspension to near “confluence” (a concentration of 1 × 106 viable cells/mL) in Corning canted neck low-profile T75 culture flasks with vent cap (Sigma-Aldrich), using Iscove's Modified Dulbecco's Medium (Thermo Fisher Scientific, Grand Island, NY) supplemented with 20% fetal bovine serum (Lonza, Walkersville, MD).
Treatment with bleomycin to mimic effects of ionizing radiation
Once KG1a cells reached the desired density, one flask was left untreated (to serve as a control), and to the other flask, bleomycin sulfate solution (Sigma-Aldrich) was added to achieve a final concentration of 10 μg/mL, to mimic an acute exposure to SEP/GCR radiation [82]. At 15 min after treatment, the drug was removed by washing the KG1a cells with 30 mL of media and centrifuging at 400 × g for 8 min. Following washing, cells from each flask were resuspended in 20 mL of fresh media. Two 5 mL aliquots of the tube of untreated (control) and of the bleomycin-treated KG1a cells were transferred into new tubes, and placed in normal gravity (1G) in an incubator at 37°C in humidified air, 5% CO2.
Culture in simulated μG
The remaining 10 mL of the untreated (control) and bleomycin-treated KG1a cells were loaded into a 10-mL chamber of a NASA-designed Rotary Cell Culture System (RCCS; Synthecon, Houston, TX), and rotated to create a state of perpetual freefall to mimic conditions of μG [83].
Analysis of DDR in 1G and μG by γ-H2AX immunofluorescence
At 1 h, the following samples were collected: (a) One 5-mL tube of untreated (control) KG1a cells grown in 1G; (b) One 5-mL tube of bleomycin-treated KG1a cells grown in 1G; (c) 5 mL of the untreated (control) KG1a cells grown in μG (replacing the missing volume with 5 mL of fresh media); and (d) 5 mL of the bleomycin-treated KG1a cells grown in μG, again replacing the missing volume with 5 mL of fresh media). At 4 h, the remainder of each cell in each experimental group was collected. After collecting cells at each time point, cells were pelleted by centrifugation at 400 × g for 8 min and analyzed by γ-H2AX immunofluorescence as we have done previously [84 –86]. In brief, cells were fixed in 4% formaldehyde in PBS at room temperature for 15 min, and again collected by centrifugation as before. Cells were then permeabilized with a solution of 0.5% Triton X-100 in PBS on ice for 10 min. Cells were collected by centrifugation, and nonspecific binding blocked by incubating the cells for 30 min at room temperature in 2 mL of PBS containing 1% BSA, 2% fetal bovine and goat sera, 0.1% Triton X-100, and 0.05% Tween-20. After blocking, cells were again collected by centrifugation, and each cell population/treatment group was then split into two tubes, and incubated in a 1 mL volume in a 37°C humidified chamber for 30 min with: mouse normal IgG-FITC isotype control (EMD Millipore, Temecula, CA) or mouse monoclonal anti-γ-H2AX pS139 (clone JBW301; EMD Millipore, Temecula, CA) primary antibody diluted 1:400 in PBS, 1% BSA. Following incubation with the primary antibody, cells were washed by adding 10 mL of PBS and centrifuging as before. Cells were then resuspended in 1 mL of Alexa Fluor® 488 goat anti-mouse H&L (Cat #A11001; Molecular Probes/Invitrogen, Thermo Fisher Scientific, Grand Island, NY) diluted 1:400 in PBS, 1% BSA, and incubated in a 37°C humidified chamber for 30 min. Cells were washed as before, and resuspended in 0.5 mL of 4% formaldehyde in PBS.
Aliquots of each cell population were then used to prepare cytospins using Shandon EZ Single Cytofunnels™ with white filter cards and caps (Thermo Fisher Scientific, Grand Island, NY). Samples were then coverslipped/mounted with ProLong® Gold Antifade Mountant with DAPI (Thermo Fisher Scientific, Grand Island, NY), and images were captured with an Olympus Fluoview 1000 confocal system (Olympus, Tokyo, Japan). Following image acquisition, ≥100 γ-H2AX foci in ≥50 cells in each sample were scored by eye, excluding nuclei with atypical size or morphology or those with very high foci counts (presumably S-phase cells). Confocal images were then subjected to minimal global processing using Adobe Photoshop CS6 (Adobe Systems, San Jose, CA). The second aliquot of each cell population was run on a FACScaliber (BDIS, San Jose, CA) flow cytometer. Data were then analyzed in FlowJo Data Analysis Software (FlowJo, LLC, Ashland, OR). For all experiments, statistical analyses were performed using Prism 6 (GraphPad, Inc. Software, La Jolla, CA). Cytospins of each cell preparation were prepared in triplicate and enumerated, to ensure significance of data generated.
Statistical methods
All experiments detailed herein were repeated a minimum of three times to ensure rigor and reproducibility of the data. In the studies examining the effects of simulated μG on the ability of primary human BM-derived CD34+ cells to differentiate into plasmacytoid and myeloid DCs, experiments were performed with three biological replicates, that is, they were repeated three times, each time using CD34+ cells from a different healthy donor, to ensure any effects observed were representative of human HSC in general, and not donor specific. For all flow cytometric analyses, 20,000 events were captured, and values obtained with the respective isotype control were subtracted from all results to obtain the antibody-specific values that are presented. Data in all figures are presented as the mean ± SEM, and were analyzed with Prism 6 (GraphPad Software, Inc., La Jolla, CA). Statistical significance of differences observed between the various experimental conditions was determined using two-way analysis of variance followed by the Bonferroni–Šidák correction for multiple comparisons. For all analyses, P ≤ 0.05 was considered to be statistically significant. Statistical significance is indicated in Figures with *.
Results and Discussion
μG impairs DNA damage repair in human hematopoietic cells
We first investigated whether conditions of μG impacted upon the ability of primitive human hematopoietic cells to repair DNA damage. To this end, the human KG1a cell line was treated with an acute dose (10 μg/mL) of the radiomimetic drug, bleomycin, to mimic the damaging effects of GCR/SEP radiation. The KG1a cell line was selected to model human HSC, because, in contrast to primary human HSC, these cells can be readily maintained and expanded in culture in the absence of exogenous cytokines/growth factors, yet they exhibit a phenotype (CD34+, Thy1 low, CD38 low, lineage negative, HLA-DR negative) [87] consistent with that of the most primitive primary HSC, and, like primary HSC, they possess self-renewal capacity. These collective properties have led to the widespread use of KG1a cells to model human HSC and response of HSC to ionizing radiation [87 –91].
The treated KG1a cells, and an identical batch of untreated cells, were split in half. Half of the cells from each aliquot were cultured under conditions of normal gravity (1G), and the other half was cultured in the NASA-developed RCCS (Synthecon, Inc.) to create a state of continual freefall, and thereby mimic μG [83]. At 1 and 4 h, half of the cells were harvested from the 1G and μG cultures, and we quantitated the extent of double-strand breaks (DSBs) and the kinetics of repair, using flow cytometry and confocal imaging to monitor the formation and disappearance of γ-H2AX foci. A diagrammatic overview of the experimental design appears in Fig. 1.

Diagrammatic overview of the design of the experiments evaluating the effects of simulated μG on the ability of human HSC-like KG1a cells to repair DNA damage induced by the radiomimetic drug bleomycin. μG, microgravity; HSC, hematopoietic stem/progenitor cells.
To determine the impact conditions of μG exerted on the ability of KG1a cells to repair DSBs induced by the radiomimetic bleomycin, we performed flow cytometric analysis and calculated the ratio of bleomycin-treated cells' median fluorescence intensity (MFI) to untreated cells' MFI under conditions of 1G versus those in μG between the 1- and 4-h time points. We reasoned that, if DNA repair proceeds normally, this ratio should decrease from the 1 to 4-h time point, since γ-H2AX foci disappear as DNA is repaired. When we examined the cells cultured in 1G, this ratio did indeed decrease, by 26%, from the 1-h time point to the 4-h time point, demonstrating successful repair of the bleomycin-induced DNA damage during this time (Fig. 2A). In marked contrast, cells maintained in μG experienced a 20% increase over this same period (Fig. 2B), suggesting they were unable to repair the bleomycin-induced DNA damage, and this damage accumulated during the 4 h incubation.
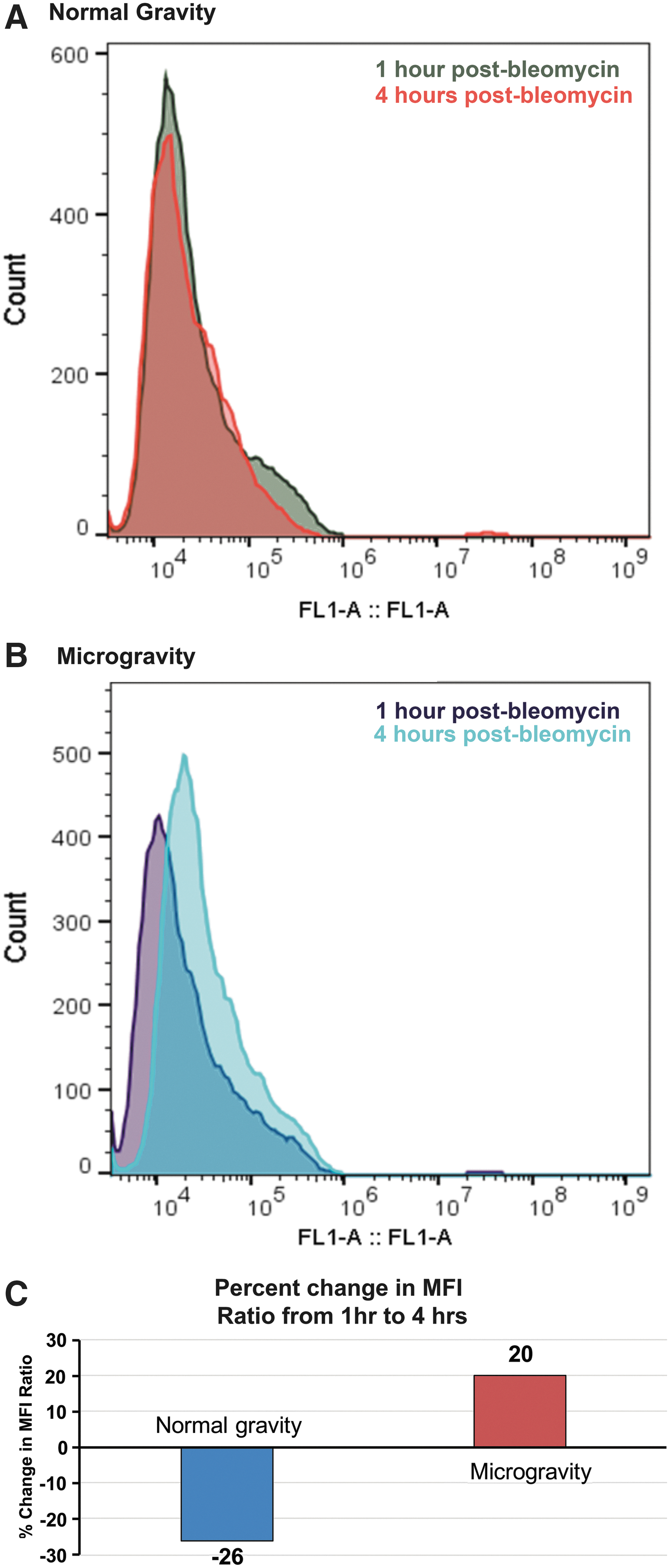
Flow cytometric analysis of γ-H2AX fluorescence in KG1 cells following acute exposure to bleomycin.
Since the “classical” method for assessing DSBs induced by bleomycin or ionizing radiation is to perform immunofluorescence microscopy on cells following staining with an antibody to γ-H2AX, we used this traditional method to confirm that fluorescence events detected by flow cytometry were indeed due to the presence of γ-H2AX foci within the nuclei of the human HSC-like KG1a cells maintained in conditions of simulated μG during the period of DNA repair following bleomycin exposure. As can be seen in Fig. 3, results obtained by confocal microscopy correlated well with the flow cytometry data, confirming a reduction in γ-H2AX foci in the HSC cultured in 1G between 1 and 4 h (Fig. 3A), and an increase during this same time period in HSC cultured in simulated μG (Fig. 3B). A graphical summary of data from these flow cytometric histograms appears in Fig. 3C. Interestingly, just the presence of conditions of simulated μG led to a 20% in the number of nuclei exhibiting γ-H2AX foci that were present in both the bleomycin-treated and untreated cells. These data suggest that μG alone may have the potential to induce DNA damage in human HSC, as has been reported in several other cell types [71,92 –94].

Representative confocal microscopy images of γ-H2AX foci in KG1a cells at 1 and 4 h postbleomycin when cultured in 1G (top) or simulated μG (bottom). γ-H2AX foci are seen in the green channel, and nuclei are counterstained with DAPI, allowing visualization in the blue channel. Magnification and scale are as indicated.
Taken together, these data suggest that HSC DNA damage repair is compromised in conditions of μG, leading to an accumulation of DSBs that cannot be resolved over time, and thus support our hypothesis that conditions of μG may enhance the genotoxic effects of space radiation.
Optimization of cell culture conditions to generate DC
Before examining the impact μG exerted upon DC generation and maturation, we first performed studies to define the optimal culture conditions to drive the differentiation of human CD34+ HSC into the two desired DC subtypes: plasmacytoid (CDw123+CD11c−) and myeloid (CD11c+CDw123−). To accomplish this objective, HSC were grown for a period of 26 days in serum-free or serum-containing medium supplemented with a cocktail of cytokines (please see Materials and Methods section for details) previously shown to promote differentiation along the dendritic lineage [95 –99]. During this time, aliquots of cells were collected and analyzed on days 3, 5, 7, 10, 12, 14, 18, and 21 for: (1) cell number; (2) viability; and (3) the presence of DC, based upon morphology and expression of DC-specific cell surface molecules. Figure 4 shows images of the physical morphology of the DC generated under these two conditions, at day 18 of culture. As can be seen, both serum-free and serum-containing culture conditions promoted the differentiation of human HSC into cells possessing the shape and numerous elongated cytoplasmic protrusions, including stellate processes with a veiled appearance, which are consistent with the published morphological description of DC [66,99,100].
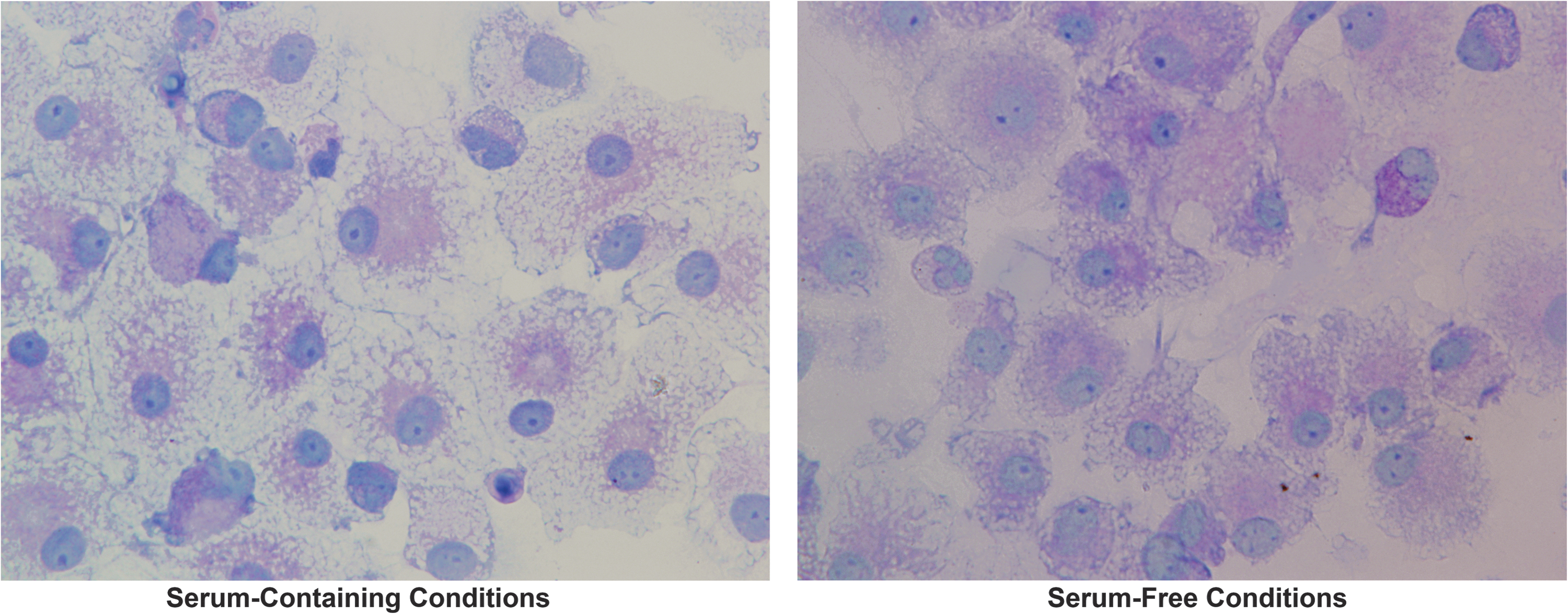
Representative transmitted light microscopy images of DCs generated in vitro from primary adult human BM-derived CD34+ HSC under conditions of normal gravity, in the presence (left panel) or absence (right panel) of fetal bovine serum. Images were acquired on an Olympus IX71 inverted microscope using a 60 × objective. DC, dendritic cell; BM, bone marrow.
Following morphological examination, flow cytometry was utilized to analyze the phenotype of the putative DC generated under these two culture conditions. DC were defined by their expression of HLA-DR, the absence of expression of any of the following lineage-associated markers: CD3, CD14, CD16, CD19, CD20, and CD56, and expression of either CDw123 (plasmacytoid DC) or CD11c (myeloid DC). The percentages of CDw123+ and CD11c+ cells were then compared between the two culture systems to determine whether serum-free or serum-containing conditions resulted in optimal induction of dendritic differentiation, and to ascertain when the peak numbers of DC were generated during the 26-day culture period in each culture system. As can be seen in Fig. 5, using the aforementioned gating criteria to define the two main DC subpopulations, serum-free conditions were significantly better than serum-containing conditions at inducing human HSC to the DC lineage (∼15% vs. ∼2% total plasmacytoid + myeloid DC in serum-free vs. serum-containing cultures, respectively).

Representative dot plots following flow cytometric analysis of DCs generated by differentiating human adult BM-derived CD34+ HSC under conditions of normal gravity, in the absence (top panels) or presence (lower panels) of serum. Cells were stained with anti-HLA-DR-PerCP (Y-axis) to mark all DCs, and PE-labeled antibody to CDw123 or CD11c (X-axis), to identify plasmacytoid or myeloid DCs, respectively.
Impact of conditions of μG on the generation of DC from CD34+ progenitor cells
Having optimized the culture conditions for efficiently generating plasmacytoid and myeloid DC, we next performed studies to test whether culture in the simulated μG conditions provided by the RCCS would affect the ability of human CD34+ HSC (from three different donors) to generate DC under the optimized culture conditions. CD34+ cells were cultured in normal gravity or in the μG, using cytokine-supplemented serum-free media (please see Materials and Methods section for details) and the cells were analyzed at various time points, using flow cytometric analysis. The percentages of CDw123+ (plasmacytoid) and CD11c+ (myeloid) DC that were generated in these two gravitational conditions were then compared, to determine what effect simulated μG had on the growth and differentiation of CD34+ progenitor cells into DCs of both the plasmacytoid and myeloid lineage. Based on these gated percentages, it was determined that higher numbers of myeloid versus plasmacytoid DC were generated, irrespective of the gravitational conditions present (Fig. 6). However, the presence of simulated μG markedly inhibited the ability of human HSC to differentiate into DC (Fig. 6). Under our optimized culture conditions, human HSC continued differentiating into both plasmacytoid and myeloid DC until the last time point assayed (day 21) in normal G. In contrast, under conditions of μG, the minimal differentiation that occurred into plasmacytoid and myeloid DC peaked at days 3 and 10, respectively, after which production of either DC lineage rapidly diminished (Fig. 6). These results thus show that μG can negatively affect the ability of human HSC to differentiate into DC, which play a critical role in the ability of the immune system to recognize and mount an effective response to tumors.

Graphical display of the effects of simulated μG on the ability of primary human adult BM-derived CD34+ HSC to give rise to plasmacytoid (left panel) and myeloid (right panel) DCs under normal gravity (green bars) or simulated μG (blue bars). Data are presented as the mean ± SEM of three independent experiments performed with HSC from three different healthy human donors. * denotes p ≤ 0.05.
A major factor limiting manned spaceflight beyond low-Earth orbit is the risk that exposure to space radiation could increase astronaut cancer morbidity/mortality. We recently demonstrated the seriousness of this risk by showing that human HSC exposed to simulated space radiation generate leukemia in vivo [12]. In addition to radiation, astronauts are exposed to μG during spaceflight, which can alter radiation response and immunity. The data from our present studies provide the first evidence that conditions of μG present during spaceflight may have the potential to act in concert with SEP and GCR radiation to produce deleterious effects on the human hematopoietic system, both by increasing the genotoxicity of SEP and GCR radiation, and by reducing the immune system's ability to recognize and clear emergent malignant cells. These data led us to hypothesize that the risk of leukemia as a result of spaceflight may be greater than previously appreciated, as a result of both increased genomic damage to HSC, and a reduced ability to generate DC needed to recognize and eliminate emergent preleukemic and/or leukemic cells. Our findings on impaired DC generation in μG agree with and extend earlier studies by Savary et al. [68], by considering both plasmacytoid and myeloid DC. Such subset delineation was not possible at the time of this early study, due to the lack of detailed phenotypic information on DC at that time. Our use of primitive CD34+ HSC as a starting population also extends the work of Monici et al. [67], by showing that μG not only impairs the final stages of differentiation of monocytes to DC, but also exerts effects much earlier at the level of stem and progenitor commitment to this important immune lineage. Our findings thus highlight the need for further studies to better define this important risk and to understand the mechanism whereby μG exerts these broad deleterious effects upon the human hematopoietic system. Multi-omics studies are currently underway in our laboratory to begin to define the molecular basis for the impaired ability of human HSC to generate DC when in conditions of μG. It is our ultimate goal to use the knowledge obtained from these omics studies to develop effective means to mitigate this risk to the hematopoietic system, and thereby ensure astronaut safety during prolonged missions.
Footnotes
Acknowledgment
This work was supported by grant # NNX17AE49G from the National Aeronautics and Space Administration (NASA) Space Biology Program.
Author Disclosure Statement
No competing financial interests exist.