
Abstract
Hematopoietic stem and progenitor cells (HSPCs) derived from human induced pluripotent stem cells (hiPSCs) hold great promise for disease modeling, drug screens, and eventually cell therapy approaches. During in vitro differentiation of hiPSCs into hematoendothelial progenitors, the emergence of CD34-positive cells indicates a critical step of lineage specification. To facilitate the monitoring of hematopoietic differentiation of hiPSCs, we established fluorescent reporter cells for the stem and progenitor cell marker CD34. An IRES-GFP (internal ribosome entry site green fluorescent protein) construct was introduced by CRISPR/Cas9 into the 3′ untranslated region of one endogenous CD34 allele. Single-cell clones were generated after excision of the floxed puromycin resistance cassette by Cre recombination and correct insertion was confirmed by genotyping polymerase chain reaction and Southern blot. To validate their functionality, the reporter hiPSCs were in vitro differentiated toward CD34+ cells using the STEMdiff Hematopoietic Kit combined with short-term inhibition of GSK3 (glycogen synthase kinase 3). All cells expressing nuclear GFP were positive for cell surface CD34, thus allowing the direct monitoring of the differentiation of hiPSCs into CD34+ cells either by flow cytometry or confocal microscopy. After fluorescence-activated cell sorting, cells displaying high GFP expression exhibited increased colony-forming potential in the MethoCult colony-forming unit assays as compared with CD34+ cells obtained by magnetic-activated cell sorting. In summary, we have generated functional CD34 GFP reporter hiPSCs, which not only permit label-free separation of HSPCs, but also tracing of the emergence and fate of CD34+ progenitors at the single-cell level.
Introduction
H
Materials and Methods
hiPSC maintenance and differentiation
hiPSCs were purchased from Thermo Fisher Scientific (A18945, hPSCreg TMOi001-A) and were approved for use by The Johns Hopkins University Institutional Stem Cell Research Oversight Committee [19,20]. hiPSCs were maintained on Matrigel hESC-qualified matrix (Corning, VWR International) in TeSR-E8 (STEMCELL Technologies) under hypoxic conditions (3% O2, 5% CO2, 37°C) and passaged with Accutase (Thermo Fisher Scientific) every 3–5 days at split ratios of 1:4 to 1:6. For the first day of culture, the medium was supplemented with 10 μM Rho-associated coiled-coil-containing protein kinase inhibitor (ROCKi) Y-27632 (STEMCELL Technologies).
For spontaneous differentiation into the three germ layers, hiPSCs were seeded in differentiation medium (Dulbecco's modified Eagle's medium, 20% knockout serum replacement, 0.1 mM nonessential amino acids, 2 mM
For hematopoietic differentiation, the STEMdiff Hematopoietic Kit (STEMCELL Technologies) was used according to the manufacturer's protocol (Document #29768) with minor changes. In brief, 4 days before differentiation, hiPSCs (2,000 per well of a Matrigel-coated 12-well plate) were seeded as small clumps in TeSR-E8 with 10 μM ROCKi. Two days later, the medium was changed to TeSR-E8 without ROCKi and the cells cultivated for another 2 days. For differentiation, STEMdiff Hematopoietic Kit Medium A supplemented with 3 μM CHIR99021 (Miltenyi Biotec) GSK3 (glycogen synthase kinase 3) inhibitor (GSKi) was applied to the hiPSC colonies. One day later the medium was changed to Medium A without GSKi. On day 3, a switch to Medium B and thereafter half media changes were performed according to the manufacturer's instructions.
MethoCult colony-forming unit (CFU) assays were performed with MethoCult H4435-enriched medium (STEMCELL Technologies) according to the manufacturer's protocol (Document #28404). Briefly, MethoCult was thawed at +4°C overnight and mixed. The HSPCs floating in the supernatant of day 12 hematopoietic differentiation cultures were harvested and dead cells were depleted using the magnetic-activated cell sorting (MACS) Dead Cell Removal Kit (Miltenyi Biotech). CD34+ cells were MACS purified with the human CD34 UltraPure MicroBead Kit (Miltenyi Biotech) and GFP negative, medium, and high cell populations were flow sorted (FACSAria Fusion; BD Biosciences). After washing with Dulbecco's phosphate-buffered saline (DPBS; Thermo Fisher Scientific) 2,000 cells were resuspended in 300 μL Iscove's modified Dulbecco's medium (Thermo Fisher Scientific) containing 2% defined fetal bovine serum (Hyclone, VWR International), gently mixed with 3 mL MethoCult and plated into six-well plates. Cord blood (CB) StemPro CD34+ cells (A14058; Gibco, Thermo Fisher Scientific) were thawed according to the manufacturer's instructions, washed in DPBS, and 2,000 cells were seeded as described above. Cells were incubated in a humidified chamber at 37°C and 5% CO2 for 13 days. Total colony numbers were compared with that of MACS CD34+ control using one-way repeated measures analysis of variance (ANOVA) with Dunnett's post hoc test in GraphPad Prism 5.
Genetic engineering
The CRISPR/Cas9 vector CRISPR 920657437-pGS-gRNA-Cas9-Neo was synthesized by GenScript (protospacer sequence: GTTCCTGTATTGCGGCAGAG). The selected single-guide RNA targeted the 3′ UTR of the CD34 locus in hiPSCs and exhibited at least four mismatches to putative off-targets in the human reference genome (
hiPSCs were electroporated using an Amaxa Nucleofector 2b (program A-023) with the Human Stem Cell Nucleofector Kit 2 (Lonza) and 2.5 μg each of the donor and the CRISPR/Cas9 vector or with 5 μg of Cre recombinase vector after selecting twice with 0.5 μg/mL puromycin (Sigma-Aldrich) for 3 days each. Following expansion of the surviving cells, 4,000 single cells were seeded on 10-cm dishes coated with Synthemax II (0.025 mg/mL in water; VWR International). Emerging colonies were picked manually and expanded. Details of targeting and cloning efficiencies for the knockin and Cre-recombination experiments are provided in Supplementary Table S1 (Supplementary Data are available online at
Genotyping
Genotyping polymerase chain reactions (PCRs) of individual clones were performed using three primers, genomic DNA lysates and HotStarTaq DNA polymerase (Qiagen). Primer sequences are listed in Supplementary Table S2. PCR products obtained from the delta allele were purified using the QIAquick PCR Purification Kit (Qiagen) and Sanger sequenced (Microsynth, Switzerland).
For Southern blotting, genomic DNA was isolated with the QIAamp DNA Blood Mini Kit (Qiagen), 10 μg of DNA was digested with EcoRI-HF (New England Biolabs), run on 0.7% agarose gels and alkaline transferred to nylon membranes (Hybond-N+; Amersham). For probe detection, the Chemiluminescent Nucleic Acid Detection Module Kit (Thermo Fisher Scientific) was used in combination with biotin-11-dUTP (Thermo Fisher Scientific)-labeled PCR probes. Probe primer sequences are listed in Supplementary Table S2.
Karyotyping (Q-banding) and short tandem repeat (STR) analysis (deposited at hPSCreg [19]) for cell authentication were performed by Labdia Labordiagnostik (Vienna, Austria) and Microsynth, respectively, according to standard procedures.
Reverse transcription quantitative PCR
Samples were collected from three consecutive passages every 3–4 days. Total RNA was isolated with TRIzol reagent (Thermo Fisher Scientific) and cDNA was synthetized from 2 μg RNA using Moloney murine leukemia virus reverse transcriptase (Promega). Reverse transcription quantitative PCRs (RT-qPCRs) were performed using iTaq SYBR Green Supermix (Bio-Rad Laboratories) on a 7500-Fast Real-Time PCR system (Applied Biosystems). Primer sequences are listed in Supplementary Table S2. Expression levels were normalized to GUSB and wild-type hiPSC and statistically analyzed using one-way repeated measures ANOVA with Dunnett's post hoc test.
Western blotting
Floating HSPCs were harvested by centrifugation of the supernatant and pooled with adherent cells that were detached by Accutase treatment for 3–10 min. The acute lymphoblastic leukemia cell line, MOLT-4 (DSMZ, German Collection of Microorganisms and Cell Cultures), used as CD34+ positive control, was harvested by centrifugation. Cells were washed once in DPBS and lyzed in high salt buffer (10 mM Tris-HCl pH 7.5, 400 mM NaCl, 0.5% NP-40, and 0.3% Triton X-100; all from Merck) containing 17.5 μg/mL phenylmethylsulfonyl fluoride, and 1 μg/mL each of aprotinin, leupeptin and pepstatin A (all from Roche). Protein lysates were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes (GE Healthcare). To control for protein loading, blots were Ponceau S (Sigma-Aldrich) stained and blocked with 1× blocking reagent (Roche) in Tris buffered saline (TBS). Antibodies (Supplementary Table S3) were diluted in TBS containing 0.5 × blocking reagent and 0.1% Tween-20, incubated overnight at 4°C, and detected using the Odyssey system (LI-COR Biosciences).
Flow cytometry (fluorescence-activated cell sorting) analysis
On days 6, 9, and 12 of differentiation, cells were harvested for fluorescence-activated cell sorting (FACS) analysis. Adherent cells (days 6 and 9) were detached by Accutase treatment for 3–10 min and HSPCs floating in the supernatant (days 9 and 12) were harvested by centrifugation. For analysis of day 9 cells, the floating and adherent fractions were pooled (bulk). Flow cytometry was performed on an LSRFortessa (BD Biosciences) and data analyzed with FlowJo V10.2 software (Tree Star). Antibodies (Supplementary Table S3) were diluted in DPBS containing 0.1% bovine serum albumin (BSA; Sigma-Aldrich) and dead cells stained with 4′,6-diamidin-2-phenylindol (DAPI; Sigma-Aldrich).
Microscopy
Confocal images were acquired on a Leica TCS SP8 equipped with an HC PL APO CS2 40x/1.10 water immersion objective. For pluripotency marker detection cells were fixed in 1% formaldehyde (Merck) in DPBS and permeabilized with 0.2% Triton X-100 in DPBS. Antibodies (Supplementary Table S3) were diluted in DPBS containing 2% BSA and 0.2% Triton X-100. Nuclei were counterstained with 2 μg/mL DAPI.
Spontaneously differentiated hiPSCs were fixed using 4% formaldehyde solution (HistoFix; Roth) for 10 min at room temperature and subsequent permeabilization was performed for 20 min (3% goat serum, 0.2% Triton X-100). Antibodies (Supplementary Table S3) were diluted in 10% goat serum.
Double positivity for GFP and CD34 was demonstrated by live cell staining of differentiation cultures on day 12. CD34 antibody (1:1,000; Supplementary Table S3), Hoechst 33342 (5 μg/mL; Thermo Fisher Scientific) and 1 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; Thermo Fisher Scientific; pH 7.2–7.5; final concentration 1 mM) were added to the culture medium, the cells incubated for 30 min, and imaged at 35°C.
Emergence of GFP+ progenitor cells during hematopoietic differentiation was monitored by confocal time-lapse microscopy between day 5 and 6 of differentiation (15-μ 24-well plate; Ibidi) in 1 mM HEPES buffered differentiation medium. Z-stack images were acquired every 4 min and time-lapse serial images of one selected layer were converted to an Audio Video Interleave (.avi) file (Supplementary Movie S1).
CFU assay colonies were counted and scored on day 13 on a 2 × 2 mm grid with a Leica M125 stereo microscope. Bright field images of colony types were taken on a Zeiss Axiovert 135 microscope equipped with a 5× phase contrast plan objective and a Canon EOS 600D camera.
Wright-Giemsa modified stain (Sigma-Aldrich) was performed on cytospins of picked CFU assay colonies on day 13 and pictures were acquired on a Nikon Labophot-2 microscope equipped with a 50 × oil immersion plan objective, a Jenoptik ProgRes C3 camera, and ProgRes CapturePro v.2.8.0 software (Jenoptik).
Results and Discussion
Generation of CD34 GFP reporter hiPSC lines
To generate reporter hiPSCs for the stem cell and progenitor marker CD34, the CRISPR/Cas9 system was used to knockin an IRES-GFP-NLS-PuroR construct into the 3′ UTR of the endogenous CD34 locus (Fig. 1A). Following puromycin selection and the generation of single-cell clones, the correct insertion of the construct was confirmed by two flanking genotyping PCRs (data not shown) in 18 of 21 clones (85%; Supplementary Table S1). A clone harboring one floxed reporter allele (fl) was subjected to Cre recombinase-mediated resistance cassette excision. Again, single-cell clones were generated and analyzed for the correct insertion of the IRES-GFP cassette by genotyping PCRs (Fig. 1B) and Southern blotting (Fig. 1C). The wild-type CD34 allele was detectable in 69 clones and thereof 38 (55%; Supplementary Table S1) also carried the expected delta reporter allele (exemplary clones are shown in Fig. 1B). Southern blotting using probes against GFP (Fig. 1C) and the ampicillin resistance present in the vectors (no band detected; data not shown) excluded random genomic integration events in several selected clones. Clone 3G6 (hPSCreg TMOi001-A-1) [19] was chosen for further analysis and PCR products targeting the delta allele were Sanger sequenced. The junction sequences between the CD34 3′ UTR and the IRES as well as the remaining loxP site are shown in Fig. 1D. The isogenicity of the parental and the reporter cell lines was confirmed by STR analysis and karyotyping; the latter also excluded the presence of gross chromosomal aberrations (Fig. 1E). Altogether, these data show that the isogenic hiPSC reporter line contains an IRES-GFP cassette knocked into the CD34 locus without off-target events.
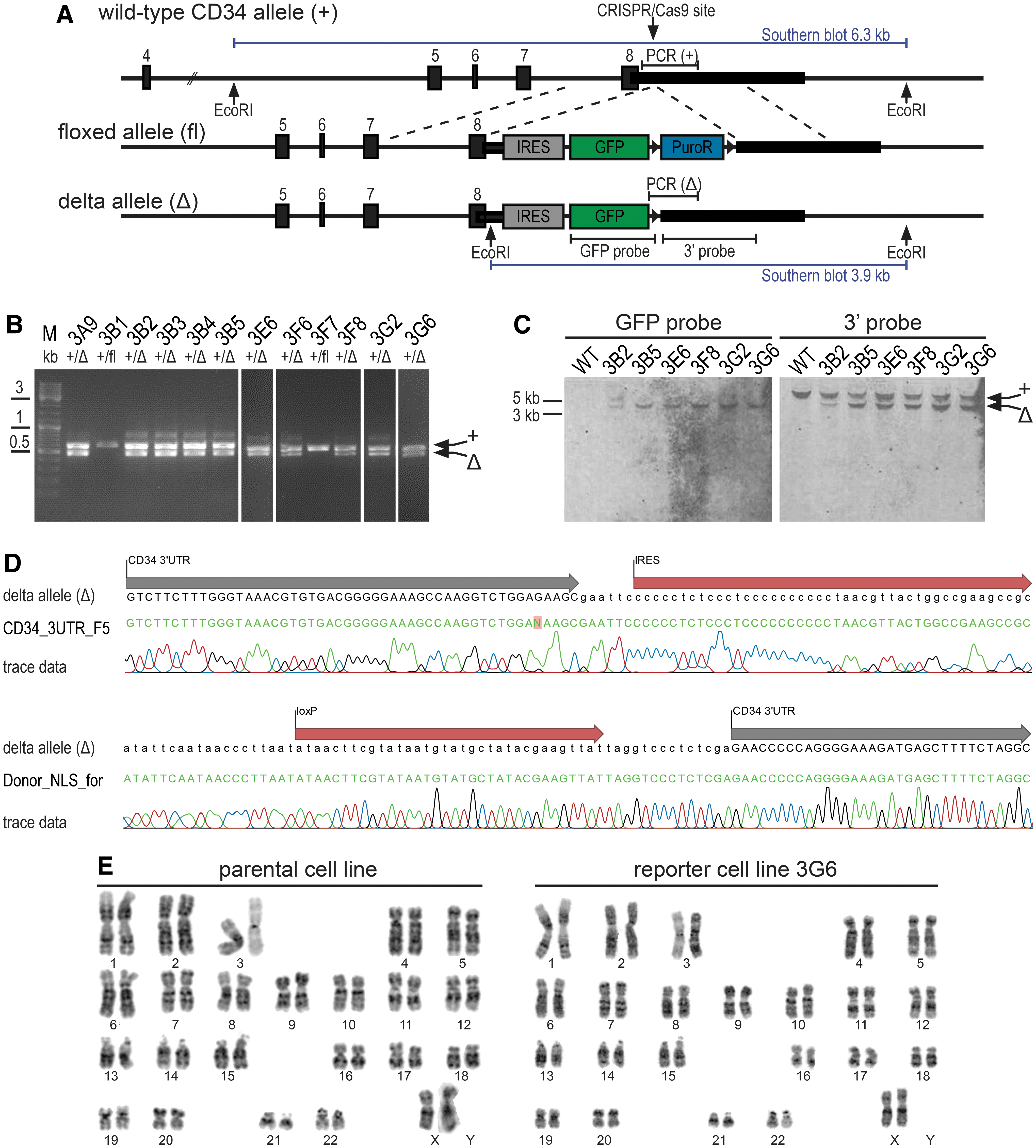
CRISPR/Cas9-mediated generation of CD34 GFP reporter hiPSCs.
Functional characterization of CD34 GFP reporter hiPSCs
To exclude detrimental effects of the genetic engineering procedure on hiPSCs, we assessed whether they remain pluripotent and are able to spontaneously differentiate [25]. First, the expression of the pluripotency markers OCT4, KLF4, NANOG, and SOX2 was analyzed by RT-qPCR, which showed that the mRNA levels of several reporter clones, measured at three different time points of cultivation, were not significantly different from those of parental hiPSCs (Fig. 2A). To confirm these results on the protein and single-cell level, OCT4, NANOG, and SOX2 were further analyzed by indirect immunofluorescence and confocal microscopy, demonstrating that these proteins were expressed at similar levels in both the nuclei of the parental hiPSC and the representative reporter line 3G6 (Fig. 2B). Upon spontaneous differentiation of the reporter hiPSCs, using embryoid body formation, cells of the three germ layers were detected by staining for endodermal, mesodermal, and ectodermal marker proteins, albumin, smooth muscle actin, and βIII tubulin, respectively (Fig. 2C).
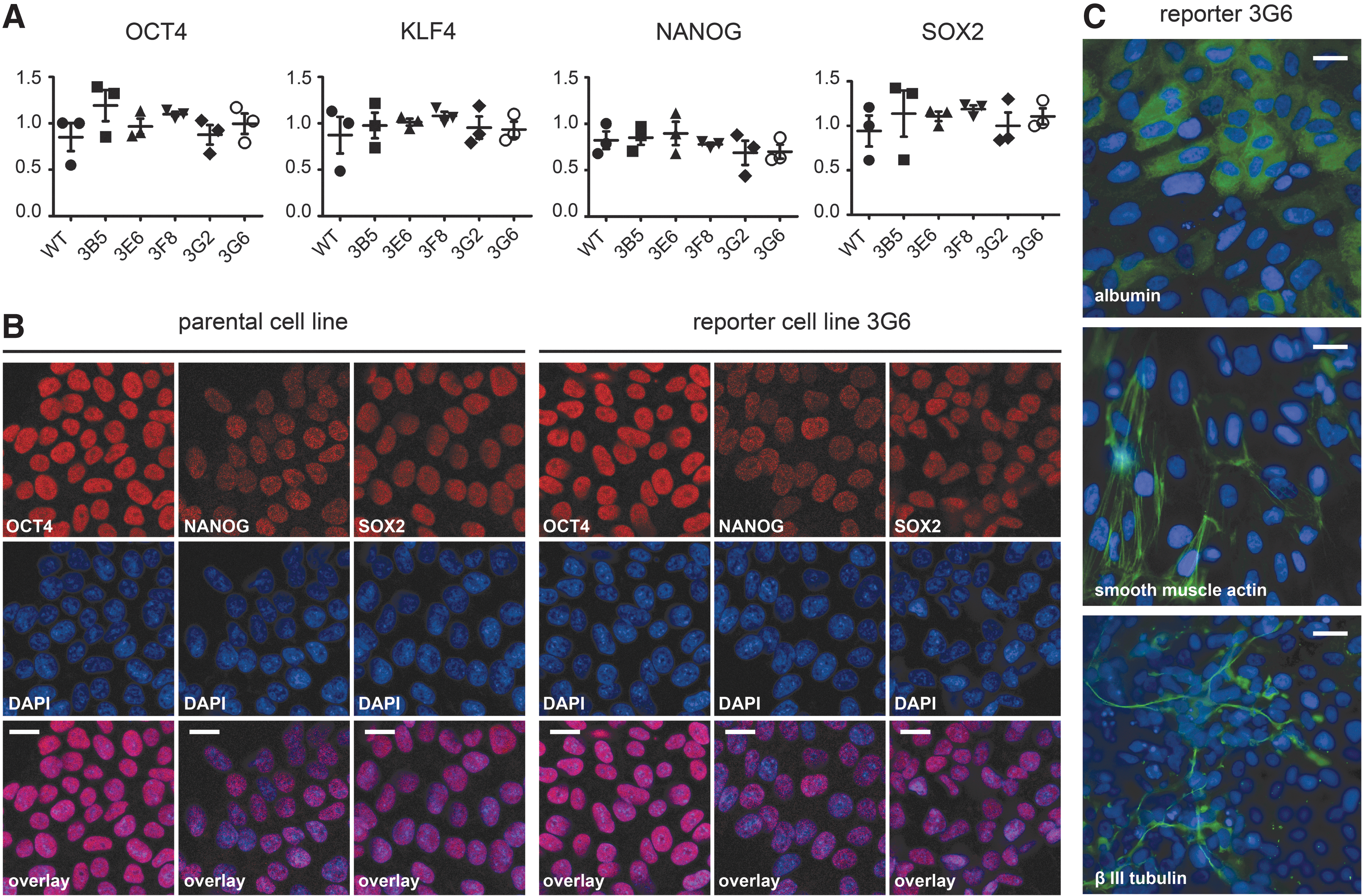
Analysis of the pluripotency and differentiation capacity of CD34 GFP reporter hiPSCs.
To validate the functionality of the reporter, the genome-edited hiPSCs were in vitro differentiated toward HSPCs expressing CD34, a commonly used marker for defining hematopoietic stem cells [11,12]. For this purpose, we employed the STEMdiff Hematopoietic Kit and to further promote hematopoiesis through stimulation of the Wnt–β-catenin signaling pathway, at the beginning of the differentiation protocol, added the GSK3 inhibitor CHIR99021 [7,13,14,26] to the cultures for 1 day (Fig. 3A). Cells were harvested at different time points of the differentiation process and CD34 protein expression was readily detectable by western blotting as early as on day 6 of differentiation in both the parental and the reporter cell line (Fig. 3B). Differentiation toward HSPCs was confirmed by flow cytometric analysis showing that the majority of the differentiated cells at day 12 were positive for cell surface CD34, CD43, and CD45 (reporter line 3G6, Fig. 3C; parental line, Supplementary Fig. S1), the latter markers being specific for hematopoietic cells [27,28].

In vitro differentiation of parental and genome-edited hiPSCs.
Furthermore, on days 6, 9, and 12 of differentiation, a fraction of the reporter line cells showed expression of GFP (Fig. 3C). More specifically, all reporter line cells exhibiting GFP expression displayed cell surface CD34, and by day 12 also CD43 and CD45 (Fig. 3C). An independent experiment using a second reporter clone (3B5) yielded similar results (data not shown). To further confirm the faithfulness of the GFP reporter, cells were stained with an anti-CD34 antibody on day 12 of differentiation directly in culture and imaged by confocal microscopy. While the parental cells were GFP negative and a proportion of the cells stained positive for CD34 (Fig. 3D, upper panel), in the reporter line GFP localized, as expected, exclusively to the nuclei of surface CD34 protein-expressing cells (Fig. 3D, lower panel) and no false GFP-positive CD34-negative cells were detected.
The presence of CD34-positive, but GFP-negative cells (Fig. 3D, upper panel, lower right quadrants) and the overall weak GFP expression may have several reasons. Since it has been demonstrated that IRES-dependent reporter gene expression is usually significantly lower than that of cap-dependent gene expression [29 –31], it may be partly explained by the attenuated IRES-mediated GFP translation from only one allele. Additionally, a lag in GFP folding and chromophore maturation [32] may cause a delay in GFP as compared with CD34 expression. However, IRES-mediated expression of the reporter has the clear advantage of leaving the preceding CD34 protein expression and its structure completely unperturbed. Other approaches, such as an exchange of the gene of interest for the reporter, and direct or 2A self-cleaving peptide-linked fusion achieve higher reporter levels, but can affect either the expression level or the structure of the targeted protein and may thereby interfere with its functionality [33,34].
Practical application of the CD34 GFP reporter
As no false-positive cells were detected, using GFP as a surrogate for CD34 expression appeared to represent a valid approach. To demonstrate the benefits of the reporter cells, we next used GFP expression to sort HPSCs (Fig. 4A) for CFU assays in MethoCult semisolid medium (Fig. 4B–D). First, the hematopoietic potential of MACS-enriched CD34+ cells obtained after 12 days of differentiation of hiPSCs was compared with that of freshly thawed CB CD34+ cells (Fig. 4B). CD34+ cells derived from the parental hiPSCs showed lower colony-forming potential than CB CD34+ cells and produced myeloid CFUs of predominant macrophage lineage at the expense of erythroid differentiation (Fig. 4B). Such lineage bias is often observed with in vitro differentiated HSPCs [35 –37].
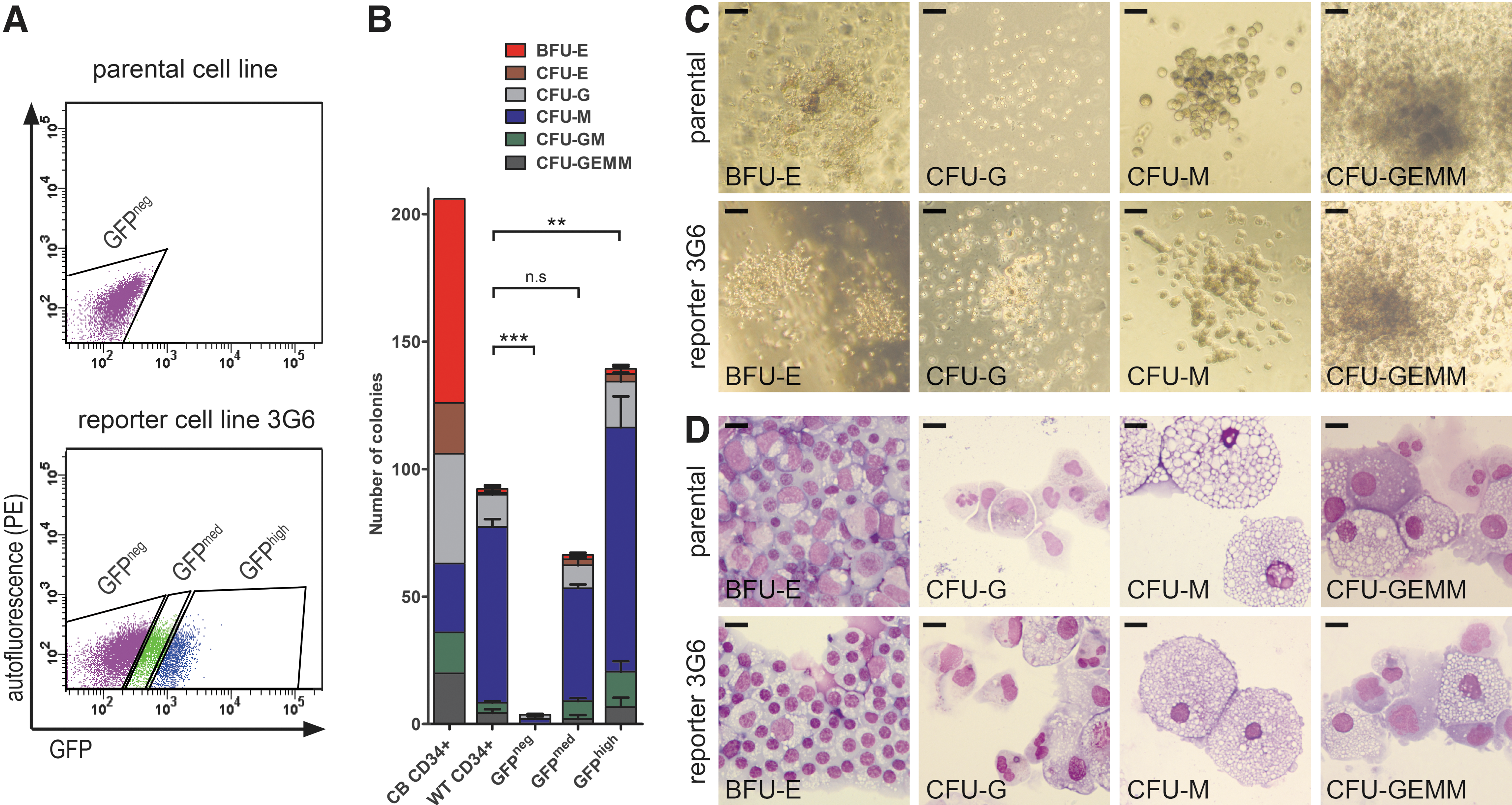
Colony-forming potential of CD34+ HSPCs.
Next, we compared the CFU potential of FACS-purified GFP+ with that of MACS-enriched CD34+ cells. While GFP-negative cells sorted from reporter hiPSCs demonstrated an almost complete lack of colony-forming potential, cells expressing medium and high levels of GFP exhibited the same bias toward macrophages as observed for parental hiPSC-derived CD34+ cells (Fig. 4A-D). Notably, a clear increase of CFUs was observed with the GFP high compared with the GFP medium population as well as the MACS-enriched CD34+ fraction, highlighting the feasibility of using GFP expression as highly specific marker for FACS-mediated hematopoietic progenitor cell enrichment.
While MACS MicroBeads should neither activate cells nor saturate cell surface epitopes, it has nevertheless been shown that iron beads may cluster at the cell surface and are sometimes endocytosed in pinocytic vesicles [38]. Moreover, cells, unspecifically binding either directly to the beads or indirectly by interacting with CD34+ cells, may be retained in the positively labeled fraction. Although negative cells can be effectively excluded by using FACS combined with antibody staining, antibody-mediated receptor crosslinking or internalization may trigger signaling events in the positive fraction and the labeling and washing steps may constitute additional cellular stress factors. In contrast, sorting of GFP+ cells circumvents such antibody-related issues, and, compared with MACS-enrichment of the entire population of CD34+ cells, permits at least an indirect distinction of CD34 expression levels and the enrichment of high-quality progenitors. Furthermore, the GFP reporter may even be utilized to identify progenitor cells within 3D structures (Supplementary Fig. S2), which are generally not accessible to direct antibody staining and to track the emergence of GFP+ cells in differentiation cultures by live cell confocal imaging (Supplementary Movie S1).
In conclusion, we generated CD34 GFP reporter hiPSCs, which considerably facilitates the straightforward monitoring and sorting of CD34+ HSPCs emerging during in vitro differentiation and we demonstrated that the GFP expression levels, as a surrogate for CD34, positively correlate with the colony-forming potential of hematopoietic progenitors.
Footnotes
Acknowledgments
The authors thank Regina Grillari for advice on hiPSC culture, Martin Distel for help with confocal microscopy, Sören Mai and Bettina Nocker for karyotype analysis, and Dieter Printz and Elke Zipperer for expert advice on flow cytometry. This work was funded by the Austrian Research Promotion Agency FFG (grant no. 843456).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.