
Abstract
Propagation of pluripotent cells from early stage embryos in mouse and human highly depend on leukemia inhibitory factor (LIF)/signal transducer and activator of transcription 3 (STAT3) and FGF2/mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) signaling pathways. However, mechanisms for maintaining pluripotency in embryonic stem cells using various combinations of growth factors (targeting LIF or FGF2 pathways) and inhibitors (targeting WNT/GSK3 or FGF2 pathways) still have to be deciphered in other models, including the domestic cat. Our objective was to understand how cytokines influence pluripotency in the cat inner cell mass (ICM) outgrowths. Cat ICM was isolated from in vitro-produced embryos and outgrowths were cultured for up to 6 days with single or combined cytokines. Cell proliferation was enhanced with almost all single growth factors and cytokine combinations. Based on gene expression and presence of NANOG, POU5F1, and Sex-determining region Y box 2 (SOX2) as cell state markers, single growth factors could not maintain similar levels in outgrowths as in the original ICMs, which is different from the response in mouse and human. In our conditions, cytokine combinations involving LIF, GSK3 inhibitor, and MEK inhibitor resulted in the most robust expression levels and allowed single-cell dissociation and propagation. However, further characterization of embryonic cells derived from ICM indicated that the pluripotent state was not fully preserved. The absence of detectable transcripts for BMP2-receptor and SMAD4, and very low levels of LIF-receptor and STAT3 in the cat ICM indicated that pluripotency regulatory machinery appear to be different in the cat from the predominant mouse and human models.
Introduction
Pluripotent stem cells have inherent capacity to differentiate into any cell type and to self-renew unlimitedly [1,2]. The embryonic stem cell (ESC) is a major pluripotent cell type that can be produced and maintained in vitro [3,4], including for biomedical research, drug testing, genetic and congenital disease modeling, and regenerative medicine. Most ESC lines and studies have focused, so far, on common laboratory rodents [3,5]. However, our research team has demonstrated the value of a larger-sized model, the domestic cat, for understanding a wide array of reproductive mechanisms analogous to the human [6]. Moreover, the cat and human share more than 250 diseases (eg, Feline Immunodeficiency Virus and human Acquired Immunodeficiency Syndrome) [7].
Other groups also have explored the development of domestic cat assisted reproduction technology that could have application to Felid species [8,9], many of which are classified as vulnerable to extinction (The International Union for the Conservation of Nature Red List;
In earlier investigations, methods developed to establish ESCs in mouse and human did not lead to stabilized, pluripotent stem cells in the cat [10,11]. This was likely due to insufficient prevention of differentiation in the culture system. From studies in the mouse model, we know how to maintain ESCs in vitro and how these cells differentiate and contribute to organ formation in chimeric embryos [12]. In addition, the core network of pluripotency in mouse [12,13], rat [14], and human [13,15] ESCs is a regulatory circuitry involving Nanog homeobox (NANOG), POU domain, class 5, transcription factor 1 (POU5F1 aka OCT4), and Sex-determining region Y box 2 (SOX2). Furthermore, four conserved pathways [leukemia inhibitory factor (LIF)/signal transducer and activator of transcription 3 (STAT3), bone morphogenetic protein (BMP), activin/nodal, mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinases (ERK)] are involved in maintaining ground state pluripotency by regulating the NANOG, POU5F1, and SOX2 expressions [16 –18].
We suspect that those same general mechanisms apply to the maintenance of pluripotency in the domestic cat. However, there are specificities in embryonic peri-implantation development process in felids [19] that may be involved. For instance, blastocyst formation occurs about 8 days after conception, it completes gastrulation, and starts neurulation by the time of implantation. This is quite different from what has been observed in mouse and human [20]. Therefore, we hypothesize that there also are species-specific variations for signaling pathways synergy in different mammalian models. To better elucidate the underlying mechanism in pluripotency, evaluation of cytokine effects on pluripotency-related protein expression in cat embryos would be very informative.
The objective of our study was to understand how the growth factors, LIF and FGF2, GSK3 inhibitor, and MEK inhibitor (known to be efficient for mouse and human ESC maintenance) [4,21] affect the pluripotency circuitry in cat embryonic cells. This could lead to the development of proper culture systems and applications for cat ESC.
Materials and Methods
In vitro maturation and fertilization of oocytes followed by embryo culture
The study did not require the approval of the Animal Care and Use Committee of the Smithsonian Conservation Biology Institute and the University of Maryland, College Park, because ovaries and testicles were collected at local veterinary clinics as byproducts from owner-requested routine ovariohysterectomies or orchiectomies. Mature cat ovaries and testes were collected from local veterinary clinics, transported, and processed within 6 hours of excision [22].
Grade 1 immature oocytes were recovered from ovarian tissue and matured for 24 hours [22] (38.5°C, 6% CO2) in 50 μL drops of human Quinn's Advantage Blastocyst Medium (Origio) supplemented with bovine FSH and LH (Sigma) at 1 μg/mL. Spermatozoa were collected from excised cat epididymis in Ham's F10 medium (Irvine Scientific) [23]. After centrifugation (350g × 8 min), spermatozoa were allowed a swim-up for 30 min (38.5°C, 6% CO2.) and oocytes inseminated by coincubation with motile spermatozoa (1 million/mL) for 24 hours. Resulting embryos with at least two blastomeres (48 hours postinsemination) were cultured under the same conditions in Quinn's Advantage Blastocyst Medium until expanded blastocysts were formed (ranging from days 7 to 9 postinsemination) [24].
Under our experimental conditions, percentage of cleaved embryos after in vitro maturation/fertilization was 46% ± 24.5% [mean ± standard deviation (SD)], and the proportion of expanded blastocysts relative to total number of two-cell-stage embryos was 15% ± 13.1%. Approximately 40% of blastocysts were formed on day 8 (postinsemination) with 30% formed on day 7 and 30% forming on day 9. Expanded blastocysts were randomly selected for RNA extraction (2 biological replicates, n = 10 and 9, respectively), immunofluorescence (n = 5), and outgrowth culturing (n = 3–5) per treatment group. A total of 212 expanded blastocysts from 150 in vitro cultures were used during the entire study.
Cat blastocyst outgrowth culture
The zona pellucida of each blastocyst was removed in 1% protease (Sigma) [25] and the whole embryo was cultured on mouse mitotically inactivated embryonic fibroblasts [mouse embryonic fibroblasts (mEFs)] grown on 0.1% gelatin coated (Sigma) in four or eight-well chamber glass slides (Millipore) for 6 days. Blastocysts outgrown for further propagation were cultured on mEFs in 96-well tissue culture plates (Olympus) until inner cell mass (ICM) expansion (considered for this investigation to be a two-fold increase in ICM diameter) [5,10].
The medium was composed of 43% KnockOut Dulbecco's modified Eagle's medium, 43% Neural Basal Medium, 5% KnockOut Serum Replacement, 2% B27 Supplements, and 1% N2 Supplements (Life Technologies).
In Study I, nine single growth factor treatments, human LIF [hLIF; (Kingfisher Biotech) at 1, 10, or 100 ng/mL; feline LIF (fLIF; Kingfisher Biotech) at 1, 10, or 100 ng/mL; FGF2 (PeproTech) at 5, 10, or 20 ng/mL], were assigned to each group (n = 4–5 blastocysts/group) from the blastocyst seeding day (day 0). The concentrations were based on previous studies using FGF2-human [18] and LIF-mouse [5]. All blastocyst outgrowths were fixed and stained (as described in Assessment of protein expression section).
The influence of cytokine supplementation in sustaining pluripotent protein markers were classified as follows: (1) “excellent” (maintaining protein concentration similar as in ICM on day 0 and higher than control on day 6); (2) “good” (maintaining protein concentration lower than day 0, but higher than control on day 6); or (3) “poor” (maintaining protein concentration lower than control on day 0 and similar to day 6). In Study II, cat blastocyst outgrowths were cultured in groups (3–4 blastocysts/group) with growth factors in combinations comprised of cytokines having “good” or “excellent” effects on at least two protein markers in Study I. GSK3 inhibitor (CHIR99021; Stemgent) [4] and MEK inhibitor (PD0325901; Stemgent) [16] were added in the treatment combination to enhance proliferation and prevent differentiation [15]. The combinations tested are numbered in Table 1.
Growth Factors and Inhibitor Combinations for Study II
LIF, leukemia inhibitory factor; MEK, mitogen-activated protein kinase kinase.
Assessment of protein expression in cat blastocyst outgrowths by immunofluorescence
Expanded blastocysts and blastocyst outgrowths cultured on mEFs on glass slides for 6 days were fixed [24] by incubating in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) at room temperature (10 min) and then rinsing twice with Dulbecco's phosphate-buffered saline (DPBS; Sigma). Fixed cells were permeabilized by incubating with 0.2% Triton X-100 and 0.1% Tween 20 in PBS at room temperature (10 min), then rinsing twice in TBS with 0.05% Tween 20.
Outgrowths were rinsed in PBS once followed by blocking with Maxblock Blocking Medium (Active Motif) at 4°C overnight. Cells were incubated with the primary antibodies [anti-human NANOG (500-P236; PeproTech), anti-human POU5F1 (MAB4401; Millipore) and anti-human SOX2 (SC1002; Millipore) at 1.5 μg/mL] in Maxbind Staining Medium (Active Motif) overnight (4°C) with gentle rocking. Cells were then incubated with the secondary antibodies [Alexa Fluor 488 goat anti-rabbit IgG (H+L), Alexa Fluor 647 goat anti-mouse IgG1, Alexa Fluor 594 goat anti-mouse IgG2a (Thermo Fisher Scientific) at 1 μg/mL] in Maxbind Staining Medium (room temperature, 1 hour, in the dark). Then cells were washed four times (5 min each) with Maxwash Washing Medium for imaging. Hoechst 33342 (Invitrogen) was added during the third wash at 1 μg/mL to stain nuclear DNA.
Imaging was done using a Leica DMI 6000 inverted fluorescent microscope. First, we evaluated background signals in blastocysts and cells stained with secondary antibodies, but without primary antibodies (Supplementary Fig. S1). Second, these primary antibodies were validated in our laboratory by staining blastocysts (stained positive for all three proteins) (Fig. 1A upper panels) and fibroblasts (stained negative for all three proteins) (Fig. 1A lower panel). We also overexpressed NANOG in cat fetal fibroblasts (CFFs) to evaluate the antibody (Supplementary Fig. S2). Cells then were washed three times (5 min each) with Maxwash Washing Medium (Active Motif).
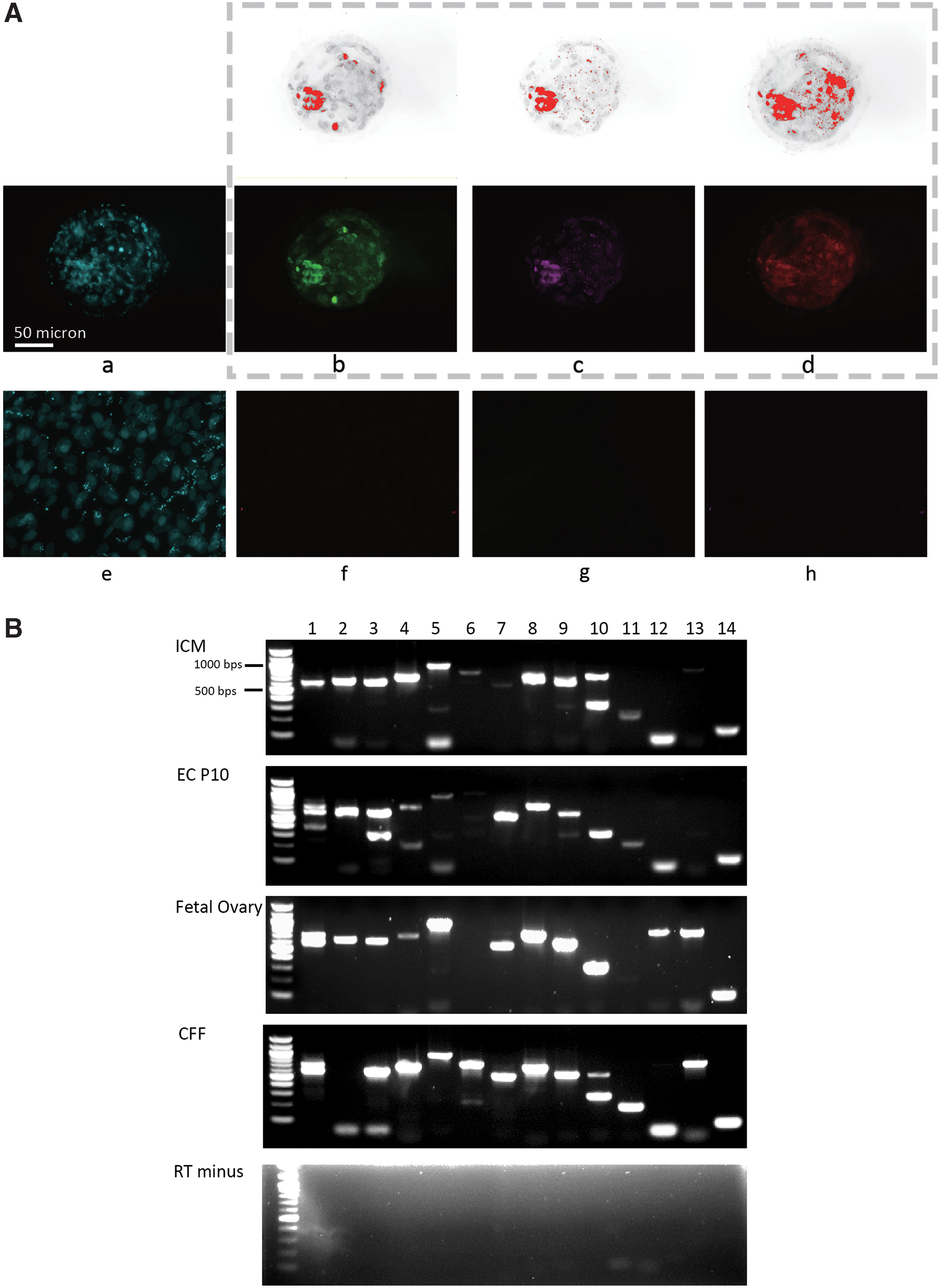
Presence of pluripotency proteins and expression of genes related to pluripotency and signaling in the ICM of cat blastocysts.
Exposure time and gain were determined initially for stained blastocysts and then were kept consistent for all subsequent images. Fluorescence intensities were analyzed using ImageJ for semiquantification of proteins (NANOG, POU5F1, and SOX2). Single-channel grayscale images were exported. Regions of interest were defined by the localization of ICM or colonies in bright field images under threshold function. The mean nucleus intensity was calculated as x = integrated density value/selected area (treatment) − integrated density value/selected area (negative control, near 0 in this study). Intensities of each stained protein were normalized to the average intensity in ICMs in days 7–9 blastocysts (control day 0).
Cat embryonic cell propagation
Based on the protein intensities measured in Study II, one cytokine combination was selected for further embryonic cell propagation for the purpose of assessing cytokine effect in synergy for long-term culture (Study III). The percentage of blastocysts (n = 41) adhering to mEF feeders in 96-well tissue culture plates and expanding to at least twice the original size was 30% ± 15.2%. Expanded ICMs (n = 13) were isolated using two 27.5-gauge needles. These were seeded on mitotically inactivated mEFs grown in 96-well tissue culture plates and the medium was supplemented with 10 ng/mL fLIF, 10 ng/mL hLIF, 3 μM GSK3 inhibitor +1 μM MEK, and 10 μM ROCK inhibitors (Stemgent). Cells were then disassociated as single cells using Accutase (Innovative Cell Technologies) and seeded onto mitotically inactivated mEFs in 24-well tissue culture plates. Cells were maintained in a humidified incubator (Thermo Fisher Scientific) (37°C, 5% CO2 in air) for 4–6 days or until colonies occupied 30%–40% of the well surface area. Colonies were then disassociated as single cells by Accutase and split (ratio of 1:3 to 1:4) on mEFs in six-well tissue culture plates. Cells were expanded in this manner until passage 10.
Evaluation of propagated embryonic cells
For transcript expression, ICMs were isolated from blastocysts using 30.5-gauge needles to separate cells that were subsequently lysed using extraction buffer (Applied Biosystems™). RNAs were isolated using the PicoCure RNA Isolation Kit (Applied Biosystems).
After being collected using cell scrapers (Thermo Fisher Scientific), embryonic cells were centrifuged (200g, 5 min) to produce a pellet that was subsequently lysed using RLT buffer (Qiagen), RNA then was isolated using the Rneasy Mini Kit following the manufacturer's instructions (Qiagen). Complimentary DNA was generated by reverse transcription of 100 ng of RNA using the SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific). Complimentary DNA samples (5 ng for each reaction) were amplified using GoTaq Green Master Mix (Promega) in an Eppendorf thermocycler.
Genes selected for amplification and primer sequences are shown in Supplementary Tables S1 and S2. The amplification was performed for 5 min at 95°C followed by 32 cycles for 30 s at 95°C, 30 s at 58°C, 1 min at 72°C, and 1 cycle for 5 min at 72°C for extension. The cycle numbers for each set of primers were determined by amplification curve by complementary DNA (cDNA) template from ICM and cat fetal ovary (Supplementary Fig. S3) [26].
The presence or absence of amplified genes was evaluated by imaging after gel electrophoresis. Fluorescence intensity of polymerase chain reaction (PCR) products were analyzed using ImageJ for semiquantification. Then 20 ng of each PCR product was amplified using the Big Dye Terminator V3.1 Cycle Sequencing Kit (following the manufacturer's instruction; Applied Biosystems) and were sequenced using a 96-capillary 3730xl DNA Analyzer (Applied Biosystems). PubMed Gene and Genome were the reference databases of sequences [27,28].
For genotyping, cells were expanded in the same medium without mEF feeders for three passages. Genomic DNA was isolated using the PureLink Genomic DNA Mini Kit (Thermo Fisher Scientific) from these embryonic cells. Genomic DNA samples (10 ng/10 μL reaction system) were amplified using AmpliTaq Gold 360 Master Mix (Applied Biosystems) in an Eppendorf thermocycler. The amplification was performed for 5 min at 95°C followed by 32 cycles for 30 s at 95°C, 30 s at 61°C, 1 min at 72°C, and 1 cycle for 7 min at 72°C for extension.
Karyotyping cat embryonic cells
Chromosome spreads (n = 21, 23, 28 for each cell line, respectively) from cat embryonic cells were prepared as previously described [29]. Chromosomal DNA was stained using Hoechst 33342 (Invitrogen) at 1 μg/mL with subsequent imaging using a Leica DMI 6000 inverted fluorescent microscope [30]. Cat genome contains 38 chromosomes [31].
Statistical analysis
Values presented for staining intensity are mean ± SD. Positive controls were ICM in expanded blastocysts, the intensities were also compared with that of ICM outgrowths cultured in the same base medium for 6 days without growth factor or inhibitor supplementation. Nine single-factor treatments and 17 combination-factor treatments were tested. Each treatment group includes three to four ICM outgrowths. Transcriptional expressions are normalized to reference gene RPL17 expression, which has been validated in the cat [26]. Relative intensity of PCR products and immunofluorescence staining were analyzed using a generalized linear mixed model [32] in SAS University Edition with statistical significance set at P < 0.05. To select the best growth factor and inhibitor cocktail, average Euclidean distance between each treatment group and the ICM control group was calculated in Perl 5.8.8. A three-dimensional scatter plot diaphragm for each ICM outgrowth was generated through an IBM SPSS program. Transcript profiles were analyzed between ICM (two biological replicates) and embryonic cells at passage 10 (three biological replicates), using generalized linear mixed model.
Results
The presence of pluripotency markers and signaling components in the ICM
The pluripotent-related proteins NANOG, POUF5F1, and SOX2 were detected in blastomeric nuclei of expanded cat blastocysts (Fig. 1A); differentiated cells like CFFs did not express these protein markers (Fig. 1A). POU5F1 was more restricted to the ICM compared with NANOG and SOX2 that were prevalent in trophectoderm nuclei as well. Transcripts of pluripotent-related genes (NANOG, POU5F1, SOX2, KLF4, and CMYC) were also expressed in the ICM (Fig. 1B). Signaling components in the FGF2 pathway (FGF2-receptor, MEK, and ERK transcripts) were highly expressed compared with low levels of LIF-receptor and STAT3 transcripts in the LIF pathway of the ICM (Fig. 1B). Activin A-receptor was modestly expressed, whereas other signaling components from the BMP and transforming growth factor (TGF)/activin pathways (specifically BMP2-receptor and SMAD4 transcripts) were nearly absent in ICM (Fig. 1B). The presence of BMP2-receptor and SMAD4 transcripts in fetal ovary validated the primers used for detecting these cat messenger RNAs (mRNAs). All PCR products were confirmed by sequencing (Supplementary Table S3). Dual bands of NANOG in embryonic cell at passage 10 (EC P10), fetal ovary, and CFF were fragments of cat NANOG transcript and predicted transcript variant, respectively, annealed with the same primers (Supplementary Table S3). Dual bands of SOX2 in EC P10 were both fragments of SOX2 transcript, the upper band was a product generated by transcript annealing with both primers, but the lower band was a product that only 5′ primers annealed with the transcript (Supplementary Table S3). Dual bands of KLF4 were both fragments of predicted KLF4 transcript, similar to SOX2 PCR, the lower band was a product that only 5′ primer annealed with the transcript. Upper band for ERK was a product only annealed with 5′-primer (Supplementary Table S3). Bands at the bottom, the lanes for CMYC and BMP2-receptor, were unspecific, and not fragments of any anticipated transcript (Fig. 1B).
Assessment of single growth factor supplementation on ICM outgrowths
Because we detected signaling components in LIF/STAT3 and FGF2 pathways, ICMs were cultured for 6 days in a medium supplemented with FGF2, hLIF, or fLIF at various concentrations. Cell numbers in ICM outgrowths were increased (P < 0.05) in most treatment groups after 6 days compared with the ICM control (day 0) or control without growth factor (day 6) (Fig. 2A). An exception that was not significant (P > 0.05) was treatment with 5 ng/mL FGF2.
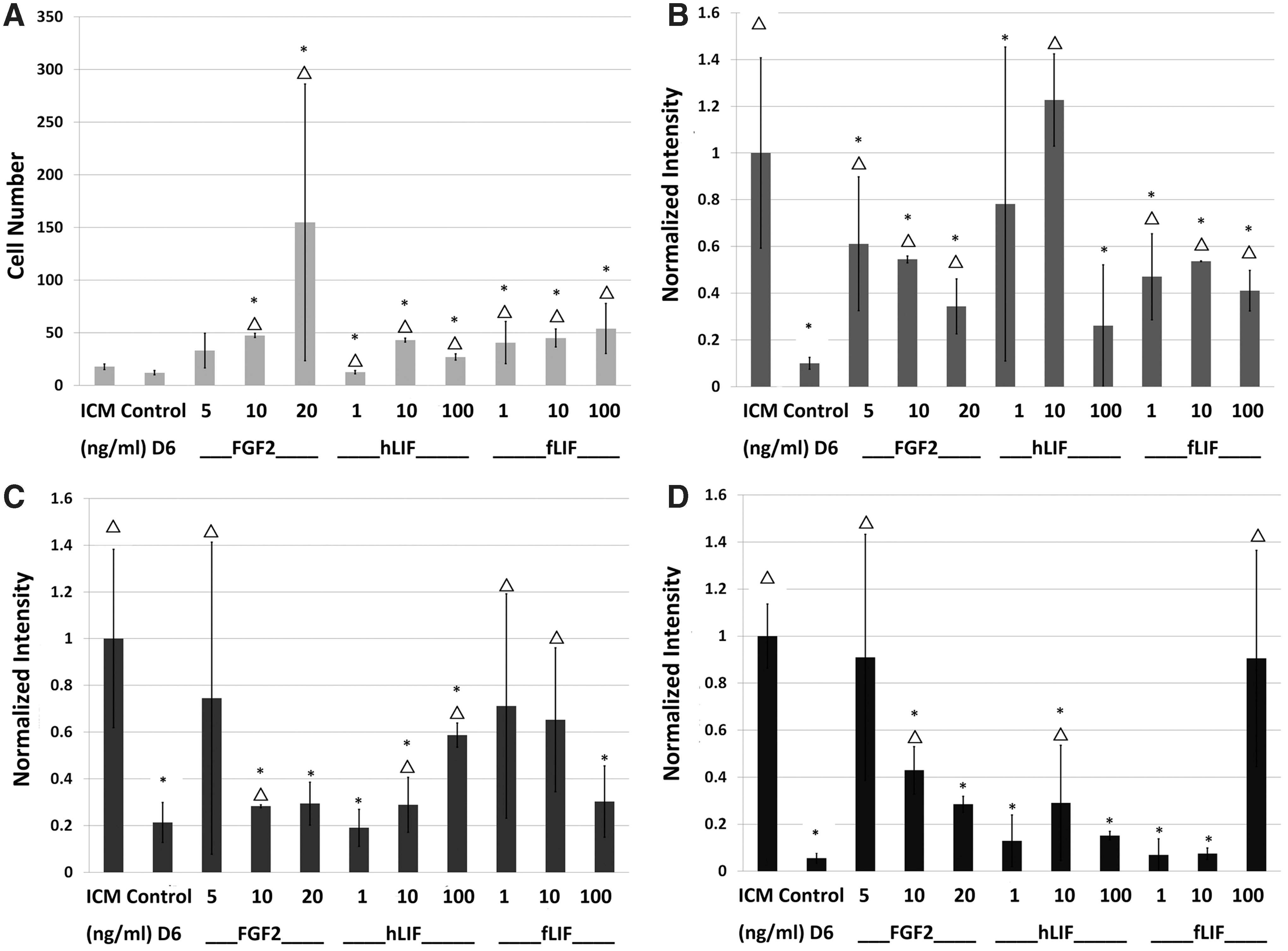
Characterization of ICM outgrowth cultured in vitro for 6 days with single growth factors (FGF2, hLIF, fLIF) at different concentrations. Positive controls were ICM in expanded blastocysts on day 0 of culture (ICM). Other control groups were ICM outgrowths cultured for 6 days without growth factor supplementation (Control D6).
With the exception of supplementing with 10 ng/mL hLIF, other posttreatment results did not meet our “excellent” classification in sustaining NANOG. However, most treatments met our “good” criteria at enhancing this protein (Fig. 2B). Three treatments (5 ng/mL FGF2, 1 or 10 ng/mL fLIF) were “excellent” at maintaining POU5F1 in ICMs, whereas several supplementations (5 or 10 ng/mL FGF2, 10 ng/mL hLIF, 1 or 10 mg/mL fLIF) were “good” at preserving POU5F1 (Fig. 2C). Treatments with 5 ng/mL FGF2 or 100 ng/mL fLIF were “excellent” at maintaining SOX2 in ICM outgrowths. Lastly, all three growth factors were capable of stimulating “good” to “excellent” results for SOX2 for at least one of the concentrations tested (Fig. 2D). However, not a single factor maintained all three markers (P > 0.05) at levels comparable to ICM controls.
Assessment of combinations of growth factors and inhibitors on ICM outgrowths
We formulated combinations of growth factor treatments based on testing of single cytokines (above) that resulted in “good” or “excellent” results. Thus, we evaluated two to four factor-containing cocktails comprised of fLIF at 1, 10, 20, or 100 ng/mL with hLIF at 1, 10, or 100 ng/mL and/or FGF2 at 5 and 10 ng/mL (Table 1). All combinations, except Treatment 2, increased (P < 0.05) cell number in ICM outgrowths (Fig. 3A) compared with control day 6. “Excellent” treatments for NANOG were combination Nos. 11, 12, and 17 (Fig. 3B). For POU5F1, “excellent” treatments were combination Nos. 4, 5, 9, and 17 (Fig. 3C). For SOX2, “excellent” treatments were combination Nos. 9, 14, and 17 (Fig. 3D). NANOG, POU5F1, and SOX2 intensities were increased (P < 0.05) compared with Control D6 in most of the treatments, although no combination sustained (P < 0.05) all three protein expressions compared with ICM (Fig. 3).
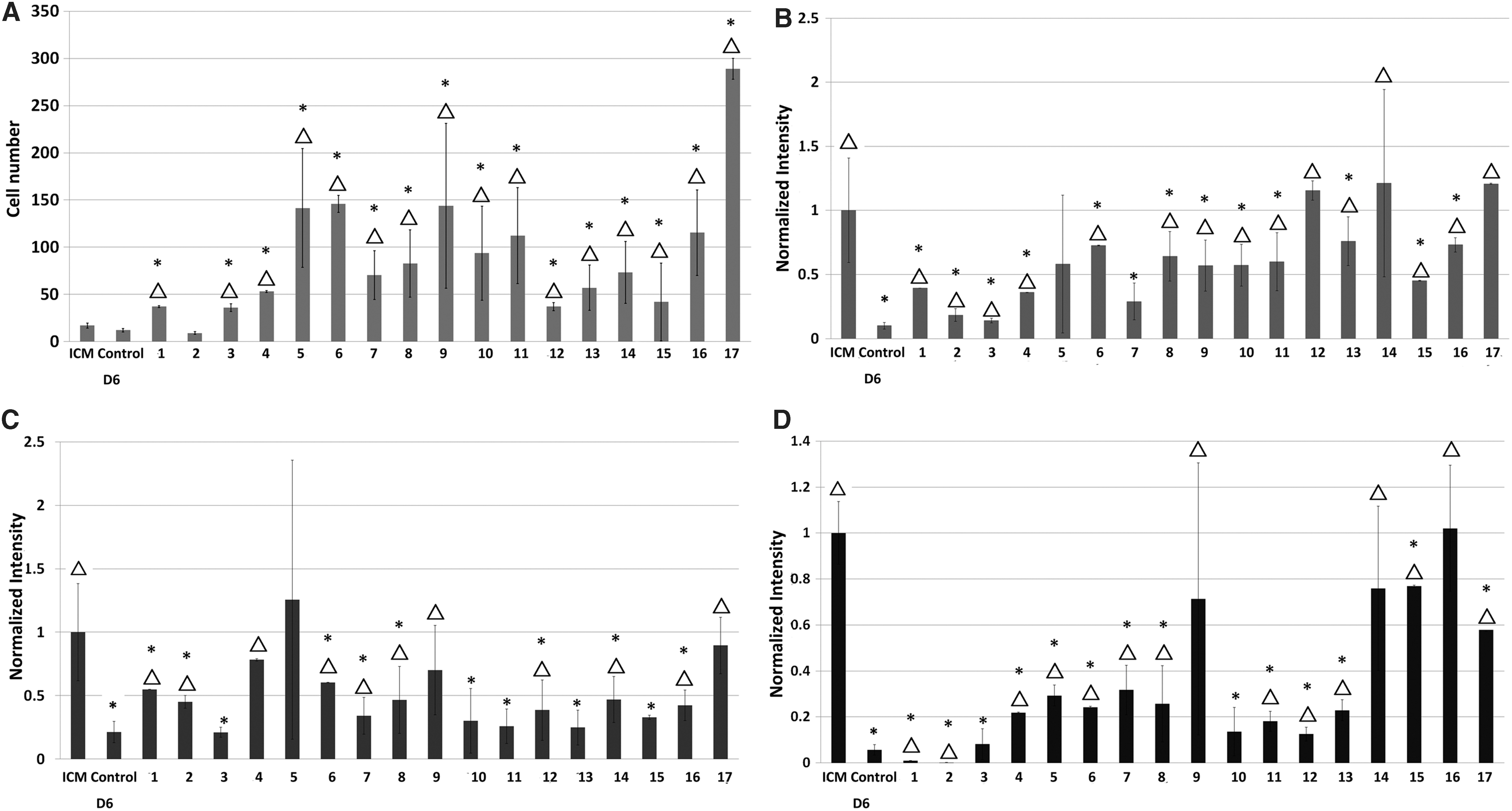
Characterization of ICM outgrowths cultured in vitro for 6 days with combinations of growth factors (FGF2, hLIF, and fLIF) at different concentrations and inhibitors (GSK3i and MEKi). Combinations are detailed in Table 1. Positive controls were ICM in expanded blastocysts on day 0 (ICM). Other control groups were ICM outgrowths cultured for 6 days without growth factor supplementation (Control D6).
To better illustrate the capacity of each treatment to collectively sustain production of all three protein markers, relative intensities of each in individual ICM outgrowths were plotted three dimensionally (Fig. 4). One treatment in particular was highly similar to the ICM control: 10 ng/mL fLIF +10 ng/mL hLIF +3 μM GSK3i + 1 μm MEKi (Treatment 17) giving the shortest Euclidean mean (Supplementary Table S4). This combination then was selected for further propagation of embryonic cells derived from ICM outgrowths.
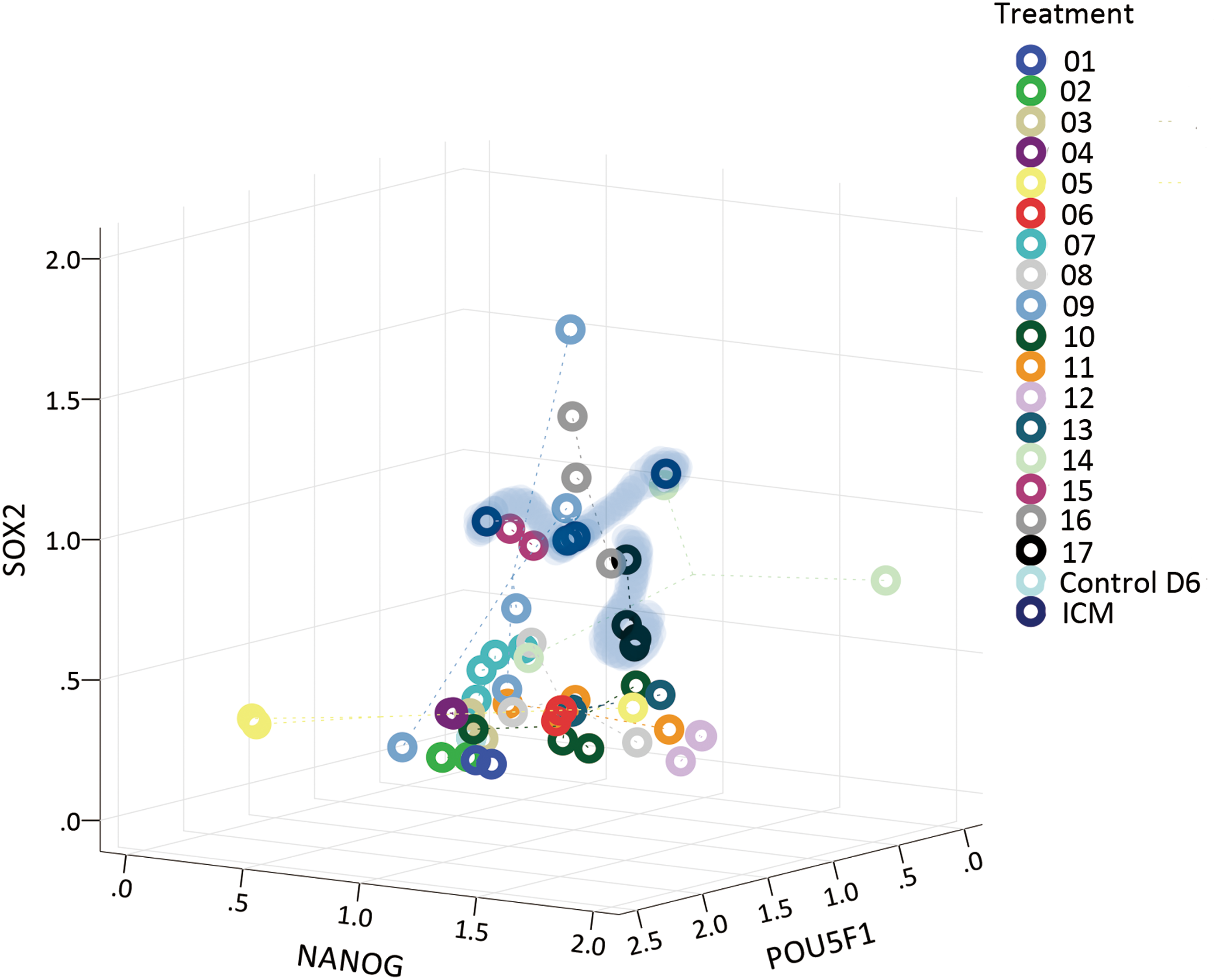
Scatter plot of individual ICM outgrowths cultured with cytokine combinations, positioned by relative intensities of proteins. Individuals in ICM group and selected treatment group No. 17 (fLIF 10 ng/mL + hLIF 10 ng/mL + GSK3i 3 μM + MEKi 1 μM) in Table 1 with smallest Euclidean mean are highlighted by blue shading. Color images are available online.
Propagation of embryonic cells with feline and hLIF plus GSK3i and MEKi
The percentage of colony formation (colony formed per ICM) after the first single-cell passage of the initial outgrowths was 10% ± 8.7%. Through 10 passages, colonies exhibited dome to flat shape morphologies (Fig. 5A) indicating that cells were stabilized. Genotyping of cells that formed colonies confirmed derivation from cat ICM outgrowths and not from mEFs used as feeder cells (Fig. 5B). More than 80% of cells from colonies (24/28, 19/23, 17/21) had normal chromosome numbers (N = 38) (Fig. 5C).

Characteristics of ICM outgrowths and derived cells at different passages.
All three proteins were present in embryonic cells after 10 passages, whereas NANOG and POU5F1 were detectable, but with lower intensity than ICM (Figs. 5D and 6B). The overall transcript profiles between ICM and embryonic cell passage 10 were similar (P > 0.05) (Figs. 1B and 6A). mRNAs of NANOG, POU5F1, SOX2, FGF2 receptor, MEK, and ERK in embryonic cells remained at comparable levels to ICM controls. KLF4, CMYC, and LIF-receptor decreased, whereas STAT3 increased (Figs. 1B and 6A). mRNA levels of Activin A-receptor, BMP2-receptor, and SMAD4 were unchanged compared with ICM (Fig. 6A and Supplement Fig. S2). However, protein intensity levels of NANOG and POU5F1 were decreased (P < 0.05) compared with the ICM control (Fig. 6B). Embryonic cells generated from expanded ICM maintained many pluripotency-related features analogous to original cells.

Gene expression in ICM outgrowths and derived cells after 10 passages (EC P10).
Discussion
Our study determined if regulation of cellular pluripotency in domestic cat ESCs diverged from previously characterized mechanisms for the mouse, rat, and human. It is known that three pluripotency factors (NANOG, POU5F1, and SOX2) are shared in the mouse [12,13], rat [14], and human [13,15] with four pathways (LIF/STAT3, BMP, activin/nodal, and MEK/ERK) playing major roles in regulating their expressions [16 –18]. In cat blastocysts, NANOG, POU5F1, and SOX2 were all detected in the blastomeres, with POU5F1 being more restricted to the ICM, which is similar to what has been observed in mouse [12,13] and human [15]. However, LIF-receptor, STAT3, and BMPR2 expression were low in cat ICM in comparison to FGFR2, MEK, and ERK, implying that FGF2/MEK signaling was already activated at this stage. We also found that cat ICMs produced in vitro expressed KLF4 at similar levels to NANOG, POU5F, and SOX2, which is similar to the observation in mouse ICM and ESCs [12,13]. These five factors appeared to constitute the foundation of pluripotency circuitry in our model species. Unlike what is found in the human ICM [33,34], transcripts of BMP2-receptor and SMAD4 were absent in the cat counterpart. All sequences of PCR products had >99% identity to the transcripts or predicted transcripts previously reported [27,28]. This finding suggested that BMP2 and activin signaling pathways might not be active in cat blastocysts cultured in vitro. Additionally, this observation indicated that cytokines tested were likely targeting both the LIF/STAT3 and MEK/ERK pathways. In turn, this supported a conclusion that a fine balance between activation/inhibition of these specific passageways could sustain and balance NANOG, POU5F1, and SOX2 expression in the ICM at sufficient levels, which do not trigger differentiation.
We then evaluated the individual influence of LIF and FGF2 on ICM outgrowths, with NANOG, POU5F1, and SOX2 expression as endpoints. As measured in the mouse and human [12,13,35], LIF sustained NANOG, POU5F1, and SOX2 in the cat ICM. Yet, the impact of LIF was insufficient to sustain transcription factors at levels similar to that observed in the ICM control. This is consistent with what has been previously reported [10,11]. While STAT3 transcript level was increased in the P10 embryonic cells as compared with ICM (Fig. 6A), the LIF/STAT3 pathway did not maintain pluripotency-related proteins, suggesting this pathway was not the dominant regulatory mechanism in ICM from expanded cat blastocysts. This finding is similar to results in other studies in which LIF did not support self-renewal of pluripotent cells in cat [8,10] even though STAT3 pathways were stimulated [36]. This poor response to LIF is contrary to stimulation of this pathway observed in mouse [5,12], which has been validated to maintain transcription factor expression in naive-state stem cells. However, we observed variable effectiveness between hLIF and fLIF on transcription factor levels. hLIF and fLIF have similar amino acid composition (164 and 180, respectively; 91.1% sequence identity); given the fact that hLIF is almost as efficient as mouse LIF in maintaining mouse ESCs (with sequence identity ∼90%) [37], hLIF and fLIF were assumed to have similar effect on cat ICM outgrowths. Yet, the responses of NANOG, POU5F1, and SOX2 were different in the treatment groups with fLIF and hLIF. This variation will require further investigations assessing binding affinity or half-liFe difference. The low levels of LIF-receptor and STAT3 expressions in cat ICMs, and the resulting limited effect of LIF in upregulating NANOG, POU5F1, and SOX2 suggests that this pathway may convey only a modest contribution to the regulation of pluripotency in the cat. Other pathways could be playing a regulatory role. For example, either an inhibitor of GSK3 or a WNT3 analog can be supportive in maintaining mouse ESC in the absence of exogenous LIF by counteracting transcription factor 3, the repressor of NANOG, POU5F1, and SOX2 [4]. WNT signaling can also upregulate STAT3 transcript [38]. In mouse epiblast stem cells [13] and human primed stem cells grown on mouse fibroblast feeder cells [18,39], FGF2/MEK control and maintain pluripotency. Interestingly, even with the high levels of transcripts of its receptor and downstream mediators (Fig. 1), FGF2 supplementation alone also was unable to maintain NANOG, POU5F1, and SOX2 expressions in cat. We also observed a trend of increasing cell number, and decreasing transcription factors in ICM outgrowths, with ascending FGF2 concentration. It is likely that FGF2 might be serving as a proliferation enhancer and a pluripotency repressor within the cat ICM. This could be consistent with what has been observed in mouse ESCs, where FGF2 is known to stimulate active MEK1 suppressing the expression of Nanog [40,41]. Single-factor treatments lead to variable responses. It is known that overexpressing POU5F1 or SOX2 will lead to cell fate decisions (POU5F1—mesodermal lineage, SOX2—neural ectodermal lineage) [42]. This unbalanced increase in either POU5F1 or SOX2 was observed in our culture system, which implied an unstable balance among NANOG, POU5F1, and SOX2 within a given ICM outgrowth, and might lead to a loss of pluripotent state [42].
Based on initial findings, we tested multiple cytokine combinations on cat ICM outgrowths over a 6-day culture. To stabilize the balance of pluripotency, we supplemented the culture medium with GSK3 and MEK inhibitors. Some treatments (Nos. 4, 5, 9, 12, 14, 16, 17) maintained pluripotency markers at levels similar to the control ICM group. Nonetheless, we did not prove our original assumption that supplementations that resulted in the highest similarity in NANOG, POU5F1, and SOX2 levels would produce normal, extended pluripotency and proliferation. The impact of adding cytokines targeting different pathways resulted in enhanced cell proliferation and transcription factor response, yet in no case supporting NANOG, POU5F1, and SOX2 at a sufficient level. However, the use of a Euclidean mean assessment assisted in identifying those treatment groups producing a result approximating the ICM control. The most promising results were generated with the supplementation of LIF combined with the GSK3 and MEK inhibitors. Specifically, we demonstrated that adding the MEK inhibitor promoted the rapid proliferation of cat embryonic cells. Similar to what has been observed in rodents [5,14]; inhibiting MEK/ERK pathway did not compromise ESC growth. The morphology of resulting cell colonies remained similar from passage 2 to 10: Taking into consideration decreased levels of NANOG, POU5F1, and SOX2, change of cell status did not lead to obvious morphological differentiation. The good expansion of stabilized embryonic cells enabled us to evaluate certain pluripotent features, including the maintenance of transcription factors and signaling components supporting pluripotency. The combined cytokine treatment boosted STAT3, which is essential in pluripotency stabilization [43]. Our observation of decreased KLF4, CMYC, and LIF-receptor transcripts in cultured embryonic cells suggested that cell status may have been compromised. Furthermore, reductions in NANOG, POU5F1, and SOX2 intensity by passage 10 was inconsistent with parallel, rising mRNA concentrations. As transcript and protein levels do not correlate well in mammalian cells [44] and posttranscriptional regulation may interfere with translation of pluripotency transcription factors in embryos [45], using the protein levels to evaluate cell status is more accurate. Mouse studies have conclusively demonstrated the need for sustained levels of NANOG, POU5F1, and SOX2 proteins to promote pluripotency [43]. In agreement, our observation in cat embryonic cells suggests a loss of pluripotency with advancing passages. Consequently, we cannot conclude that our selected cytokine combinations had the capacity to sustain embryonic cells to a state analogous to ESCs reported in other studies [5,14,34].
Our cat model confirms that there is complexity in regulating NANOG-POU5F1-SOX2 circuitry, not only due to the doses needed to target hundreds of ESC hallmark genes [15], but also to their positive/negative feedback loop in regulating each other [46]. Certainly, the dynamism we observed in the cat ESC system indicated that there was no simple cytokine formula for perpetuating cell pluripotency. Deviations in signaling pathways and cellular fate may be related to natural differences among species in modes of embryo development preimplantation. This already is apparent from the notable disparities in timing and signaling requirement in mouse versus human [5,34]. Whereas a mouse blastocyst implants at approximately embryonic day 4.5 (E4.5) [47], the human counterpart embeds at E7–10 [48]. Then, more specifically in terms of mechanisms, the human epiblast experiences a highly expressed transforming growth factor-β (TGFβ) pathway [34] (eg, nodal) that, if inhibited, downregulates the core pluripotency factor NANOG [49]. However, this identical manipulation in the mouse has no discernible influence on the same metrics [49], again demonstrating a remarkable species variation in pluripotency regulation. Therefore, given that the cat embryo has a comparatively long preimplantation embryo development period and that the cat blastocyst proceeds to gastrulation by implantation (at E13–14) [19], these differences probably explain why the mouse and human protocols for sustaining pluripotency in embryonic cells were not translatable to the cat.
Our findings also help to explain the loss of pluripotency observed in previous studies attempting to create viable cat ESCs [10,11]. We demonstrated that, unlike for the mouse and human, LIF or FGF2 solely could not support NANOG, POU5F1, and SOX2 expressions. Those findings, the absence of detectable transcript for BMP2-receptor and SMAD4, and very low levels of LIF-receptor and STAT3 in the cat ICM indicated that pluripotency regulatory machinery may be unique to this model system. To sustain pluripotency in cat ESCs, the nature of cat early embryos needs to be further investigated. For instance, the dynamics of LIF-receptor, STAT3, and the Interleukin 6 (IL6) family need to be drawn, and the controlling elements on these factors have to be revealed. The relation between featured signaling pathways (LIF/IL6/STAT3, FGF2/MEK/ERK, etc.) and embryonic stage (four-cell, morulae, and ICM) would contribute to a deeper understanding of pluripotency. Furthermore, time-lapse transcriptome and proteomes analysis in cat embryos up to blastocyst stage might be necessary to finely tune the culture conditions.]
Footnotes
Acknowledgments
The authors thank Dr. Brent Whitaker (Animal Rescue, Inc.) and Dr. Keiko Antoku, and their staff for providing domestic cat ovaries. This research was supported by a JoGayle Howard Graduate Fellowship in Felid Reproduction to the first author (R.Z.) and a Smithsonian, University of Maryland seed grant for supplies and equipment.
Presented at the 50th Annual Meeting for the Society for the Study of Reproduction, “Specific combinations of cytokines enhance cell proliferation and expression of pluripotency-related proteins in inner cell mass outgrowths in the domestic cat model,” in Washington, DC. Abstract No. 244, Poster No. P66.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.