
Abstract
Mesenchymal stem cells (MSCs) are widely investigated as potential therapeutic agents due to their potent immunomodulatory capacity. Although specific mechanisms by which MSC acts on immune cells are emerging, many questions remain, including the potential of extracellular vesicles (EVs) to mediate biological activities. Canine MSCs are of interest for both veterinary and comparative models of disease and have been shown to suppress CD4pos T cell proliferation. The aim of this study was to determine whether EV isolated from canine Wharton's jelly-derived MSC (WJ-MSC EV) suppresses CD4pos T cell proliferation using biochemical mechanisms previously ascribed to soluble mediators [transforming growth factor beta (TGF-β) and adenosine]. WJ-MSC EV exhibited mode of 125 nm diameter, low buoyant density (1.1 g/mL), and expression of EV proteins Alix and TSG101. Functionally, EVs inhibited CD4pos T cell proliferation in a dose-dependent manner, which was absent in EV-depleted samples and EVs from non-MSC fibroblasts. EV suppression of CD4pos T cell proliferation was inhibited by a TGF-βRI antagonist, neutralizing antibodies to TGF-β, or A2A adenosine receptor blockade. TGF-β was present on EVs as latent complexes most likely tethered to EV membrane by betaglycan. These data demonstrate that canine WJ-MSC EV utilizes TGF-β and adenosine signaling to suppress proliferation of CD4pos T cell and will enable further investigation into mechanisms of immune cell modulation, as well as refinement of WJ-MSC and their EVs for therapeutic application.
Introduction
Connective tissue surrounding the umbilical cord vasculature, known as Wharton's jelly (WJ), is a rich source of mesenchymal stem cells (WJ-MSCs) that show promise for regenerative medicine applications [1 –5]. The immunomodulatory capacity of WJ-MSC has served as the major premise for the development of WJ-MSC for cell therapy [4]. WJ-MSC exhibits comparable or superior immunomodulatory potential to that of adipose tissue-derived MSC (AT-MSC) and bone marrow MSC (BM-MSC) [6 –13], although these results are not universal [24]. A wide range of immunologic processes are mitigated by WJ-MSC, including suppression of T cell proliferation [9 –11,15 –17], promotion of a T regulatory cell phenotype [7,13,14], and polarization of macrophages toward an anti-inflammatory M2 phenotype in vitro [18] and in vivo [19 –21]. Immunomodulation has been observed with or without direct contact between MSC and immune effector cells [10,15].
The realization that many biological activities reside in cell-free preparations of MSC supernatant has ignited interest in components of the MSC secretome. Included within the MSC secretome are extracellular vesicles (EVs), nanoscale cellular products secreted by all cell types that contain RNA, protein, and lipids that recapitulate many biological properties previously attributed to their parent cells or the cells' soluble secretions [22 –24]. Recent studies have focused on the potential for MSC EV rather than the cells themselves as therapeutic agents or vectors [19,25 –31], including one clinical application of MSC EV for treatment of severe graft-versus-host disease [32]. Recent studies have demonstrated that EVs released from WJ-MSC efficiently suppress T lymphocyte proliferation [17,33] and polarize macrophages toward an anti-inflammatory cell type [19,34]. However, these findings were not corroborated by other studies [18,35 –37], and treatment of MSC with interferon gamma (IFN-γ) was required to induce production of EV with immunomodulatory properties in one report [38]. To better understand the therapeutic potential or limitations of MSC, there is a need to further evaluate the mechanisms of immunomodulation in different types or subtypes of MSC EV.
There is insufficient knowledge about the molecular (biochemical and genomic) mechanisms by which MSC EV exerts putative immunomodulation; in contrast, a number of immunosuppressive protein ligands have been identified from tumor-associated EV [39 –45], which may serve as a framework to understand MSC. Similar to tumor EV, immunomodulatory proteins are associated with MSC EV (IDO, NO, PGE2, TGF-β1, adenosine, IL-10) [29,38,46] although their functional relevance in association with immune cells or disease states remains unclear. Certain ligands (PD-L1, TGF-β1, and Galectin) can be transferred by murine MSC-derived EV to lymphocytes inducing autocrine production of immunosuppressive interleukin-10 (IL-10) and transforming growth factor beta-1 (TGF-β1) [47,48]. For example, BM-MSC-derived EVs were shown to act as vectors for mature protein and mRNA of TGF-β1 to peripheral blood mononuclear cells (PBMCs) from type 1 diabetes, reducing antigen-specific T cell responses [49]. In contrast, embryonic stem cell-derived MSC EV only weakly suppressed T cell function, rather exerting their effects indirectly by inducing an M2 phenotype in cultured monocytes [28]. Hence, the paradigm of MSC EV immunomodulation may also involve networks of signaling between immune effector cells, and these mechanisms may also diverge for MSC versus their shed EVs [50]. These observations are particularly germane to testing hypotheses using in vitro immune assays that rely upon PBMCs to elucidate EV mechanisms of immunosuppression [17].
This study investigates canine WJ-MSC and their secreted EVs. Canine spontaneous models of disease are an important bridge between laboratory and human studies of therapeutic mechanisms [51,52]. Refinement in our understanding of MSC mechanisms in the canine species will further enable their application as preclinical models.
In this study, we hypothesized that canine WJ-MSC EV suppresses peripheral CD4pos T lymphocytes consistent with the effects of their parent WJ-MSC. Furthermore, we predicted that WJ-MSC EV utilizes TGF-β or adenosine signaling, both of which are known to be important components to the immunomodulatory mechanisms of canine AT-MSC and BM-MSC [53,54]. Using WJ-MSC isolated from tissues of healthy canine patients undergoing Cesarean section, EVs were found to suppress mitogen-activated canine CD4pos cells, with the effect partially mediated by TGF-β and adenosine. Findings herein confirm a role for immunomodulation by WJ-MSC EV and will enable investigation of detailed mechanisms and utility of WJ-MSC EV for treatment of canine diseases.
Materials and Methods
WJ-MSC: isolation and characterization
Tissue collection, MSC isolation, and culture
The study was approved by the Clinical Sciences Research Committee of Cummings School of Veterinary Medicine of Tufts University. Privately owned healthy female dogs between 1 and 10 years old from various breeds (Supplementary Table S1; Supplementary Data are available online at
Quantitative real-time polymerase chain reaction
Total RNA was isolated from WJ-MSC using the RNAeasy Kit (Qiagen) as per manufacturer's instructions. RNA concentrations and quality were determined with the RNA 6000 Nano Assay Kit and the Bioanalyzer 2100 (Agilent Technologies). Complimentary DNA was generated with the RT2 First Strand Synthesis Kit (Qiagen) and heat cycled at 42°C for 15 min followed by 95°C for 5 min before placing on ice. Five microliters of cDNA was then mixed with RT2 SYBR Green Mastermix, RNase free water, and 10 μM primer. mRNA expression of CD73 (PPF01104A; Qiagen), CD44 (PPF00491A; Qiagen), MHCII (PPF01028A; Qiagen), CD45 (PPF10210A; Qiagen), CD34 (PPF00586A; Qiagen), and CD90 and CD105 (Invitrogen) (Supplementary Table S2) was measured. HPRT (hypoxanthine phosphoribosyltransferase 1 gene) and RPS19 (ribosomal protein S19 gene) were measured as housekeeping genes, using the mean Ct for these two genes for normalization of Ct data.
Flow cytometry
Cells were incubated with primary antibodies in 5% FBS for 30 min on ice. Antibodies used were anti-CD34-PE (mouse anti-dog clone 1H6; AbD Serotec), CD44-APC (rat anti-dog clone YKIX337.8.7; AbD Serotec), CD45-APC (rat anti-dog clone YKIX716.13; AbD Serotec), MHCII-FITC (rat anti-dog clone YKIX334.2; AbD Serotec), and CD90-APC (rat anti-dog clone YKIX337.217; eBioscience). 7-AAD (Becton Dickinson) was applied to all samples for exclusion of dead cells. After gating on viable cells, phenotype was determined by comparing histograms of the stained samples to an isotype control. Samples were evaluated using an Accuri C6 (BD Biosciences), with a minimum of 100,000 events analyzed using CFlow Plus v. 1.0.208.2.
Trilineage differentiation
Cells were plated at passage 4 at 1.6 × 104 cells/cm2 in cAlpha-MEM and changed to differentiation or control media upon reaching 80% confluence. Dulbecco's modified Eagle's medium (DMEM) high glucose (Gibco) with 2 mM
WJ-MSC EV: isolation and characterization
EV isolation from serum-free culture by stepwise ultracentrifugation
WJ-MSC was thawed and seeded at low density (∼6,000 cells/cm2) in cAlpha-MEM. Once 70% confluent, cells were transitioned to serum-free defined chemical media (DCM) modified from Lai et al. [67], composed of DMEM supplemented with 25 μM HEPES (Life Technologies), penicillin–streptomycin and
Particle enumeration and size distribution using nanoparticle tracking analysis
Samples were analyzed using a NanoSight N300 unit (Malvern) equipped with a 488 nm (blue) laser module and Nanoparticle Tracking Analysis (NTA) 3.0 software. Samples were diluted in sterile PBS to a concentration of 1–10 × 108 particles/mL for analysis. Specific NTA settings were optimized for each sample, with fixed settings of temperature (23°C), screen gain (1.0), infusion flow rate (5 μL/min), and camera level set at 12–14 depending on sample characteristics. Five videos were recorded for each sample (30–120 s video length) with all settings remaining constant within each sample source to minimize variation. Detection threshold was set to 5 using auto blur and auto max jump distance settings, with a minimum analysis of 200 valid tracks per video and a minimum of 1,000 valid tracks per sample. The NTA unit was periodically evaluated for accuracy of size determination using polystyrene beads of known size (100 and 200 nm).
Transmission electron microscopy
EVs were diluted 1:100 in PBS and adhered to copper mesh SPI 200 SuperGrids™ (SPI). Uranyl acetate (1%) in deionized water was used for negative staining. Images were obtained at 87,000× and 135,000× magnification using an FEI Tecnai™ Spirit 12 electron microscope (Thermo Fisher Scientific).
Density gradient separation of WJ-MSC EV
Gradients were constructed with iodixanol (OptiPrep™ Density Gradient Medium, 60% aqueous preparation; Sigma) diluted in gradient buffer containing 0.25 M sucrose (Sigma), 10 mM Tris (Fisher Scientific), and 1 mM EDTA (MediTech), at pH 7.4. EV samples were prepared to a final concentration of 0.25 M sucrose and 1 mM EDTA in a final volume of 500 μL and mixed with 1 mL of 60% iodixanol to yield 1.5 mL of sample in 40% iodixanol and loaded in the bottom of Ultra-Clear™ 0.5- by 2-inch centrifuge tubes (Beckman Coulter). Iodixanol solutions were layered on top as follows: 1.2 mL of 30%, 1.2 mL of 20%, and 1.4 mL of 10%. A control gradient (using gradient buffer in place of the EV sample) was prepared in the same manner. Gradients were ultracentrifuged at 350,000 g for 2 h at 4°C using a SW 55 Ti rotor (k-factor 50.3) to float EV up to their buoyant density. Following centrifugation, 8 fractions of 625 μL were removed from the top of the tube. Fraction density was calculated by measuring absorbance at 340 nm and comparing the values to a standard curve prepared using varying iodixanol concentrations in gradient buffer. Fractions were concentrated to a volume of 175 μL with Ultracel®-10K regenerated cellulose 10,000 kDa molecular weight cutoff (MWCO) centrifugal filters (Amicon). Particle counts (NTA) and protein concentration (bicinchoninic acid assay [BCA]; Pierce) were established for the concentrated fractions. Immunoblot to detect TSG101 was performed by loading equal volumes of the concentrated fractions using the protocol described below.
Immunoblots
WJ-MSCs were lysed, and proteins were solubilized with m-PER (Thermo Fisher Scientific). Protein from cell extracts or EV was quantified by BCA as per manufacturer's protocol (Pierce). Immunoblotting was performed using the iBlot™ system (Thermo Fisher Scientific) according to manufacturer's instructions. Proteins were electrophoresed in Bolt™ 4%–12% Bis-Tris gels (Invitrogen) with Bolt MES SDS (sodium dodecyl sulfate) running buffer (Invitrogen) and transferred onto polyvinylidene fluoride membranes. The iBind™ Flex system and solution (Thermo Fisher Scientific) was used for blocking and antibody application. Antibodies used were mouse anti-TSG101 (clone 51/Tsg101; BD Biosciences) at 1:1,000, rabbit polyclonal anti-PDC6I (Alix; Abcam) at 1:1,000, and rabbit polyclonal anti-calnexin (Abcam) at 1:1,000 dilutions. Biotinylated conjugated horse anti-mouse antibody (Vector Laboratories) or biotinylated goat anti-rabbit (Vector Laboratories) at 1:40 dilution was used as a secondary antibody. Detection was performed using the VECTASTAIN ABC Kit (Vector Laboratories) followed by the DAB Peroxidase Substrate Kit (Vector Laboratories). Controls included lysates of HeLa cells, Mardin-Darby Canine Kidney (MDCK) cells, and dog brain tissue (isolated from donated tissue from euthanized animals following client approval), as well as MDCK EVs isolated and prepared in the same manner as the WJ-MSC EVs.
PBMC suppression assays
PBMC isolation and proliferation assay
Twelve healthy adult purpose-bred Beagle dogs (five neutered males, seven spayed females, ages 1–4) housed at the Laboratory Animal Medicine department at the Cummings School of Veterinary Medicine at Tufts University served as blood donors under an approved protocol through the Institutional Animal Care and Use Committee at Tufts University. Healthy status was confirmed by clinical examination plus normal results of hematological and serum biochemical testing within 6 months of blood sampling. All dogs were fasted for 12 h before peripheral blood sampling. Peripheral blood samples were obtained through jugular venipuncture using a 21-gauge needle; blood was immediately placed into an EDTA collection tube and gently mixed. PBMCs were harvested through density-gradient centrifugation using Ficoll-Paque (density 1.077; GE Healthcare Life Sciences). PBMCs were washed in RPMI (Life Technologies) supplemented with 10% heat-inactivated FBS (Gibco), 25 μM HEPES (Life Technologies), 100 μM β-mercaptoethanol (Sigma-Aldrich), 100 U/mL penicillin–streptomycin, 2 mM
Dose–responses of WJ-MSC EV
WJ-MSC was plated in cRPMI with 1 mM ATP, with or without 5 μg/mL concanavalin A (ConA; Sigma-Aldrich). PBMC was incubated with WJ-MSC EV in a 1:102, 1:103, or 1:104 (PBMC:EV) ratio (n = 3 technical replicates per sample). After 72 h of incubation, the PBMCs were collected and surface stained for CD4 (rat anti-canine CD4-Alexa 647, clone YKIX302.9; AbD Serotec). Cells were stained for 30 min, washed twice in cold PBS with 5% FBS, and resuspended in PBS with 5% FBS and 7-AAD for analysis by flow cytometry (Accuri C6). The effects of WJ-MSC or WJ-MSC EV on CD4pos T cell (T lymphocyte) proliferation were determined as percentage of cells proliferating and number of divisions for proliferating cells by measuring CFSE fluorescence and utilizing the FlowJo® proliferation platform (FlowJo v7) after gating on the viable CD4pos lymphocyte population.
EV depletion and non-MSC EV negative controls
The supernatant resulting from EV pelleting and collection by ultracentrifugation of conditioned medium (100K supernatant) was recovered as an EV-depleted control sample. WJ-MSC EVs were also compared to EVs from conditioned medium of non-MSC fibroblasts (canine left ventricular cardiac fibroblasts; 104 EV:1 PBMC) for suppression of PBMC.
For EV depletion, WJ-MSC collected using stepwise ultracentrifugation was filtered through a 10 or 50 kDa MWCO filter (Sartorius Vivaspin 4) as per the manufacturer instructions. The concentrated EV-containing retentate was resuspended in PBS, and total particle content was evaluated by NTA. The volume of retentate needed to yield sufficient particles to generate a ratio of 104 EV:1 PBMC, or an equivalent volume of EV-depleted filtrate, was cocultured with PBMC as described.
Chemical inhibition of EV biogenesis was achieved by treating cultured WJ-MSC with 6 μM neutral sphingomyelinase inhibitor GW4869 [58] for 48 h. GW4869-treated or nontreated WJ-MSC was plated across from PBMC in a 1:10 ratio (PBMC:WJ-MSC) using a 0.4 μm Transwell and was maintained for 72 h before collection of PBMC and evaluation of lymphocyte suppression.
To disrupt EV membranes and remove extravesicular RNA and/or extravesicular proteins, EVs were treated with Triton-X (0.1%; Thermo Fisher Scientific), RNase A (2 μg/mL; Qiagen), Proteinase K (2 μg/mL; Qiagen), RNase A, and proteinase K or left untreated and incubated for 30 min at 37°C. Samples were subsequently washed by dilution and centrifugation at 100,000 g for 70 min. The resultant pellet was resuspended in PBS, and particle numbers were quantified using NTA. A ratio of 1 PBMC to 104 EV was cultured in cRPMI with 1 mM ATP, with or without 5 μg/mL ConA for 72 h (n = 3 technical replicates per sample).
Functional evaluation of WJ-MSC EV TGF-β and adenosine
PBMC in coculture with WJ-MSC or with WJ-MSC EV added was incubated for 72 h with the following: a specific TGF-βRI inhibitor [59] (SB4312 10 μM; Tocris); a specific adenosine2A receptor inhibitor (ZM241385 50 μM; Tocris); or TGF-β (1, 2, 3) neutralizing antibody (0.1 μg/mL; R&D Systems). Separately, recombinant human TGF-β1 (R&D Systems) or TGF-β3 (Sigma) at 5, 10, or 50 ng/mL was added to ConA-stimulated PBMC cultures. For disruption of the heparan sulfate side chains on TGF-βRIII (betaglycan), WJ-MSC EVs were incubated with heparinaseIII (Sigma) or heat-inactivated heparinase (control) at 0.006 U/mL for 3 h at 37°C (5% CO2) according to Webber et al. [60] and then washed in PBS and ultracentrifuged at 100,000 g for 70 min before application in PBMC coculture.
Identification of TGF-β receptor
CD4pos T cells were sorted from PBMC using magnetic enrichment (AutoMACS; Miltenyi). Detection of TGF-βRI protein on PBMC (unsorted) and CD4pos T cells was performed by immunoblot as described for the WJ-MSC, but using nitrocellulose membrane, 2.6 μg protein per lane, and rabbit anti-TGF-βRI polyclonal antibody (Abcam) at 1:500. HEK293 cell lysate was used as a positive control for protein detection.
Enzyme-linked immunosorbent assay
The Quantikine Mouse/Rat/Porcine/Canine TGF-β1 Enzyme-Linked Immunosorbent Assay (ELISA) Kit (R&D Systems) was used to determine concentration of TGF-β1 in WJ-MSC EV samples, using manufacturer instructions with slight modification. Briefly, samples underwent acid activation of TGF with 1 N HCl (Sigma) for 10 min room temperature (RT) (volume of acid added was 1/5 that of sample volume) and then neutralized with 1.2 N NaOH (Sigma) +0.5 M HEPES (volume added half that of the acid). This ratio was found to bring samples into target pH range for the assay. Samples were then diluted in the provided calibrator diluent to read within the standard curve range. Controls for the assay were as follows: nonacid activated samples with water substituted for the acid/neutralization solutions; PBS as a negative control (both activated and nonactivated); and rTGF-β1 positive control (included in kit). Reagents were prepared as per manufacturer's instructions. Samples, controls, and standard curve were run in duplicate according to the protocol for cell culture supernatants.
Statistical analyses
ANOVA was used to compare three or more groups of cell suppression data, followed by Tukey multiple means comparison post hoc. Analyses were performed using SPSS (Version 24; IBM). For all analyses, statistical significance was set at P < 0.05. Values are expressed as mean ± standard deviation unless otherwise indicated.
Results
WJ-MSC exhibits MSC phenotype and differentiation capacity
The MSC isolated from canine WJ exhibited characteristics that are used to define MSC. Specifically, WJ-MSC retained a typical MSC-like (spindle shaped, elongated, and fibroblastic) morphology through at least passage 6 (Fig. 1A). WJ-MSC expressed genes encoding phenotypic markers of MSC (CD44, CD90, CD105), but not the non-MSC marker CD45, or MHCII (Fig. 1B) which were corroborated by flow cytometry (Fig. 1C). CD34 gene expression was evident on quantitative real-time polymerase chain reaction (qPCR) but undetectable by flow cytometry, and CD73 concordant with qPCR was not detected at the protein level by flow cytometry. Commensurate with the designation as MSC, WJ-MSC showed the capacity for differentiation to osteocytes, chondrocytes, and adipocytes (Fig. 1D).
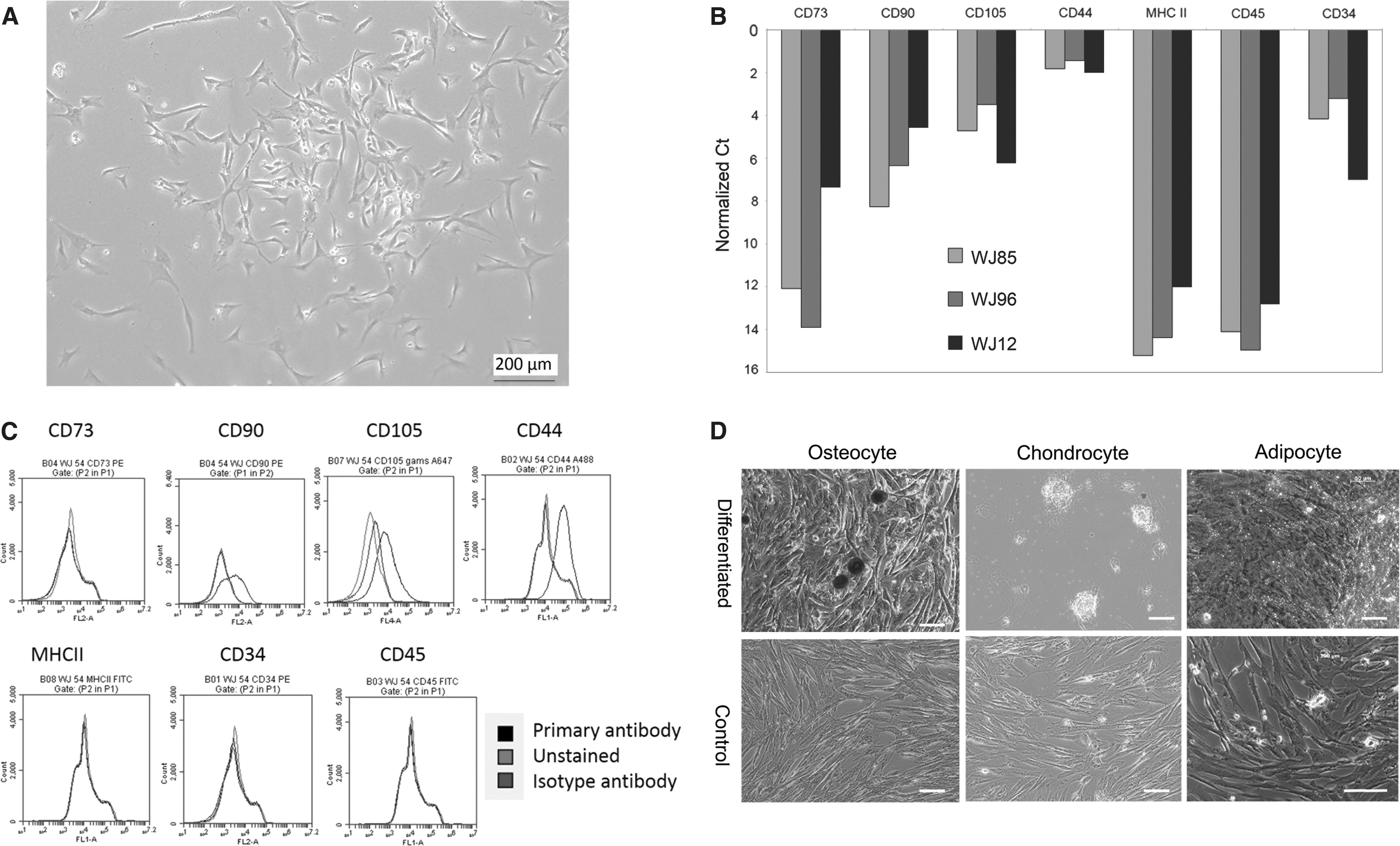
WJ-MSCs in culture display typical stromal cell phenotype.
WJ-MSC EV size distribution and morphology
The mode and mean particle size derived from composite data of five WJ-MSC EV lines was 125 and 199 nm, respectively, with 76% of all EVs falling within the 50–250 nm size range (Fig. 2A). The ultrastructural morphology of WJ-MSC EV (cup shaped) by transmission electron microscopy (TEM) included EVs in the size range of 50–100 nm and included many smaller spherical or torus-shaped structures (∼20 nm), adding to the diversity of vesicles produced by a single cell type (Fig. 2B) [71]. WJ-MSC EVs, isolated from cell culture supernatant by stepwise ultracentrifugation and applied to a density gradient, were observed to be concentrated in fractions 1, 2, and 3 out of 8 fractions based on particle distribution (NTA) and protein content (BCA) (Fig. 2C) and fractions 2 and 3 (corresponding to buoyant density of 1.094–1.105 g/mL) based on TSG101 content (Fig. 2D). On immunoblots, WJ-MSC EVs were enriched for TSG101 and Alix (proteins associated with exosome biogenesis) relative to parent WJ-MSC, whereas calnexin (an endoplasmic reticulum protein) was detected in parent WJ-MSC but not in respective WJ-MSC EVs, demonstrating EV enrichment of samples (Fig. 2E).
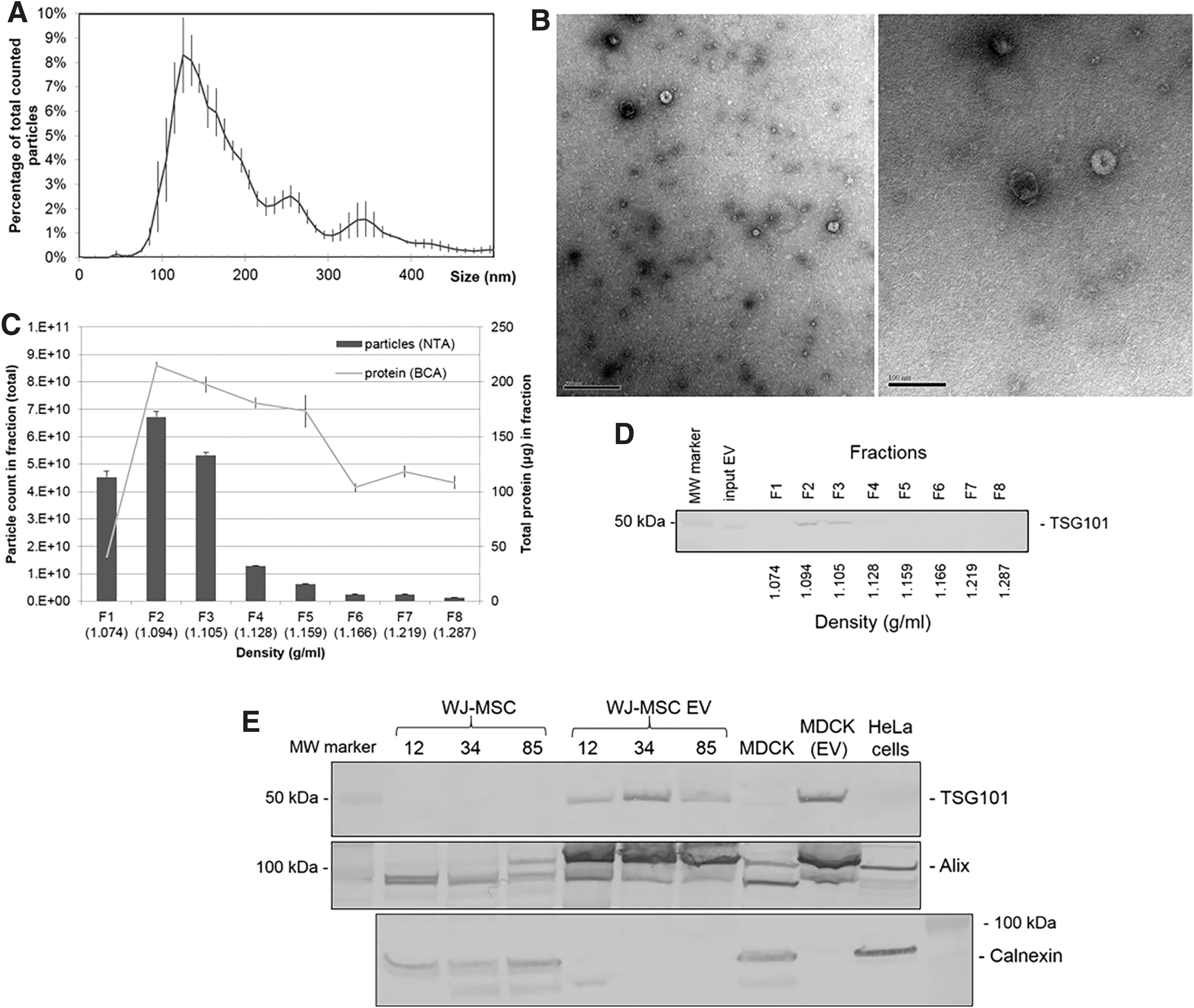
EVs derived from WJ-MSC through stepwise ultracentrifugation demonstrate typical morphology consistent with exosomes through NTA, electron microscopy, buoyant density, and immunoblot.
WJ-MSC and WJ-MSC EV suppress CD4pos T cell proliferation
The WJ-MSC EV immunomodulatory capacity was demonstrated to be dose dependent through coculture of EV with ConA-stimulated PBMC (Fig. 3A). Dilutions of 102 and 103 EV:PBMC resulted in suppression of CD4pos T cell proliferation relative to PBMC with ConA alone (43.7% ± 12.8% and 42.9% ± 8.2% vs. 59.2% ± 8.7%, respectively, P < 0.01), and 104 EV:PBMC suppressed T cell proliferation further (30.8% ± 13.2%, P < 0.01), to an extent that was equivalent to WJ-MSC at 1 MSC to 10 PBMC across transwell (32.7% ± 15%) (Fig. 3A). Both WJ-MSC (across transwell) and WJ-MSC EV suppressed the percentage of CD4pos T cells that proliferated in response to mitogen, but not the number of cell divisions for dividing cells (data not shown). Hence, the percentage of CD4pos T cells that proliferated in response to mitogen was utilized as the end point in PBMC responder assays. EV-depleted supernatant generated during ultracentrifugation (equivalent v/v) did not suppress CD4pos division, implying that the effect is derived specifically from the EV-enriched sediment fraction; similarly, there was no effect of EV derived from non-MSC cardiac fibroblasts on T cell proliferation (Fig. 3A) suggesting that the effect was unique to MSC-derived EVs. Based on these data, a concentration of 104 WJ-MSC EV per PBMC was utilized for subsequent experiments.
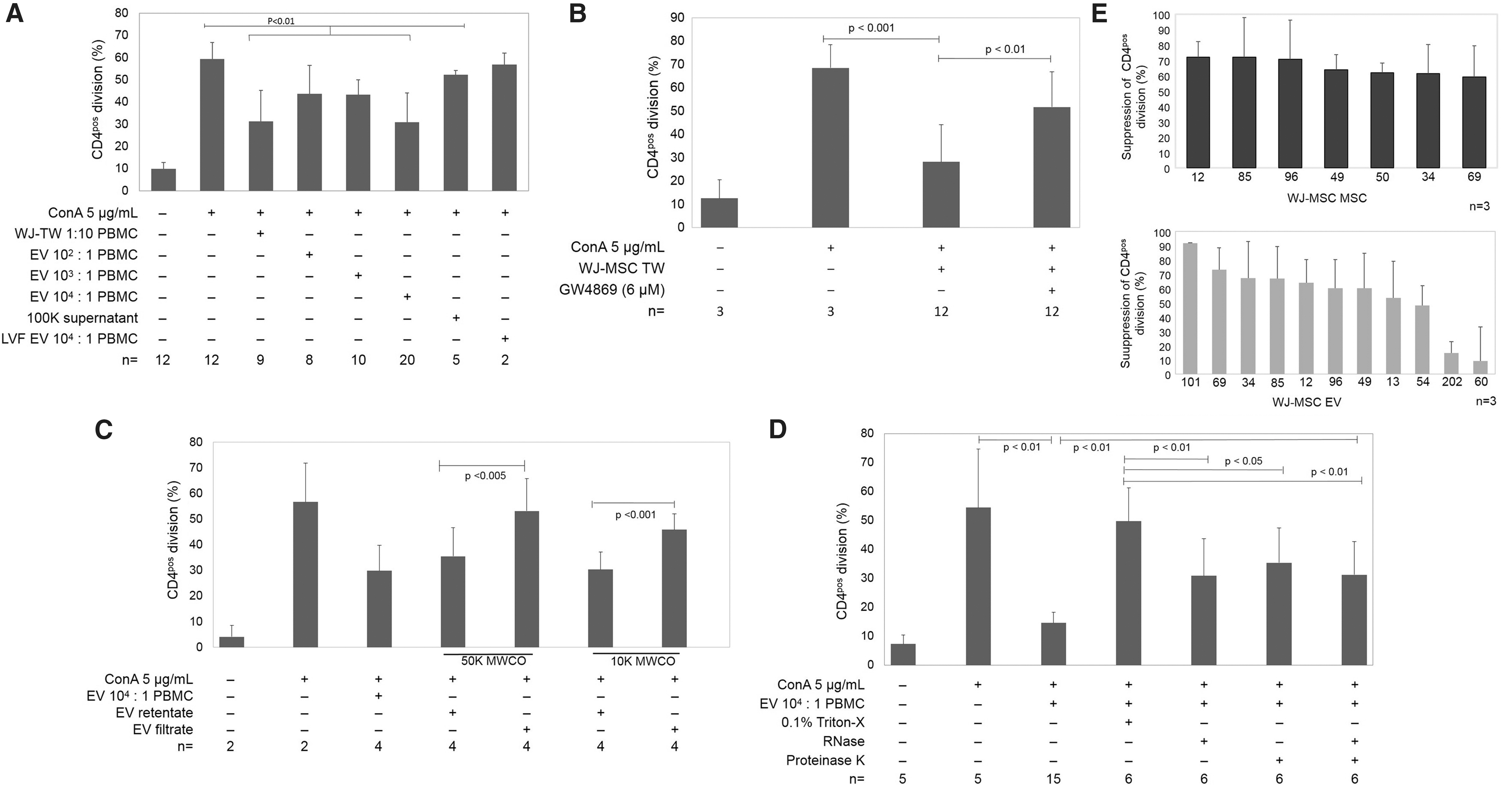
WJ-MSC EVs suppress CD4pos T cell division.
Depletion of WJ-MSC EV reduces CD4pos T cell suppression
Increasing doses of GW4869 were applied to the WJ-MSC to determine a concentration that would reduce EV output maximally without unacceptable loss of cell viability; optimal concentration was found at 5–10 μM (Supplementary Fig. S1). Pretreatment of MSC with 6 μM GW4969 to inhibit EV release yielded MSC that failed to suppress T cell proliferation (51.7% ± 13.5% for GW4969-treated WJ-MSC, compared to 28.2% ± 1.4% for nontreated WJ-MSC, P < 0.01) (Fig. 3B). WJ-MSC EV filtered through 10 or 50 kDa MWCO filters allowed evaluation of the role of non-EV fraction in suppression of CD4pos T cells. The EV-depleted filtrates of both 10 and 50 kDa MWCO filtered samples were inactive; however, the EV-enriched retentates reduced CD4pos division (Fig. 3C) consistent with the major effect on proliferation arising from the WJ-MSC EV themselves rather than from a soluble component co-sedimented during EV preparation. Triton-X, which destroys cell or EV membranes, completely extinguished the suppressive effects of EV (Fig. 3D) consistent with the role of WJ-MSC EV. To a lesser extent, exposure to RNase and proteinase K also significantly reduced WJ-MSC EV-induced suppression of cell division (Fig. 3D).
Reproducibility of WJ-MSC and WJ-MSC EV activity
Across all cell lines, WJ-MSC and WJ-MSC EVs consistently suppressed ConA stimulated T cell proliferation, with exception of EVs from two WJ-MSC lines (60 and 212) (Fig. 3E). Activity of WJ-MSC EV to suppress CD4pos T cell division was also consistent within WJ-MSC donor cell lines (Supplementary Fig. S1). The number of EVs released by each WJ-MSC (estimated by NTA) varied between cell lines. On average, WJ-MSC generated 5,780 ± 3,291 EVs per cell (Table 1).
Number of Extracellular Vesicles Released per Wharton's Jelly Mesenchymal Stem Cell
EV, extracellular vesicle; WJ, Wharton's jelly.
IFN-γ preconditioning of WJ-MSC does not increase suppression of T cell division by WJ-MSC or WJ-MSC EV
We explored whether WJ-MSC pretreatment with 500 ng IFN-γ for 48 h before collection of WJ-MSC EVs would augment either WJ-MSC (across transwell) or WJ-MSC EV activity. While there were trends in further suppression by WJ-MSC or WJ-MSC EV following preconditioning, these effects were not statistically significant (Supplementary Fig. S2).
TGF-β and adenosine signaling are mechanisms of WJ-MSC EV-mediated suppression of CD4pos T cells
To determine if TGF-β1 or adenosine contributes to EV-induced immunomodulation, WJ-MSC EV and PBMC were cocultured with TGF-β (1, 2, 3) neutralizing antibody, pharmacological inhibitors of TGF-βRI, adenosine 2A receptors, or both. Neutralization of TGF-β (1, 2, 3) with functional antibody significantly reduced suppression of CD4pos cell division (Fig. 4A). Similarly, blockade of TGF-βRI or the adenosine 2A receptor significantly reduced the effect of WJ-MSC EV to suppress T cell division (Fig. 4A).
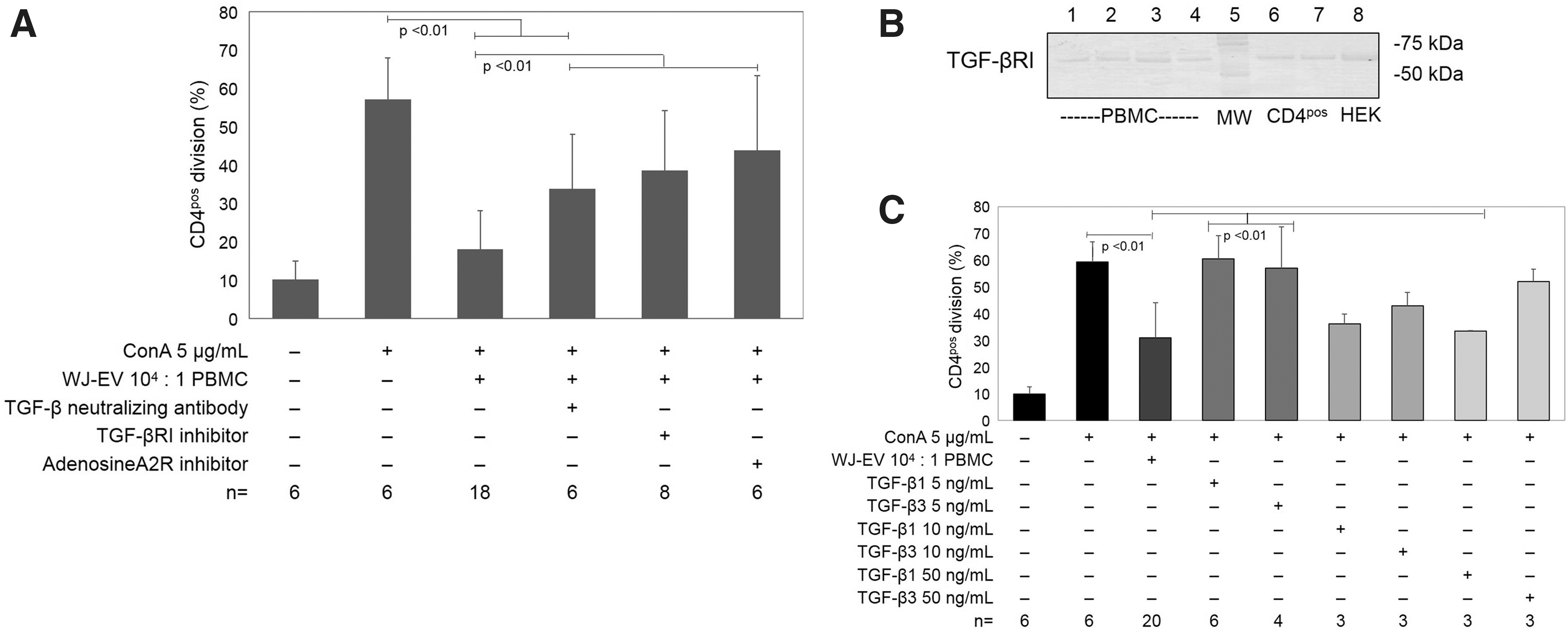
TGF-β and adenosine contribute to WJ-MSC EV suppression of CD4pos T cell division.
Exogenous soluble TGF-β suppresses EV
Presence of TGF-βRI on both PBMC and magnetically isolated CD4pos T cells was demonstrated with immunoblot (Fig. 4B), indicating a capacity for TGF-β interaction. In support of this, addition of 10 ng/mL (but not 5 ng/mL) TGF-β1 or TGF-β3 to PBMC suppressed mitogen-driven CD4pos T cell division (Fig. 4C).
TGF-β observed in latent form on WJ-MSC EV
Using ELISA, WJ-MSC EVs were found to possess TGF-β1 only after acid activation, consistent with the bulk of TGF-β1 in the latent form associated with EVs (Fig. 5A). The amount of TGF-β1 ranged from 0.1 to 1.0 ng per 5 × 109 EV (the number of EVs used in each standard PBMC assay well for a 104 EV:PBMC ratio) (Fig. 5A). The minimum concentration of exogenous rhTGF-β1 that was effective to suppress CD4pos T cell proliferation to an equivalent degree was 10 ng/mL (equivalent to 2 ng per assay well), suggesting that EV-associated TGF-β1 exerts greater biological activity w/w than rhTGF-β1. To further understand the structure of TGF-β on EVs, we evaluated whether the TGF-βRIII, which requires heparan side chains for activity, serves as a functional co-receptor to membranous WJ-MSC EVs. WJ-MSC EVs were pretreated with heparinase, which reduced EV suppression of T cell proliferation, compared to control heat-inactivated heparinase (Fig. 6).
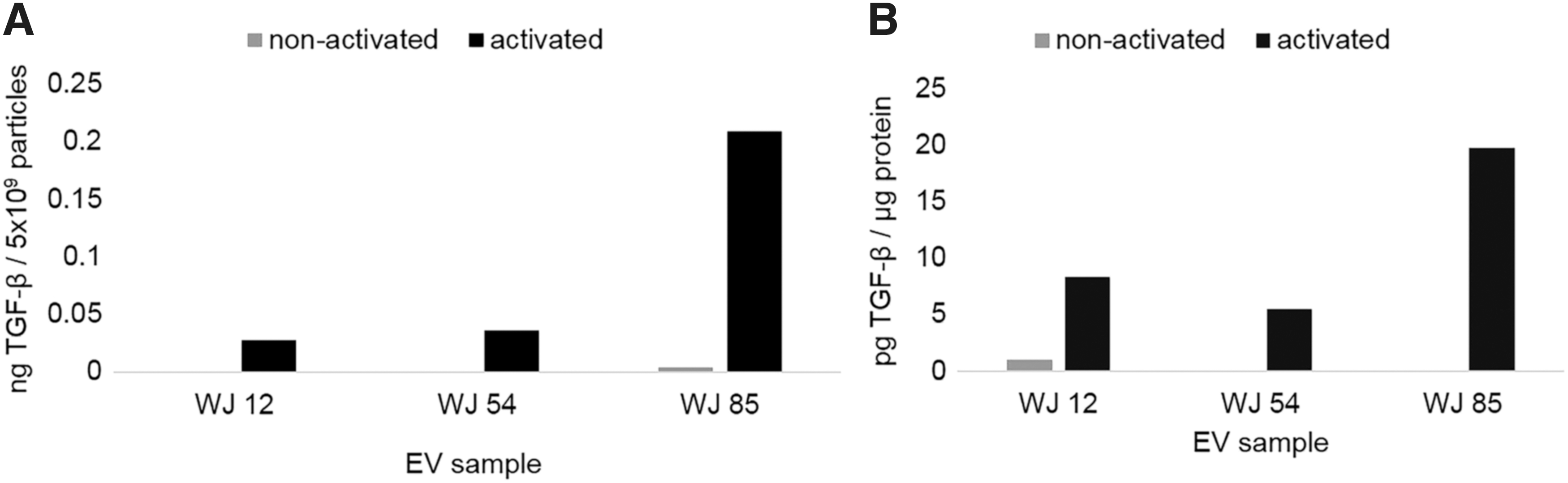
WJ-MSC EVs contain latent TGF-β. TGF-β1 ELISA before (nonactivated) or after acid activation (activated), of WJ-MSC EV TGF-β from latent form. Data expressed as a function of
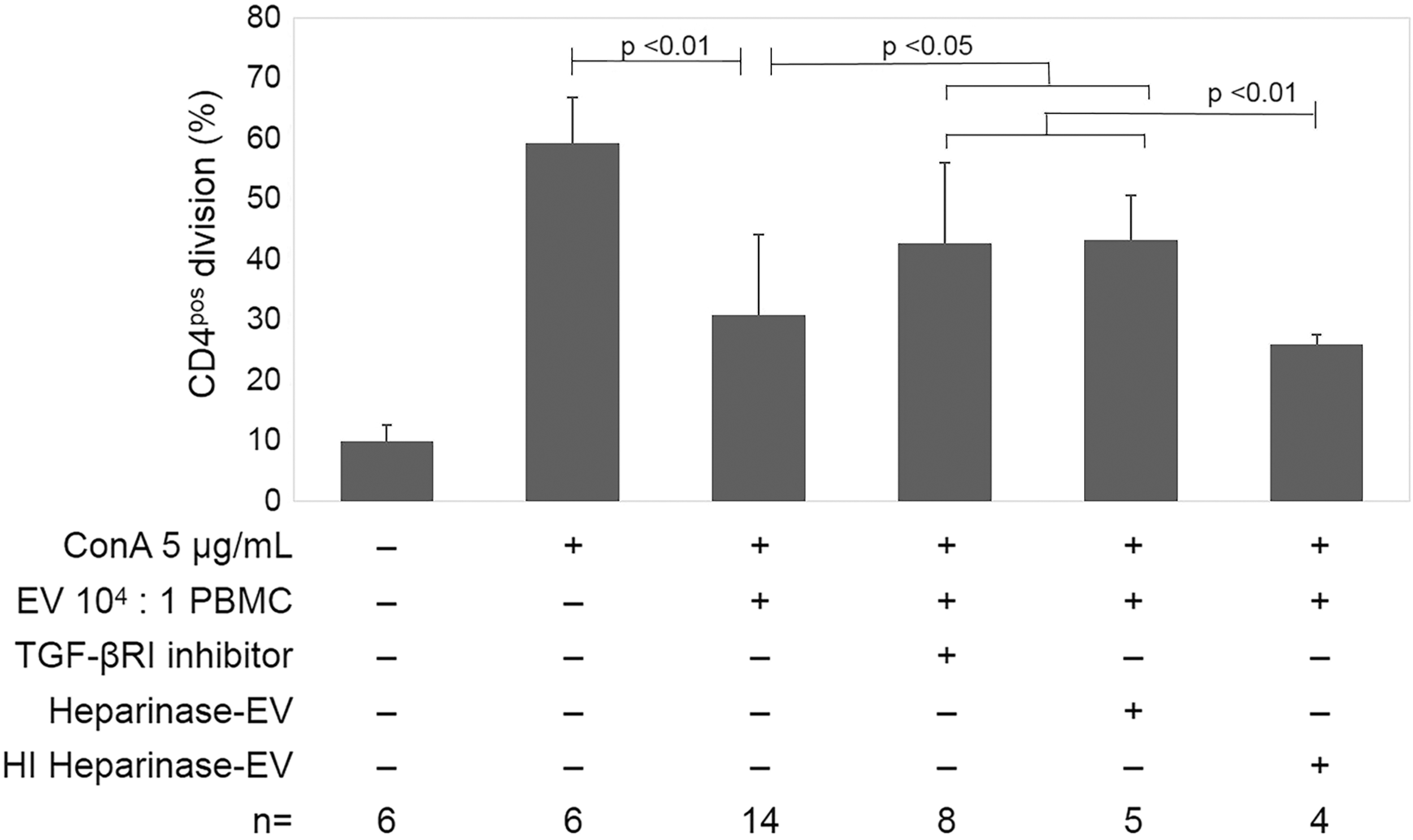
TGF-β signaling has a role in EV-mediated suppression of CD4pos T helper cell division. Inhibition of TGF-βRI (10 μM) and heparinase treatment (0.006 U/mL) of EVs both reduced EV-mediated suppression of CD4pos cell division, an effect which is abolished with pretreatment of EVs with heat-inactivated heparinase (HI heparinase).
Discussion
Despite the huge promise that stem cells, including MSCs, have for regenerative medicine, there has been surprisingly limited success and no FDA-approved MSC products in the United States to date. Moving forward, it is important to undertake further mechanistic studies to improve and refine cell based therapies [62]. To advance EV therapeutics, it will be critical to link mechanisms of action of MSC and MSC EVs to putative therapeutic benefits [25].
In this study we rigorously demonstrate that EVs from canine WJ-MSC significantly disrupt mitogen-activated CD4pos T cell proliferation through signaling pathways long held as soluble mediators secreted by MSCs. Mechanisms of WJ-MSC EV-mediated suppression of immune effector cell functions are largely unknown. Studies in AT-MSC revealed TGF-β1, HGF, PGE2, and IDO to be relevant paracrine factors in immunomodulation in the canine species [54]. Additional studies in canine AT-MSC and BM-MSC determined that TGF-β1, adenosine, and cyclooxygenase were key mediators [64]; however, neither of these studies evaluated MSC EVs. Importantly, the data presented herein suggest that a substantial fraction of the effects of MSC in PBMC assays may arise from EV-associated mediators such as TGF-β and adenosine.
WJ-MSC phenotype
The cells isolated from WJ (WJ-MSC) isolated in this study exhibited surface markers and gene expression that are typical for MSCs, including CD44, CD90, and CD105, and the absence of CD45 and MHCII [55,63 –67]. However, the authors acknowledge that the expression of CD90 and CD105 was lower than expected for MSC; in addition, CD34 gene expression and, to a limited extent, protein expression were detected in WJ-MSC in this study. The latter is consistent with low-level protein expression in past descriptions of canine WJ-MSC [63], but inconsistent with criteria for human MSC [68]. The method used to isolate WJ-MSC before expansion in culture can influence the phenotype of the resulting cell population. In retrospect, the collagenase digestion method of WJ-MSC isolation used may not have been ideal for isolation of WJ-MSC as there is some evidence to suggest that the explant method yields more MSCs with greater expansion potential and retention of MSC markers [6]. This may have influenced the consistency of phenotypic markers (eg, CD90, CD105) on the surface of WJ-MSC in this study. Further comparison of isolation methods and culture conditions will shed more light on these phenomena, as comparatively little is known regarding heterogeneity of canine MSC phenotype and subpopulations.
Multipotency of the WJ-MSC was evident, as they robustly differentiated into adipocytes and osteocytes consistent with established criteria for MSC [68]. Differentiation to chondrocytes was inconsistent although Alcian blue staining was evident in a few cells.
WJ-MSC EV phenotype
The particle size distribution and ultrastructural morphology of WJ-MSC EV were consistent with small EV (range ∼50–100 nm) but included smaller vesicle-like structures that were not specifically identified or characterized. The wide range of EV sizes detected by NTA and TEM in this study is consistent with the diverse repertoire of vesicles from a single source [61]. The low buoyancy and detection of TSG101 and Alix imply that canine WJ-MSC EVs isolated here consist mainly of small EVs [24]. Canine WJ-MSC EVs were physically larger than exosomes isolated from human WJ-MSC that similarly expressed both TSG101 and Alix [69], although differences in isolation and size measurement techniques make comparisons between studies difficult. Whether the very small structures (∼20 nm) observed using TEM are EVs, lipoproteins, small micelles, or protein aggregates in serum-free medium used for EV isolation awaits further analysis using additional techniques such as density gradient separation and cryo-EM.
WJ-MSC EVs are responsible for suppression of CD4pos T cell proliferation
Minimal criteria for characterization of EVs were put forth by a consortium from the International Society of Extracellular Vesicles in 2014 (MISEV) [70]. In this study we adhered to MISEV guidelines by detailing specific EV isolation methods, performing characterization (EV-specific and non-EV cellular proteins by immunoblot, buoyant density measurement, single vesicle characterization using two methods, including TEM and NTA, and functional assays, including dose–response, response to EV depletion, and controls for the source of EV. We showed that the method of stepwise ultracentrifugation yields an EV enriched population of particles, and the data in this study rigorously demonstrate that the biological activity measured in PBMC assays is intrinsic to WJ-MSC EVs.
We observed striking T cell suppression as a function of WJ-MSC EV dosage, a finding that was absent in EV-depleted fractions or using EV from non-MSC fibroblasts. Similarly, it has been reported that human MSC EVs (including WJ-MSC EVs) were strongly suppressive of mitogen-stimulated CD4pos T cells but not those stimulated by mixed lymphocyte reaction [17]. However, such findings about MSC or MSC EV are not universal in the literature; for example, MSC or MSC-conditioned medium was effective to suppress T cell proliferation due to mitogen stimulation, but MSC EVs isolated by stepwise ultracentrifugation, even at 10-fold higher concentrations than used in our assays, were not immunosuppressive in another study [37]. In addition, suppression of CD3pos or CD4pos T cell proliferation was not observed in response to MSC EVs; rather, increased apoptosis was observed, and increased numbers of T regulatory cells and IL-10 in supernatant were noted [50]. Cell death of PBMC was not evident in response to WJ-MSC or WJ-MSC EVs in this study.
The specific observation that canine WJ-MSC EVs (or parent WJ-MSC) suppress the percentage of dividing CD4pos T cells (responders) but not the number of cell divisions of responders is consistent with arrest at G0/G1 for suppressed T cells [71]. WJ-MSC EVs affected CD4pos cells to a greater extent than CD4neg cells, which may reflect greater proportions of CD4pos than CD4neg mitogen responders in our assay, not necessarily the specific impact on CD4neg cells, and should be investigated further.
Intrinsic variation in the effectiveness (potency) of WJ-MSC to modulate immune effector cells is a confounding aspect of MSC therapies in regenerative medicine. We explored the range of responses observed across 11 cell lines (where 2 pairs were littermate donors). Overall, the responses ranged from 9% to 92% mean suppression of CD4pos T cells across all WJ-MSC EV lines tested. The major variation was due to two cell lines (9.1%, 14.2% suppression) versus the range across the other nine cell lines (48%–92% suppression). The magnitude of suppression observed, under the conditions of 1 WJ-MSC to 10 PBMC across a transwell, is consistent with the literature [71]. The magnitude of suppression by WJ-MSC EVs (50%–75%) observed in the present study is comparable to past studies using canine AT-MSC or BM-MSC [53,73], although direct comparisons are confounded by different subsets of lymphocytes stained in these studies. Further analysis of mechanisms of suppression with respect to cell lines with reduced activity will be critical to our understanding of MSC selection.
“Priming” MSC with a pro-inflammatory stimulus has been shown to increase their immunosuppressive ability, even increasing the capacity of MSC EVs to suppress T, B, and NK cells [38]. However, pretreatment of WJ-MSC with IFN-γ did not enhance the antiproliferative activity of WJ-MSC or WJ-MSC EVs in this study. Similarly, it was found that immunosuppression by human umbilical cord matrix MSC is not enhanced by IFN-γ [13,72]. This may imply that WJ-MSC and EV biogenesis are uniquely unresponsive to IFN-γ, and IFN-γ stimulation may not be an effective strategy in this cell type or in the canine species. Alternatively, longer exposures or alternative forms of IFN may influence WJ-MSC EVs in ways that were not explored in this study.
Addition of high concentrations (10–50 ng/mL) of soluble TGF-β1 or TGF-β3 suppressed mitogen-driven CD4pos T cell division, while the amount of EV-derived TGF-β1 that needed to achieve equivalent suppression was ∼10% of that. This suggests that EV associated TGF-β1 may have higher or qualitatively unique biological activity compared to soluble TGF-β1 or that it acts in combination with other factors on or in the EVs. Similar observations were made in past studies comparing EV membrane-associated TGF-β1 (derived from cancer-associated fibroblast and dendritic cells) versus soluble TGF-β1 [45,74]. However, it is probable that multiple mechanisms aside from solely EV-associated TGF-β are contributed by WJ-MSC EVs, such as stimulation of autocrine production of TGF-β1. That adenosine had similar effects supports the idea that WJ-MSC EVs introduce a combination of mechanisms, similar to the repertoire of growth factors attributed to parent MSC. A possible role for TGF-β1 in immunomodulation by MSC-derived EVs was described by Mokarizadeh et al. [48] and Favaro et al. [49], showing expression of TGF-β1 on EV membrane using bead-assisted flow cytometry or western blots. Human BM-MSC EVs were shown to suppress IFN-γ production by PBMC in the latter study [49], which was partly reversed by neutralizing antibodies to TGF-β1, and to a lesser extent IDO and PGE2 inhibitors. Our studies augment these observations by showing the specific and dose-related activity of TGF-β produced by canine WJ-MSC EVs in the mitogen-stimulated T cell proliferation assay.
Furthermore, the participation of TGF-βRIII (betaglycan) as a membrane coreceptor for TGF-β was explored [60,75]. Betaglycan can increase affinity for TGF-β to its receptors TGF-βRI and TGF-βRII. Indeed, TGF-β-induced fibroblast differentiation and angiogenesis by cancer EVs require the interaction of betaglycan with TGF-β1 and intact heparan sulfate side chains [60]. It was observed that heparinase (but not heat inactivated heparinase) reduced EV suppression, supporting a potential role for betaglycan in facilitating WJ-MSC EV TGF-β signaling. This also suggests a membrane (as opposed to luminal) topology of latent TGF-β on EVs. However, the mechanism by which the latent form of TGF-β is activated to release the mature form of TGF-β in the PBMC assay is unresolved by our study, as are potential interactions and effects of other immune cells in the PBMC mix. Proteases may arise from serum or be released from bystander cells (monocytes, residual neutrophils, and lymphocytes). Alternatively, EV-derived TGF-β1 may not be the sole source of this cytokine in the assays; rather, TGF-β1 may be generated de novo in the assay following EV interactions with monocytes [18] or T cells as an autocrine loop [49]. To resolve this question, further cell depletion assays are needed.
In summary, these data demonstrate that WJ-MSC EVs possess intrinsic immunomodulatory mechanisms previously attributed to soluble factors released by MSC and involve interactions with TGF-β and adenosine. In future studies, it will be important to expand the studies to explore the role of additional EV proteins or RNA cargo to fully appreciate their interactions with immune effector cells in the PBMC responder assay. To understand why results in the literature have been inconsistent with respect to biochemical immunomodulatory mechanisms exerted by EVs, it will be necessary to further explore mechanisms by which EVs act, understand the heterogeneity of MSC-EVs, and detail differences in the protocols used to isolate EV and study EV-immune effector cell interactions in vitro. Finally, the data are relevant to advancement of MSC line selection and development of MSC EVs as a therapy for canine disease.
Footnotes
Acknowledgments
The authors thank Drs. Cynthia Webster, Nicholas Frank, and Mary Labato, for their insight and support. The authors thank the veterinarians and staff of Slade Veterinary Clinic (Natick, MA) and Danvers Animal Hospital (Danvers, MA) for their donation of specimens for isolation of MSC. The authors also thank Lyndah Chow (Colorado State) for insight into protocols for pharmacologic inhibition in immune assays. This project was funded by the Shipley Foundation (award to A.M.H.), with additional salary support for S.K.C. by the Anne Engen Foundation (award to Dr. Mary Labato).
Data presented in this article were presented in part at the American Society of Exosomes and Microvesicles (2017) and International Society of Extracellular Vesicles (2017).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.