
Abstract
How mesenchymal stem cells (MSCs) interact with tumor cells and promote tumor growth is not well understood. In this study, we demonstrate that when naive MSCs and malignant breast cancer cells (MDA-MB231) were injected into opposing mammary glands of an immunodeficient nude mouse, both cell types formed tumor-like masses within 8 weeks at the injected site. Surprisingly, MDA-MB231 cells were detected in the opposing mammary gland injected with the naive MSCs, indicating migration and crosstalk between naive MSCs and MDA-MB231 cells. Furthermore, when naive MSCs preexposed to MDA-MB231-derived conditioned medium (CM; MSCCM) or purified exosomes (Exo; MSCExo) were injected into mammary glands of nude mice, they too formed a tumor-like mass with stromal tissue within 14 weeks. Interestingly, cells dissociated from these primary explants also formed tumor-like masses. Finally, injecting MSCCM or MSCExo and naive MSCs into opposing mammary glands formed tumor-like masses on the naive MSC-injected side, suggesting migration and crosstalk between MSCCM or MSCExo with naive MSCs, similar to that observed between malignant MDA-MB231 cells and naive MSCs. Importantly, molecular analysis of MSCCM and MSCExo revealed DNA hypermethylation. These data demonstrate that MSCs and breast cancer cells communicate, resulting in the transformation of naive MSCs into cells capable of forming explants in nude mice. Our data also suggest that DNA hypermethylation might have contribute to their migration. Understanding the crosstalk between MSCs and tumor cells, and identifying the players involved in their interaction, will help us develop novel therapeutics for breast cancer regression and elimination.
Introduction
Breast cancer is responsible for nearly one third of all newly diagnosed cancer cases, and a leading cause of cancer deaths in women [1,2]. Breast cancer comprises of both parenchyme and mesenchyme, and interactions between tumor cells and the surrounding stroma result in increased expression of multiple protumorigenic inflammatory mediators, including cytokines, chemokines, adhesion molecules, and growth factors [2 –4]. Importantly, this inflammatory state drives the recruitment of nonmalignant cells, including macrophages, myeloid-derived suppressor cells, and mesenchymal stem cells (MSCs) into the tumor microenvironment (TME) [5 –7], further promoting tumor growth and dissemination.
MSCs, as pluripotent stem cells, are present in all tissues that contain blood vessels. They can be identified by specific markers, including Oct4 (Organic Cation/Carnitine Transporter 4), CSPG4 (Chondroitin Sulfate Proteoglycan 4; or NG2), Nestin, SSEA-4 (Stage-Specific Embryonic Antigen-4), CD29, and CD49, and later express CD34, CD44, CD73, CD90, and CD105 as progenitor cells [8,9]. Depending on the microenvironment, MSCs have the potential to differentiate into cells of all three germ layers, including osteocytes, chondrocytes, neurons, cardiomyocytes, myocytes, hepatocytes, and cells of the hematopoietic system [10,11]. Based on their homing capacity to injured tissue, several reports have proposed a reparative function for MSCs in various organs and tissues, including skin, lung, liver, brain, heart, joints, tendons, and bone [12 –14].
We have previously demonstrated that MSCs migrate toward breast cancer cell-derived conditioned medium (CM) in vitro [15,16]. Migration and integration of MSCs into breast cancer tissue are critical in promoting tumor growth [17]. In fact, the crosstalk between cancer cells and the cells in the surrounding stroma has been shown to promote tumor growth by creating a dynamic extracellular matrix that favors invasion of tumor cells [5,18,19]. Conceivably, an important characteristic of MSC–tumor interactions is the development of a TME that forms an integral part of breast cancer progression.
The TME contains both tumor cells and nontumor cells, including vascular progenitors, activated fibroblasts, immune cells, and undifferentiated early MSCs, all of which contribute to stromal structure that takes part in tumor cell invasion, growth, malignant transformation, and metastasis [20]. Changes in gene expression of cellular components within the TME may also promote tumor growth and dissemination [21,22]. In fact, transcriptome analysis revealed a higher degree of tumor-related gene expression in stromal cells associated with aggressive tumors [16]. The TME also consists of an acellular component (e.g., soluble cytokines, chemokines, and growth factors, as well as exosomes [18,23,24]) that forms part of the stromal structure [20,25], enabling MSCs to specifically home to tumor tissues while avoiding peritumoral normal tissues [26]. Thus, in addition to playing a role in the communication between malignant and nonmalignant cells, the TME also contributes to tumor growth and malignant transformation. Importantly, aside from the primary tumor site, the tumor–host communication also occurs in metastatic niches, such as liver, lung, brain, and bone [5,27].
Although MSCs are identified in the TME, their role in tumor growth remain controversial. While some studies reported that MSCs suppress tumor growth and thus play a protective role, a greater body has reported their potential in promoting tumor growth in various preclinical animal models [22,28]. In this study, we investigated how the acellular fraction of the TME, specifically proinflammatory cytokines and exosomes, alters the characteristics of naive MSCs into tumor-forming cells both in vitro and in vivo.
Materials and Methods
Cell culture
Human adipose tissue-derived MSCs
Following approval by the Institutional Review Board (IRB #140571) at Tulane University School of Medicine, New Orleans, Louisiana, and informed written consent, adipose tissue specimens were collected from healthy subjects undergoing cosmetic surgery. The MSCs were isolated from ∼50 g of tissue by collagenase digestion (Collagenase Type I, Cat# A1064401; Invitrogen, Carlsbad, CA) as previously described [29]. The cells were plated at a density of 1,000 cells/cm2 in cell culture plates (Nalgene, Nunc, Rochester, NY) and cultured in Minimal Essential Medium Alpha (MEM-alpha; CellGro, Manassas, VA) containing 20% heat-inactivated fetal bovine serum (FBS; Atlanta Biologicals, Lawrenceville, GA), 1% Penicillin/Streptomycin (CellGro), and 1% L-Glutamine (CellGro) at 37°C in a humidified atmosphere containing 5% CO2. The cells were used up to passage 3. MSCs were characterized by analyzing for the surface expression of CD4, CD11b, CD34, CD45, CD44, CD90, and CD105 by flow cytometry using a Beckman-Coulter Epics FC500. The results showed that MSCs were negative for CD4, CD11b, CD34, and CD45, but positive for CD44, CD90, and CD105 (data not shown).
Green fluorescent protein (GFP) expressing human malignant breast cancer cell line MDA-MB231 (MDA-MB231/GFP, Cat# AKR-201) was purchased from Cell Biolabs, Inc. (San Diego, CA). This cell line was originally obtained from a patient in 1973 at the M. D. Anderson Cancer Center in Houston, TX. The vendor has authenticated the cells were positive for KRAS mutation (heterozygous DNA change: c.38G>A; correlates to protein sequence p.G13D), sterile (aerobic and anaerobic), and pathogen-free (PCR-based assay for HIV, HepB, HPV, EBV, and CMV). They have epithelial-like morphology and appear as spindle-shaped cells. During the experiments, the morphology of all cell lines was checked routinely under phase contrast microscope. The cells were able to grow on agarose (anchorage independence), an indication of transformation and tumorigenicity, and display a relatively high colony-forming efficiency. We monitored their tumorigenic potential by intramammary injection (5 × 105 cells in Matrigel®) every 6 months. The cells were cultured in MEM-alpha growth medium containing 10% FBS, 1% Penicillin/Streptomycin, and 1%
Conditioned medium
The CM is the supernatant of a cell culture medium without the FBS, and was collected according to a published method [30]. Briefly, MDA-MB231 cells were grown to 85% confluency in 10-cm dishes (Nalgene, Nunc, Rochester, NY), washed twice with phosphate-buffered saline (PBS; CellGro), and incubated at 37° for 72 h in serum-free MEM-alpha medium with supplements. The culture supernatants were then collected, centrifuged (300 g for 10 min followed by 1,200 g for 10 min), filter sterilized through a 0.2 μm filter, and stored at −20°C until use.
Exosome enrichment
Exosomes are cell-derived vesicles present in various bodily fluids [31]. To concentrate exosomes from the MDA-MB231 cell-derived CM, we used a series of ultracentrifugation steps as previously described [24]. The CM was spun at 115,000 g for 1 h at 4°C in a SW41Ti ultracentrifuge (Beckman Coulter, Fullerton, CA). After discarding 90% of the supernatant, 3.6 mL of PBS was added and layered onto a 30% sucrose/D2O density cushion and spun at 115,000 g for 1 h at 4°C. The supernatant (700 μL) was collected and spun again at 115,000 g for 1 h at 4°C. The pellet was then resuspended in PBS and centrifuged at 115,000 g for 1 h at 4°C. Following one last wash, the pellet was resuspended in PBS and stored at −80°C. The concentration of exosomes was determined by quantifying the protein content using NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA).
Treatment of MSCs with MDA-MB231-derived CM or exosomes
MSCs were plated (5 × 105 cells) in MEM-alpha medium containing 20% FBS, 1% Penicillin/Streptomycin, and 1%
Animal experiments
All animal protocols were approved by the Animal Care and Use Committee at the Tulane University School of Medicine in New Orleans, LA, and conformed to the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health (DRR/National Institutes of Health, 1996). Female 6–8-week-old immunodeficient NIH-III nude mice (hereafter referred to as nude mice) were purchased from Charles River Laboratories, Inc. (Wilmington, MA), and maintained in a 12-h light/12-h dark cycle barrier facility, with food and water available ad libitum.
All cell types injected into nude mice were suspended in Matrigel (Cat#354234; Corning, NY) in a final volume of 100 μL. MDA-MB231/GFP cells (5 × 105 cells in 100 μL Matrigel) were injected subcutaneously into the fourth left mammary gland of nude mice. Simultaneously, the opposing mammary gland (fourth right mammary gland) was injected with naive MSCs (5 × 105 cells in Matrigel) (Fig. 1. I: Schematic experimental design). After 8 weeks, the mice were euthanized and tumor-like masses collected for further analysis. Four groups of animals served as controls (n = 3/group): (i) naive MSCs in Matrigel injected on one side and PBS into the opposing mammary gland, (ii) MDA-MB231 cells in Matrigel injected on one side and PBS into the opposing mammary gland, (iii) Matrigel on both sides, and (iv) PBS on both sides (Supplementary Fig. S1). All animals were monitored for 14 weeks (Supplementary Fig. S1A–D).
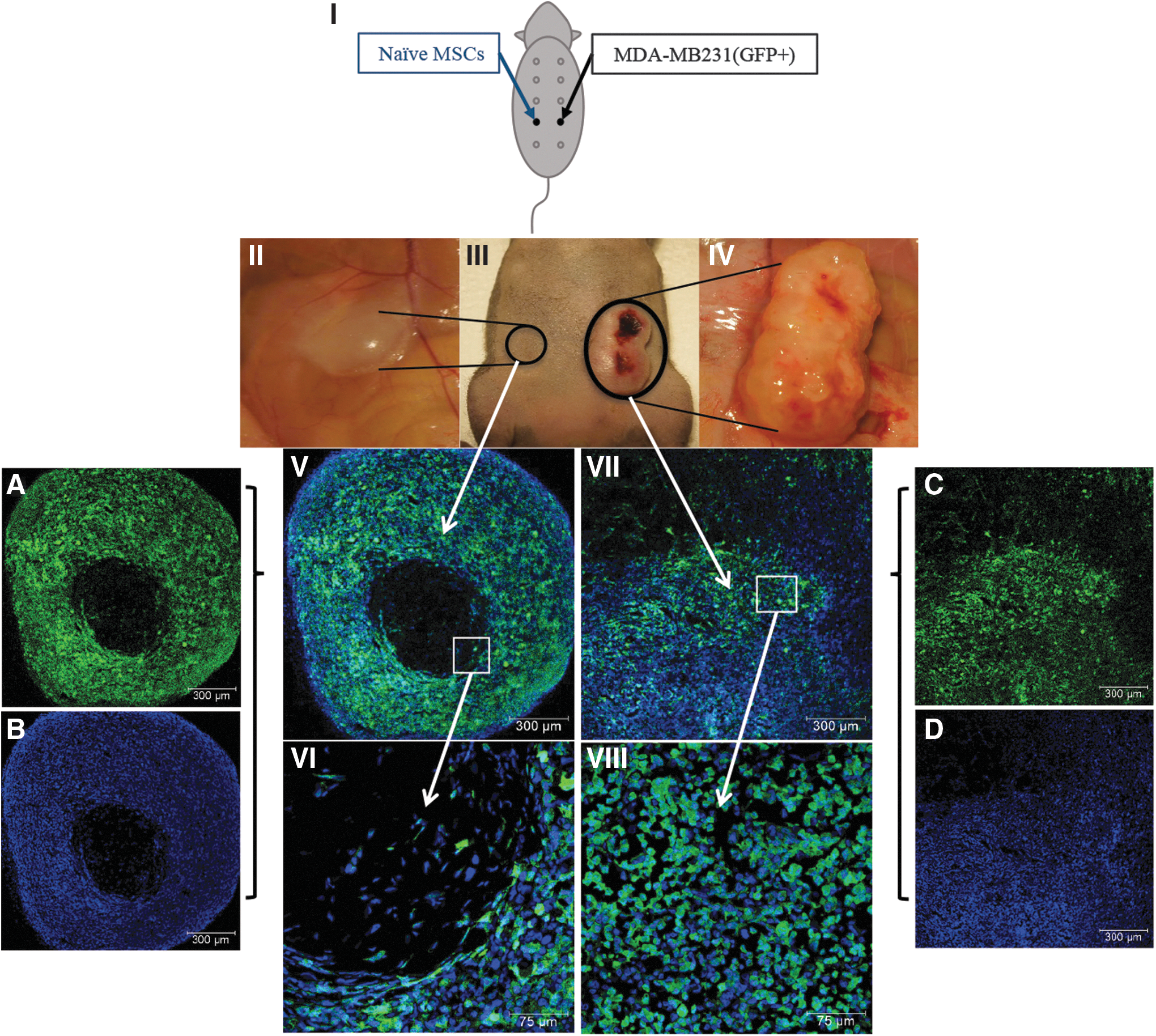
Formation of tumors due to migration of tumor cells toward MSCs in vivo (Experimental series I):
Administration of MSCs exposed to MDA-MB231-derived CM or purified exosomes
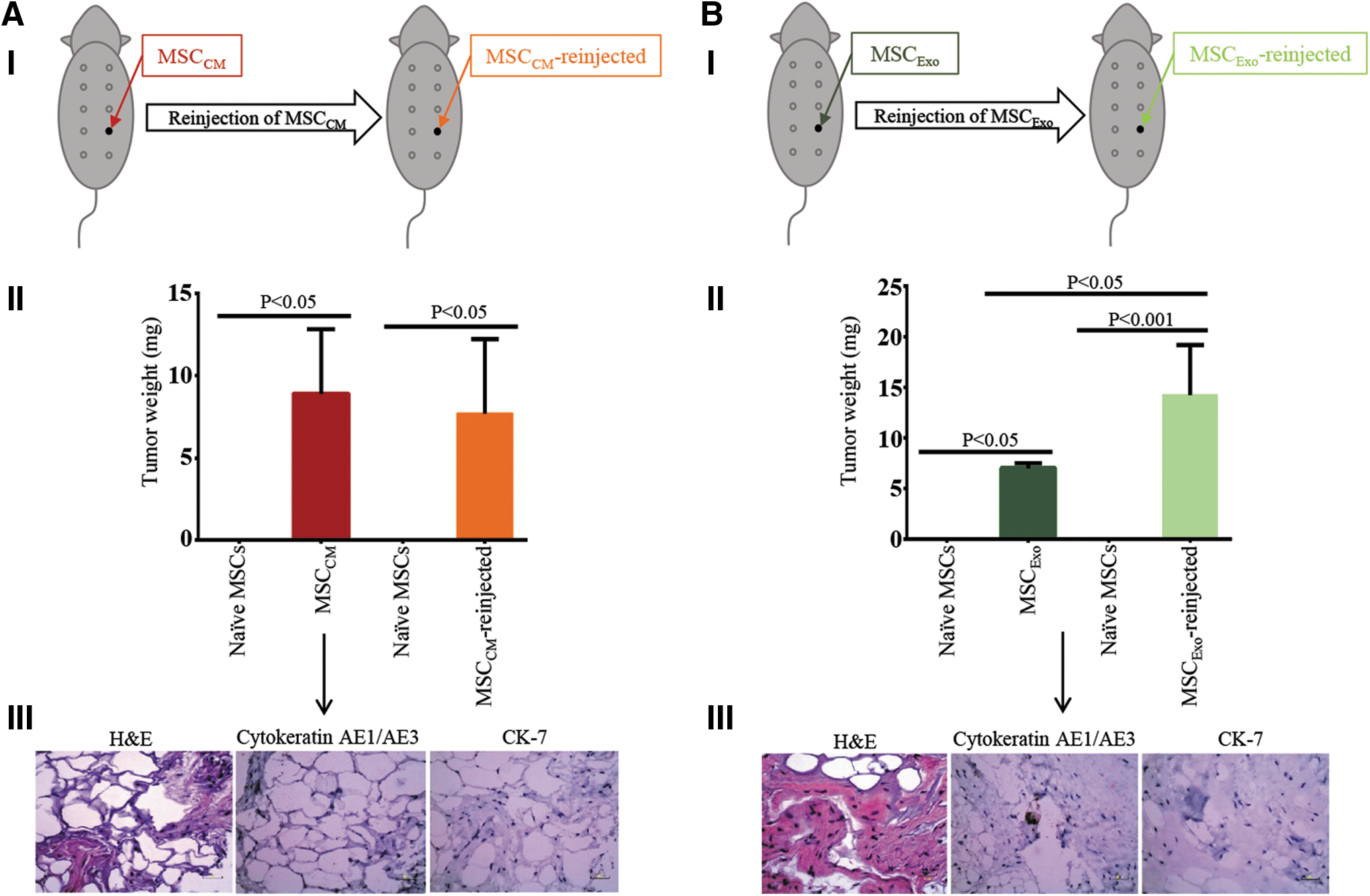
Naive MSCs (5 × 105 cells) exposed to MDA-MB231-derived CM (MSCCM) or purified exosomes (MSCExo) were injected into the fourth mammary gland on the left side of nude mice (n = 3 mice/group). After 14 weeks, the growing tumor-like masses were harvested, dissociated into single cells, and injected (5 × 105 cells) subcutaneously into the fourth left mammary gland of a new batch of nude mice (n = 3 mice/group). Animals were monitored for growth for 14 weeks. Since two out of three mice reinjected with MSCCM grew tumor-like masses, the third mouse was excluded from statistical analysis to prevent a false report of low average weight of the tumor-like masses grown. For better understanding, please see Figs. 2AI, BI and 4. Control animals (Supplementary Fig. S2A–C) were similarly monitored for 14 weeks.
Intramammary engraftment of MSCCM and MSCExo (Experimental series II):
Administration of MSCCM or MSCExo with naive MSCs into opposing mammary glands (Experiments series III):

Experimental Design-Injection Strategy. This schematic summarizes the experiments conducted under Experimental series I, II, and III including the injected sites and the results. The column on the left indicates the experiments previously described in Figs. 1 –3 and Supplementary Figs. S3 and S4. The middle column describes the cells injected subcutaneously in the mice as displayed in the right column. The color pattern of the middle and the right columns correlate. +, growth of a tumor-like mass, −, no growth.
Administration of MSCCM or MSCExo and naive MSCs into opposing mammary glands
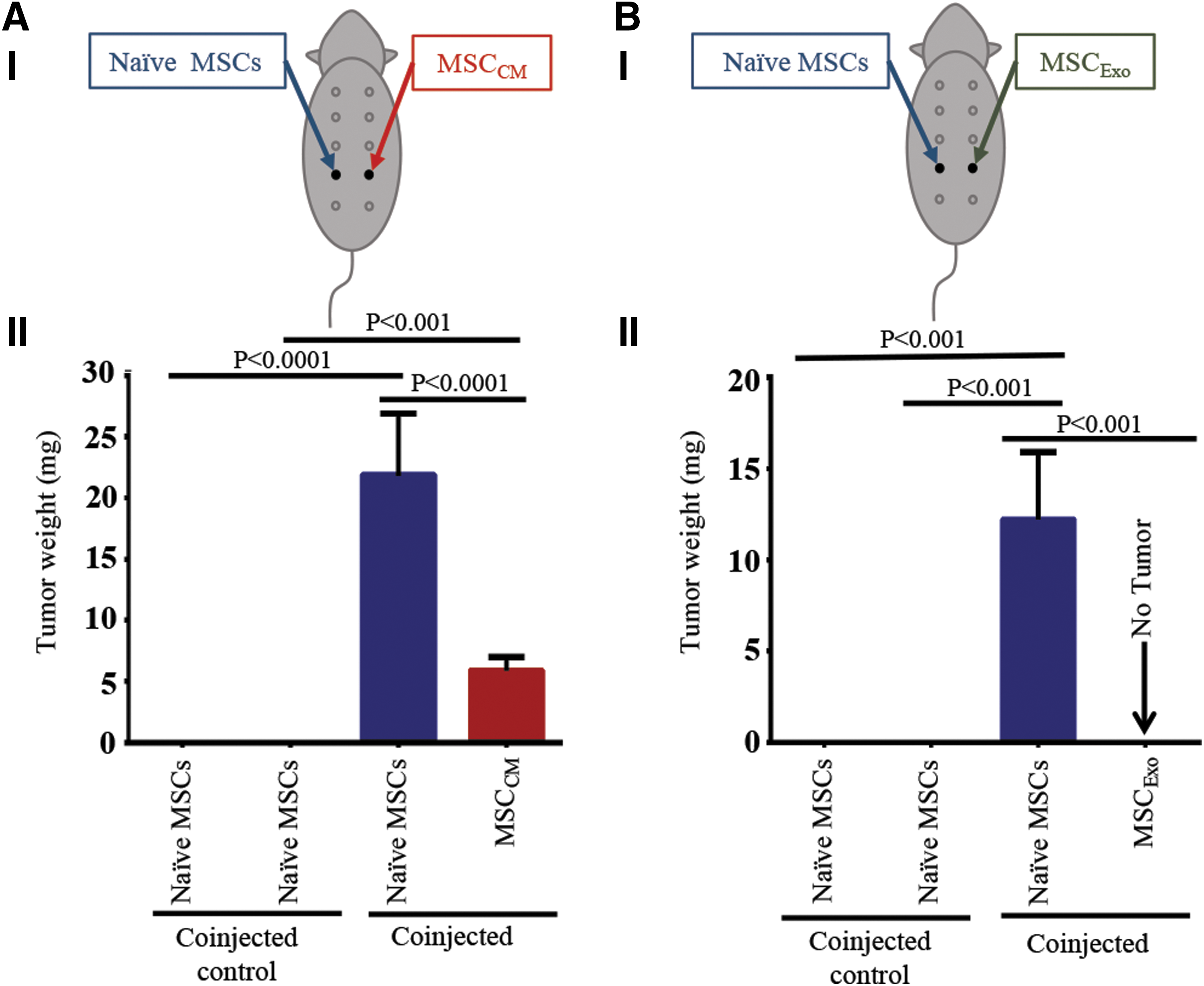
To determine the crosstalk between MSCCM or MSCExo and naive MSCs, we performed additional in vivo experiments. In this study, MSCCM or MSCExo (5 × 106 cells) were injected into the fourth left mammary gland of nude mice (n = 3). Naive MSCs (5 × 106 cells) were injected into the opposing mammary gland (schematic experimental design Fig. 3AI, BI). After 14 weeks, growing tumor-like masses were harvested for analysis. In MSCCM and naive MSCs injected animals, only two out of three mice developed tumor-like masses in the MSCCM-injected side, whereas tumor-like masses grew in all naive MSC-injected sides. Again to prevent a false report, the outlier was excluded from statistical analysis. The tumor-like masses derived from naive MSCs and MSCCM as well as naive MSCs and MSCExo were harvested, dissociated, and injected into new groups of nude mice (n = 3) to confirm their potential to form tumor-like masses again (Fig. 4 and Supplementary Fig. S4AI, BI). Once again, the animals injected with PBS, Matrigel, or naive MSCs served as controls (n = 3 mice/group) (Supplementary Fig. S5A–C).
Immunofluorescence and histology
The tumor-like masses were fixed overnight in 4% paraformaldehyde, treated with a cryoprotectant (30% sucrose/1× PBS; Sigma-Aldrich, St. Louis, MO), frozen in OCT (Optimum Cutting Temperature) compound (Sigma-Aldrich), and stored at −80°C. Twenty micrometer-thick sections were stained with DAPI (Molecular Probes, Eugene, OR). The GFP expression indicates the presence of MDA-MB231 cells. Images were obtained using a Leica TCS SP-2 confocal microscope equipped with Argon (457–477 nm; 488 nm, 514 nm) and HeNe lasers (543 nm; 633 nm).
For histological analysis, the tumor-like masses derived from MSCCM and MSCExo were fixed in 10% neutral buffered formalin for 24 h, embedded in paraffin, cut into 4 μM-thick sections and stained for H&E, Cytokeratin AE1/AE3 or CK-7 according to the manufacturer's protocol (Agilent, CA). The entire stained slide was evaluated for immunostaining by light microscopy. Photomicrographs were obtained with a Leica Microscope.
Quantification of global methylation level
Genomic DNA was extracted using the GenElute™ Mammalian Genomic DNA Miniprep Kit (Cat. # G1N70–1KT; Sigma-Aldrich). DNA levels were quantified photometrically using NanoDrop 2000. Changes in global DNA methylation levels were analyzed by an ELISA (Imprint® Methylated DNA Quantification Kit, Cat. # MDQ1; Sigma-Aldrich). A plate reader at 450 nm was used for signal detection. A minimum of three independent experiments was performed for each condition.
Methylation array
Genomic DNA was extracted using the GenElute Mammalian Genomic DNA Miniprep Kit (Sigma-Aldrich). Methylation levels of 96 tumor suppressor gene promoters were analyzed in naive MSCs and MSCExo using EpiTect® Methyl II PCR array Human, Breast Cancer, Complete Panel (Cat. # EAHS-3010Z; Qiagen, Valencia, CA). The array was performed using a ICycler MyIQ (BioRad, Hercules, CA). CT values were generated using iQ5 software. The data were then exported and analyzed with an Excel-based data analysis template (
Statistics
Data are presented as mean ± SD in each figure. Comparisons between values were analyzed using analysis of variance (ANOVA) and Tukey multiple comparison test for post hoc analysis. The results are expressed as the mean difference in variables together with 95% confidence intervals, as indicated in the Results section (the numbers are indicated in parenthesis). All mean difference hypotheses were tested at 5% level of significance throughout the analysis and significant results reported at P < 0.05 (*P = 0.05–0.01; **P = 0.01–0.001; ***P = 0.001–0.0001; ****P < 0.0001).
Results
Experimental series I: interactions between malignant breast cancer cells and MSCs in vivo
We previously reported that MSCs migrate toward CM derived from MDA-MB231 cells in vitro [16]. However, interactions between breast cancer cells and MSCs in vivo have not been investigated thoroughly. To investigate this interaction, we injected GFP-labeled MDA-MB231 cells into the fourth mammary gland of nude mice. At the same time, a similar number of naive MSCs were injected into the opposing mammary gland (Fig. 1-I). The results demonstrated that while MDA-MB231-injected mammary glands showed tumor growth starting from 2 weeks postinjection, the MSC-injected side showed visible growth starting from week 6 postinjection (Fig. 1).
Histological analysis of the tissues demonstrated the presence of GFP-positive MDA-MB231 cells on both sides (Fig. 1V, VII). The tumor-like masses formed on the MSC-injected side primarily contained naive MSCs at the center surrounded by a dense layer of GFP-positive MDA-MB231 cells (Fig. 1V and A, B). However, tumors on the MDA-MB231-injected side contained mainly MDA-MB231 cells (Fig. 1VII and C, D), indicating that MDA-MB231 cells migrated toward naive MSC-injected side. Among the four control groups, tumors were developed only in the MDA-MB231-injected group, whereas the others showed no growth even after 14 weeks (Supplementary Fig. S1). These results indicate that although breast cancer cells and MSCs are in distinct anatomical sites, MDA-MB231 cells migrate toward MSC-injected site, indicating a crosstalk between cancer cells and naive MSCs.
Experimental series II: naive MSCs transform into cells capable of growing tumor-like masses in nude mice when exposed to breast cancer cell-derived CM or exosomes
To understand the effects of TME on MSCs, we next examined whether exposure of naive MSCs to MDA-MB231-derived CM or purified exosomes transforms them into cells that can grow tumor-like masses in nude mice. Therefore, naive MSCs incubated with MDA-MB231-derived CM (MSCCM) or purified exosomes (MSCExo) for 14 h were mixed with Matrigel and injected into the fourth mammary gland of nude mice (Fig. 2AI, BI). The animals that received naive MSCs in Matrigel, Matrigel alone, or PBS alone served as controls (Supplementary Fig. S2). A tumor-like mass appeared beginning 8 weeks postinjection in animals injected with MSCCM or MSCExo. After 14 weeks, tumor-like masses were harvested, measured, and weighted (Fig. 2AII, BII). Both groups of animals showed consistent growth of tumor-like masses and of similar size (Fig. 2AII, P < 0.05, [4.085, 13.715] & Fig. 2BII, P < 0.05 [2.347, 11.653], respectively; Supplementary Fig. S3AI, BI). In these animals, no metastasis was detected in other organs. Control groups also showed no growth 14 weeks postinjection (Supplementary Fig. S2A–C).
Histological analysis showed formation of stromal structures in tumor-like masses formed by both cell types, MSCCM and MSCExo. However, no markers of malignancy, such as Cytokeratin AE1/AE3 and CK-7, were detected in these masses (Fig. 2AIII, BIII). These data indicate that exposure to breast cancer-derived CM or exosomes can transform naive MSCs into cells capable of growing tumor-like masses in nude mice.
We have demonstrated that both MSCCM and MSCExo form tumor-like masses (Fig. 2); however, it is not known whether the cells derived from these primary tissue masses retain the potential to form tumor-like masses again in nude mice. Therefore, cells were dissociated from the primary tumor-like masses formed by the MSCCM and MSCExo and injected into the fourth mammary gland of a new batch of nude mice. Once again, mice developed a visible tumor-like mass beginning 8 weeks postinjection (Fig. 2AII; P < 0.05, [2.640, 13.760] & Fig. 2BII P < 0.001, [9.581, 18.886], Supplementary Fig. S3). Interestingly, a significant difference in size was only found between explants of MSCExo and MSCExo-reinjected (Fig. 2BII, P < 0.05, [1.861, 12.606]). Again, no markers of malignancy, such as penetration of the peritoneum or visible growth in other major organs, were detected in these MSCCM-reinjected and MSCExo-reinjected explants. These results are summarized in Fig. 4.
Experimental series III: naive MSCs communicate with MSCCM and MSCExo
To evaluate possible communication between naive MSCs and MSCCM or MSCExo, MSCCM or MSCExo cells in Matrigel were injected into the left fourth mammary gland of nude mice, whereas naive MSCs in Matrigel were injected into the opposing mammary gland (right fourth mammary gland) (Fig. 3AI, BI). Surprisingly, significant growth was observed on both sides 14 weeks postinjection in two out of the three mice. However, a growth on the side injected with naive MSCs was consistent in all three mice (Fig. 3AI, AII, MSCCM vs. naive MSCs, P < 0.001, [4.2540, 7.5460]; naive MSCs coinjected with MSCCM vs. naive MSCs, P < 0.0001, [17.3540, 20.6460]). However, in mice injected with MSCExo and naive MSCs in opposing mammary glands, growth of a tumor-like mass was seen only on the naive MSC-injected side (Fig. 3BI, BII, P < 0.001, [8.8262, 15.6405]). Since we have shown that a single injection of MSCExo, but not naive MSCs alone, can form tumors in mice, we hypothesize that MSCExo cells have migrated to the naive MSC-injected side, enabling naive MSCs to also grow explants in nude mice (Fig. 3AII, BII).
In the next series of experiments (Supplementary Fig. S4), we evaluated whether cells dissociated from explants derived from MSCCM and naive MSCs can grow tumor-like masses again. Therefore, cells derived from MSCCM and naive MSC explants were injected into opposing mammary glands of a new batch of nude mice. While there was no detectable growth on the side injected with cells derived from MSCCM explants (Supplementary Fig. S4 AIII.3), the side injected with cells derived from naive MSC explants showed a significant growth (Supplementary Fig. S4-AIII.4, P < 0.05, [2.999, 15.668]). These results support our previous studies in mice injected with MSCExo and naive MSCs (Fig. 3). Regarding the explants developed by the coinjection of MSCExo and naive MSCs, we saw a growth only on the naive MSC-injected side (Fig. 3BII). Therefore, only cells of naive MSCs were dissociated and reinjected in three new nude mice (Supplementary Fig. S4BI). In accordance to the previous experiments, a growth was detected on the side injected with naive MSCs (Supplementary Fig. S4BIII.3, P < 0.05, [0.1872, 4.679]). Once again, no metastasis in major organs, such as liver, kidneys, lung, peritoneum, etc., was visible. These results are summarized in Fig. 4.
Exposure to MDA-MB231-derived CM or exosomes alters methylation status in MSCs
We have demonstrated that MSCs exposed to MDA-MB231-derived CM (MSCCM) or exosomes (MSCExo) can transform into cells capable of forming tumor-like masses in nude mice (Fig. 2). Since methylation status of the genome plays an important role in breast cancer development and progression, and as global methylation levels directly correlate with invasiveness of a subtype of breast cancer [32], we analyzed the methylation status in MSCCM, MSCExo, and control naive MSCs using an ELISA-based technique. The analysis revealed that both MSCCM and MSCExo cells show a significant increase in global DNA methylation levels compared with naive MSCs (MSCCM vs. naive MSCs, P < 0.05; MSCExo vs. naive MSCs, P < 0.0001) (Fig. 5). Interestingly, MSCExo showed markedly higher levels of methylation compared with MSCCM (MSCExo vs. MSCCM, P < 0.01).

Effect of MDA-MB231-derived CM or purified exosomes on methylation levels in MSCs: Global methylation levels in naive MSCs, MSCCM, and MSCExo were analyzed and quantified using an ELISA-based technique. Global methylation levels increased significantly in both MSCCM and MSCExo compared with naive MSCs. The methylation levels of naive MSCs are considered baseline in the graph (n = 3/cell type).
Since MSCExo showed markedly higher levels of methylation than MSCCM, we next analyzed the methylation status of specific gene promoters specifically in MSCExo cells. The analysis showed that the promoter methylation status of nine genes was altered in MSCExo cells compared with naive MSCs (Fig. 6). According to the specifications of the assay, promoter methylation between 10% and 20% results generally in the downregulation of a specific gene, and upregulation when the methylation levels are below 10%. For example, the methylation level of CXCL12 (C-X-C Motif Chemokine Ligand 12) was decreased from 98.13% (naive MSCs) to 0.77% in MSCExo. CXCL12 promotes tumor progression in adenocarcinoma of the esophagogastric junction as well as proliferation, migration, and metastasis of ovarian cancer cells [23,33]. The methylation level of CYP1B1 (Cytochrome P450 Family 1 Subfamily B Member 1) decreased from 45.18% to 1.86%. CYP1B1 plays a role in hormone-induced cancer. CYP1B1 is also expressed in MDA-MB231 cells and plays an important role in responsiveness of tumors to chemotherapeutic drugs [34]. On the other hand, HOXA5 (Homeobox A5; naive MSCs: 43.37% vs. MSCExo: 3.39%) as well as PER2 (Period Circadian Regulator 2; naive MSCs: 20.00% vs. MSCExo: 1.03%) showed a significant reduction in methylation after exposure to exosomes (Fig. 6). HOXA5 plays a role in dedifferentiation of cells [35] and PER2 is a key regulator of circadian rhythm in human bone marrow MSCs, and therefore involved in increasing the migration potential if expressed [36]. For tumor suppressor genes such as PLAGL1 (PLAG1-Like Zinc Finger 1) [37], GADD45A (Growth Arrest And DNA Damage-Inducible Alpha) [38] or CST6 (Cystatin E/M) [6], higher promoter methylation levels have been reported in malignant cells. Our results confirmed those reports, and demonstrated that the promoter methylation level of PLAGL1 increased from 3.31% to 37.69%, that of GADD45A from 0.57% to 23.05%, and that of CST6 from 0.03% to 13.18%. The highest methylation level after exposure to exosomes was found for MSX1 (Msh Homeobox 1; 0.06% to 97.82%), a gene involved in apoptosis and inhibition of migration [39,40]. These data indicate that tumor-derived exosomes exert a tumor-enhancing effect in MSCs through a change in the methylation status of specific genes.
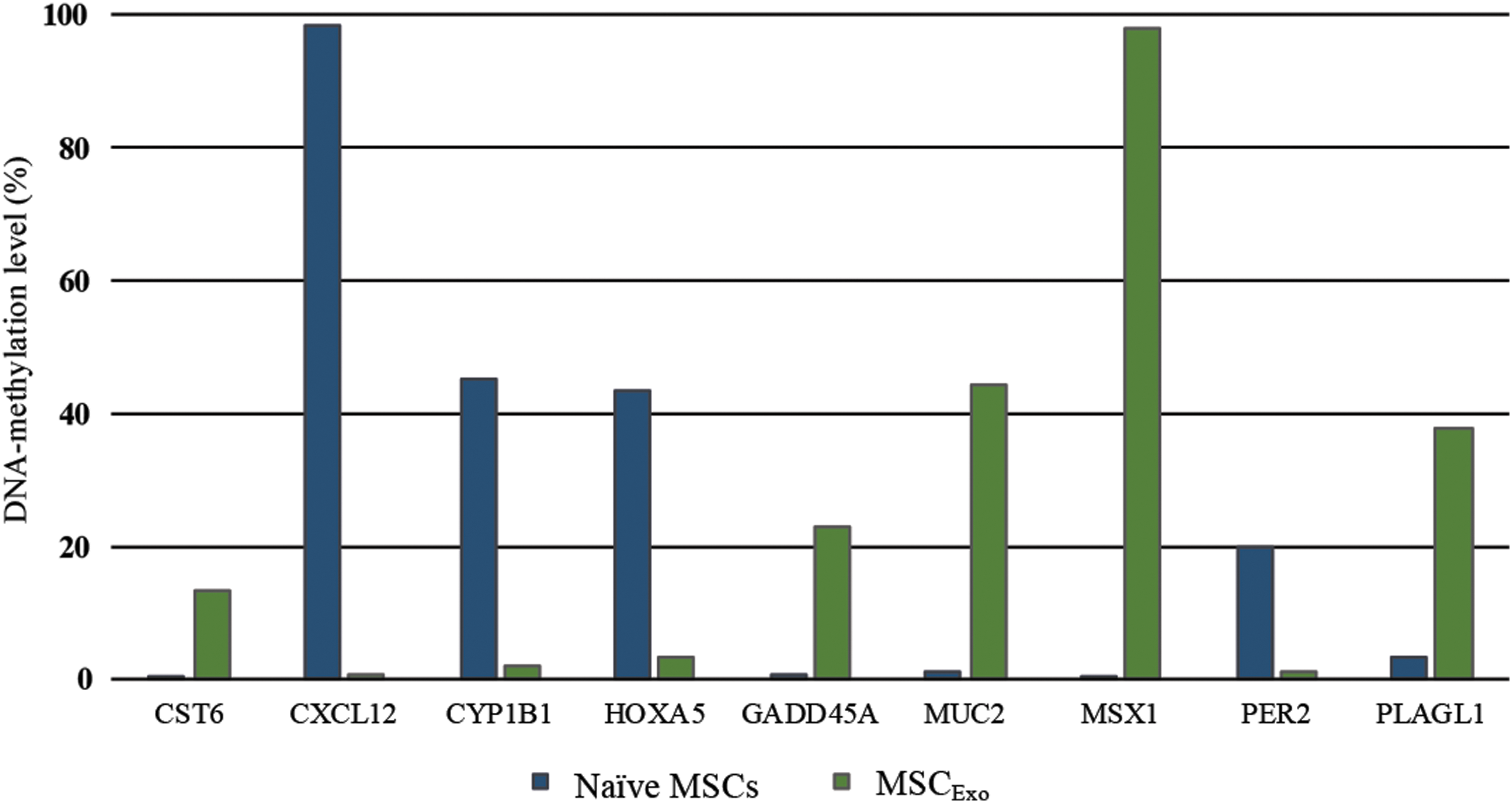
MDA-MB231-derived purified exosomes modulate methylation levels of specific tumor-suppressor gene promoters in naive MSCs: The promoter methylation levels of 96 tumor-suppressor genes were analyzed in naive MSCs and MSCExo using a qPCR Array. A higher methylation level of GADD45A, PLAGL1, CST6, MUC2, and MSX1 promoters was detected in MSCExo compared with naive MSCs. However, the methylation levels of CXCL12, CYP1B1, HOXA5, and PER2 promoters were decreased (P < 0.05).
Discussion
Here we report several novel findings; (i) the breast cancer TME can significantly modify the biological characteristics of recruited MSCs, and these effects are mediated through a crosstalk between malignant cells and MSCs by means of elements within the TME. The results of our previous publications [5,16], when viewed alongside our novel data in the present study, suggest that malignant breast cancer cells and MSCs exhibit bidirectional migration affinities (Fig. 1), (ii) exposure to tumor cell-derived CM or exosomes can transform naive MSCs into cells capable of forming tumor-like masses in nude mice (Fig. 6), (iii) cells derived from MSCCM and MSCExo retain the potential to grow explants, indicating that the changes observed in MSCs exposed to CM or exosomes are stable, and (iv) when naive MSCs and MSCCM or MSCExo were injected into the opposite mammary gland of a nude mouse, a tumor-like mass was formed at the naive MSC site (Fig. 3). Our results also suggest that (v) MSCExo exhibit higher migratory potential than MSCCM, possibly due to higher DNA methylation. Altogether, these data indicate that naive MSCs can transform into cells capable of forming explants in nude mice when exposed to tumor cell-derived CM or exosomes (Fig. 7). Therefore, it is likely that a similar mechanism may be operative in breast cancer contributing to tumor growth.
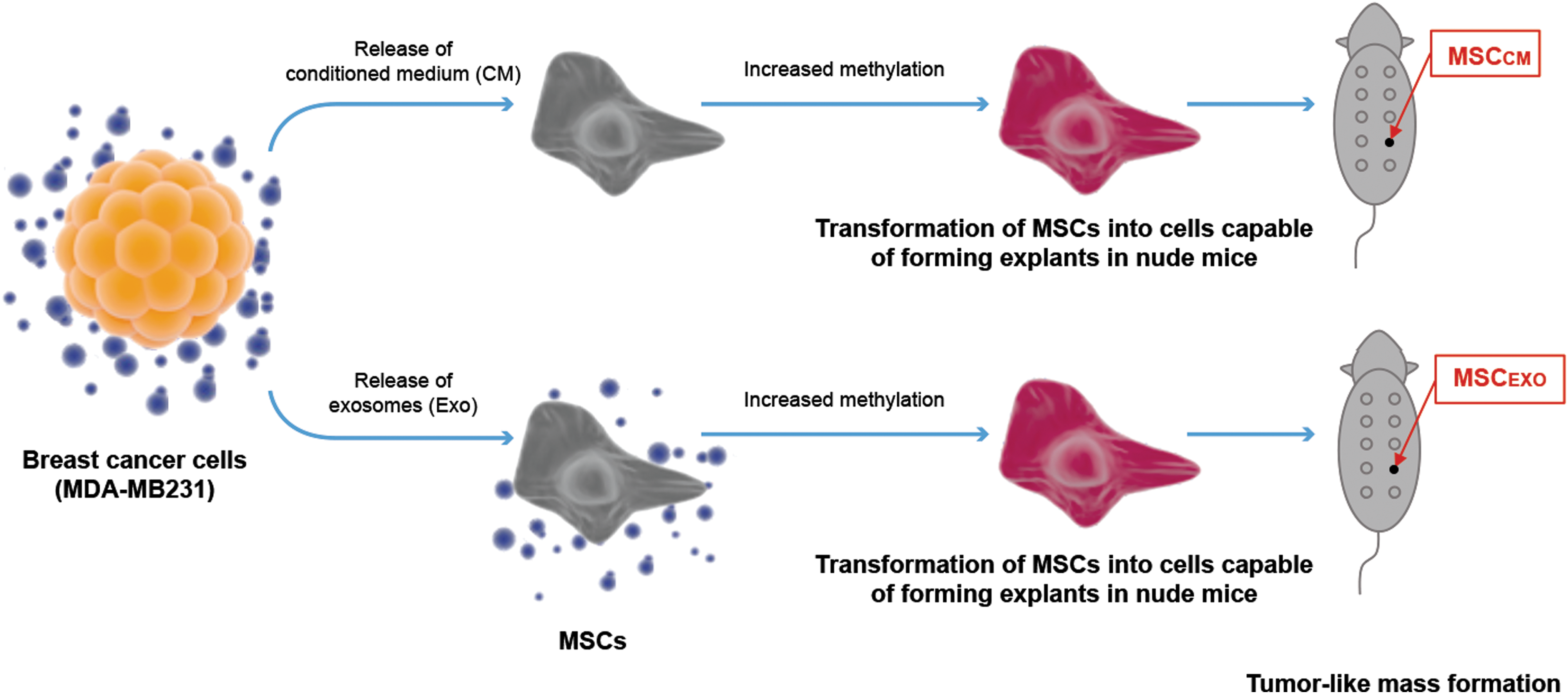
Schematic showing that malignant breast cancer cell-derived CM or purified exosomes transform naive MSCs into cells capable of forming tumor-like masses in nude mice. CM, conditioned medium.
In addition to modulating gene expression in surrounding stromal cells, tumor cell-derived exosomes also play a role in communicating with nonmalignant cells [41,42]. In fact, exosomes derived from tumors, such as breast, mesothelioma, gastric, and prostate cancers, can induce the differentiation of stromal cells into tumor-associated myofibroblasts that support tumor growth, neoangiogenesis, and metastasis [41,43]. A particularly effective way of communication between exosomes and recipient cells is through surface receptor interaction or internalization [41]. Exosomes possess surface receptors, such as tetraspanins, CD63, CD9, major histocompatibility complex I and II, and lipid raft-associated and adhesion proteins [41]. Exosomes can also stimulate the release of MMPs to promote migration [44]. Exosomes released by breast cancer-associated fibroblasts also stimulate cancer cell motility and metastasis through activation of the autocrine WNT planar cell polarity pathway [45].
In addition to the effects of tumor cell-derived purified exosomes on MSCs, we also investigated the effects of CM on MSCs. The CM contains microRNAs, mRNAs, cytokines, and exosomes [24,46]. Evidence suggests that microRNAs and mRNAs can be transferred between cells through exosome uptake and modulate gene expression [47]. As shown in Fig. 1, injection of MDA-MB231 cells and naive MSCs into opposing mammary glands formed tumors on the MDA-MB231 side as expected, but also enabled tumor-like masses to grow on the naive MSC-injected side. However, when injected alone, naive MSCs failed to form any tumor-like structures (Supplementary Fig. S1). Previously, we reported that malignant MDA-MB231 cells can transform naive MSCs into tumor-supporting myofibroblasts [43,48]. In this study, we show that MDA-MB231 cells can transform naive MSCs into tumor-like cells with no malignant characteristics.
Gene mutations and DNA hypermethylation are two key cellular changes that allow cancer cells to survive and proliferate. Such changes also allow cancer cells to resist death normally triggered by DNA damage. Thus, gene mutations allow cancer cells to survive and grow rather than die by apoptosis and/or senescence [32,49]. Our data showed significant changes in DNA methylation levels in MSCs exposed to breast cancer cell-derived CM or exosomes (Fig. 5). Noticeably, MSCExo showed higher methylation level, suggesting that purified exosomes rather than conditioned media had more pronounced effects on methylation level. Furthermore, hypermethylation of specific genes can enhance the migration potential of tumor cells [50]. Our data suggest that CXCL12, PER2, and MSX1 might have also contributed to the migration potential in MSCExo cells. This could possibly explain our findings where no tumor growth was detected on the side of MSCExo, but significant growth on naive MSC-injected side. Studies are ongoing to further understand and identify specific genes and their protein products that contribute to transformation of MSCs. In addition to DNA hypermethylation, direct cell–cell communication between tumor cells and MSCs, as well as exposure of MSCs to the TME might have contributed to the potential of MSCs to grow explants, as has been reported before [51,52]. These data suggest a novel supportive role for MSCs in tumor development. These findings also might provide enhanced insight into understanding and designing novel cancer therapeutics.
In summary, the TME significantly alters the biological characteristics of MSCs as reported previously [16]. Specifically, exposure of MSCs to tumor cell-derived CM or purified exosomes can transform MSCs into cells capable of forming explants. Importantly, these changes in MSCs appear to be stable as seen by the formation of secondary explants. Histological analysis showed development of stromal structures in tumor-like masses derived from MSCCM and MSCExo, confirming that transformed MSCs can take part in tumor growth. Our data also suggest that DNA hypermethylation might play a role in higher migratory potential of transformed MSCs. Elucidating the molecular mechanisms and pathways underlying stroma–tumor interactions will help us better understand carcinogenesis and identify novel and potential targets to inhibit tumor growth. One of the limitations of the study is the lack of analysis showing direct methylation levels in the tumor-like masses. The second limitation of the study is that a mouse had to be excluded in studies involving MSCCM reinjected (Fig. 2AII) and the coinjection of MSCCM with naive MSCs (Fig. 3AII). However, we repeated the statistical analysis and included the 95% confidence intervals. In conclusion, our data show that the acellular compartment of the TME transforms the migrated naive MSCs into tumor-forming cells, possibly contributing to further tumor growth.
Footnotes
Acknowledgments
This work was supported by a grant from the Alliance of Cardiovascular Researchers (to EA). RI is supported in part by Elsa U. Pardee Foundation. BC is a recipient of the Department of Veterans Affairs Research Career Scientist award (#IK6BX004016-01), and is supported by the U.S. Department of Veterans Affairs, Office of Research and Development-Biomedical Laboratory Research and Development (ORD-BLRD) Service Award VA-I01-BX004220. The authors would like to specially thank and express their gratitude to Drs. David Jansen, Abigail Chaffin, and to Hamid Izadpanah for their support in this study. In addition, they would like to thank the Department of Pathology and Cancer Center at Tulane University School of Medicine for histological analysis and flow cytometry.
Author Disclosure Statement
The authors have declared no conflict of interest.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.