
Abstract
Mesenchymal stem cells (MSCs) have been considered promising tools for tissue engineering and regenerative medicine. However, the optimal cell source for bone regeneration remains controversial. To better identify seed cells for bone tissue engineering, we compared MSCs from seven different tissues, including four from dental origins, dental pulp stem cells (DPSCs), periodontal ligament stem cells (PDLSCs), gingival MSCs (GMSCs), and dental follicle stem cells (DFSCs); two from somatic origins, bone marrow-derived MSCs (BM-MSCs) and adipose-derived stem cells (ADSCs); and one from birth-associated perinatal tissue umbilical cord (UCMSCs). We cultured the cells under a standardized culture condition and studied their biological characteristics. According to our results, these cells exhibited similar immunophenotype and had potential for multilineage differentiation. MSCs from dental and perinatal tissues proliferated more rapidly than those from somatic origins. Simultaneously, DPSCs and PDLSCs owned stronger antiapoptotic ability under the microenvironment of oxidative stress combined with serum deprivation. In respect to osteogenic differentiation, the two somatic MSCs, BM-MSCs and ADSCs, demonstrated the strongest ability for osteogenesis compared to PDLSCs and DFSCs, which were just a little bit weaker than the formers. However, GMSCs and UCMSCs were the most pertinacious ones to differentiate to osteoblasts. We also revealed that the canonical intracellular protein kinase-based cascade signaling pathways, including PI3K/AKT, MAPK/ERK, and p38 MAPK, possessed different levels of activation in different MSCs after osteoblast induction. Our conclusions suggest that PDLSCs might be a good potential alternative to BM-MSCs for bone tissue engineering.
Introduction
B
Mesenchymal stem cells (MSCs) are multipotent adult connective tissue cells that originate from the mesoderm, and are involved in development, wound healing, and replacement of cells lost by exfoliation or in pathological conditions [2]. They can be obtained from various sources of tissues such as bone marrow, adipose tissue, heart, skin, birth-associated tissues, and tissues of the orofacial area [3 –8]. Due to their availability from different tissues, rapid proliferation, multipotent differentiation abilities, and immunomodulatory properties [9], MSCs have been used as the ideal seed cells for tissue engineering and regenerative medicine therapies [1].
Distinct from the hematopoietic stem cell population, bone marrow-derived MSCs (BM-MSCs) were first described by Friedenstein et al. [10] and are still the most frequently studied type of cell and often regarded as the gold standard for MSC research [11]. Recent studies have revealed that dental stem cells are powerful tools used in studying early human developmental biology to improve tissue engineering and the potentials of regenerative medicine [12].
Although there are many alternative cells, the biological characters of MSCs from different tissue origins vary. Differences in the extracellular circumstance such as the presence of interacting adjacent cells, exposure to proteases, or a hypoxic microenvironment contribute to the diversity of developments within MSC populations originating from different tissues. In addition, intracellular conditions such as the expression levels of certain noncoding RNAs can also impact MSC peculiarity [11]. Although many alternative types of MSCs have been studied, the most suitable origin of MSCs used for bone tissue engineering, especially in the oral and maxillofacial area, remains inconclusive.
To clarify this issue, this study investigated MSCs derived from seven different kinds of tissues, including four dental origins, dental pulp stem cells (DPSCs), periodontal ligament stem cells (PDLSCs), gingival MSCs (GMSCs), and dental follicle stem cells (DFSCs); two somatic origins, BM-MSCs and adipose-derived stem cells (ADSCs); and one from birth-associated perinatal tissue umbilical cord (UCMSCs). The morphology, immunophenotype, proliferation, antiapoptosis, and osteogenic differentiation abilities of the different cells were compared under an identical condition, and some canonical cascade signaling pathways were detected after osteogenic induction to explore the internal difference between different MSCs.
Materials and Methods
Isolation and cultivation of cells
All protocols involving human subjects were approved by the Ethics Committee of Shandong University and informed consent was obtained from all donors. The donors were between 15 and 25 years of age with no systemic disease. Each kind of MSC had three samples from three independent individuals and the cells were randomly used for the experiments.
Isolation and culture of MSCs from dental origins
MSCs from dental origins were isolated by the method of enzymatic digestion as previously described [13 –16].
Briefly, third molars extracted for impacted or orthodontic reasons were collected from donors in Stomatological Hospital of Shandong University. For DPSCs, after washing with phosphate-buffered saline (PBS), the teeth were cut open and the pulp was gently separated, and one-third parts of the apical region were removed and cut into small pieces. For PDLSCs, teeth were rinsed with PBS. Afterward, the periodontal ligament tissues were scraped off from the middle third of the root surface and minced into small patches. The gingival tissue was washed in PBS, and the epithelial layer was removed and minced into small pieces. For DFSCs, the attached dental follicles were separated from the impacted third molars. The separated dental follicle tissues were then washed in PBS and sliced into small patches.
All cut tissues were digested in a solution of 3 mg/mL collagenase type I (Sigma) and 4 mg/mL dispase (Sigma) for 60 min at 37°C. Single-cell suspensions were obtained by passing the digested tissues through a 70 μm cell strainer (BD Falcon). Cell suspensions were seeded into 25 cm2 culture flasks and cultured in the basal culture medium (BCM), which consisted of α-minimum essential medium (α-MEM) (Corning) supplemented with 10% fetal bovine serum (FBS) (Gibco) and 100 U/mL penicillin/streptomycin (Beyotime, China), at 37°C in 5% CO2, respectively.
Isolation and culture of human BM-MSCs
BM-MSCs were isolated using a plastic-adherent method as previously described [17]. Briefly, patients with alveolar clefts need to be repaired with iliac bone grafting. During this process, excess bone marrow was harvested for the cell culture. BCM was used to resuspend the aspirate and plated in tissue culture flasks. The medium was changed every 3 days. Nonadherent hematopoietic cells were removed during medium replacement after 10–12 days of culture.
Isolation and culture of human ADSCs
Adipose tissue samples were obtained from the abdomen of donors who underwent caesareans as described previously [18]. In brief, adipose tissues were washed with PBS and were then treated with 0.15% type I collagenase (Sigma) for 60 min at 37°C with gentle shaking. After inactivating the collagenase with BCM, the mixture was centrifuged for 5 min at 1,000 rpm. The top lipid layer was discarded and the cellular pellet was resuspended in BCM and seeded into 25 cm2 culture flasks.
Isolation and culture of human UCMSCs
Wharton's jelly was obtained from the umbilical cords as previously described [19]. The umbilical cord tissues were washed with PBS several times and the blood vessels were removed. Wharton's jelly tissues were chopped into small pieces and digested in the solution containing 1 mg/mL collagenase type I (Sigma) at 37°C for 8 h with gentle shaking. The filtrate of the suspension was centrifuged for 5 min at 1,000 rpm and resuspended in BCM before seeding.
For all cell types, the BCM was changed every 3 days and the cells were passaged at the ratio of 1:3 by trypsin when they reached 85%–90% confluence. A differential digestion method was used to separate the mixed epithelial cells.
Immunophenotype analysis
BD Stemflow™ hMSC Analysis Kit (BD Biosciences) was used to identify the immunophenotype of cells according to the manufacturer's instructions. Briefly, after being trypsinized and washed with PBS, cells were incubated with monoclonal antibodies conjugated with fluorescent dyes in the dark at 4°C for 20 min. The following antibodies were used: CD90 FITC, CD44 PE, CD105 PerCP-Cy, CD73 APC, and PE-negative cocktail (CD34PE, CD11b PE, CD19 PE, CD45 PE, and HLA-DR PE). Respective positive isotype control and negative isotype control cocktails were used as systemic negative controls. Then the cells were washed with PBS and analyzed by flow cytometry (BD Biosciences). The results were analyzed by software FlowJo.
Multilineage differentiation
For osteogenic differentiation assays, cells were exposed to an osteogenic induction medium, which contained α-MEM (Corning) supplemented with 10% FBS (Gibco), 10 nM dexamethasone (Solarbio, China), 10 mM β-glycerophosphate (Solarbio, China), and 50 mg/L ascorbic acid (Sigma). The medium was refreshed every 3 days. After 4 weeks, the mineralized nodules were detected by Alizarin Red staining. The intensity of the Alizarin Red stain was quantified by a microplate reader (SPECTROstar Nano; BMG Labtech, Germany) at 562 nm after solubilizing in 10% cetylpyridinium chloride (Sigma) for 10 min at room temperature [20].
For adipogenic differentiation assays, cells were exposed to an adipogenic medium; the medium was α-MEM (Corning) containing 10% FBS (Gibco), 2 μM dexamethasone (Solarbio, China), 0.2 mM indomethacin (Sigma), 0.01 g/L insulin (Sigma), and 0.5 mM isobutyl-methylxanthine (Sigma). The adipogenic medium was also refreshed every 3 days. After 4 weeks, lipid droplets were detected by Oil Red O staining.
For chondrocyte differentiation, the medium contained α-MEM (Corning) supplemented with 10% FBS (Gibco), 2 ng/mL transforming growth factor-β1 (TGF-β1; R & D Systems, Minneapolis, MN), 1 × ITS + Premix (BD Biosciences, San Jose, CA), 50 mg/mL L-ascorbic acid (Sigma Aldrich, St Louis, MO), 100 mg/mL sodium pyruvate (Wako, Japan), 100 nM dexamethasone (Solarbio, China), and 100 U/mL penicillin/streptomycin. Approximately, 2.5 × 105 cells were placed into 15 mL polypropylene culture tubes containing 0.5 mL chondrogenic medium, centrifuged at 1,000 rpm for 5 min at room temperature, and cultured at 37°C, in a humidified atmosphere of 5% CO2. After 4 weeks, chondrogenic differentiation of the cells was assessed by Alcian Blue staining of sulfated proteoglycans.
Cell proliferation assay
The proliferation of cells was detected by Cell Counting Kit-8 (CCK-8) (Dojindo Laboratories, Japan) and 5-ethynyl-20-deoxyuridine (EdU) (Ribobio, China) labeling assay according to the manufacturer's instructions. In brief, for CCK-8 assay, cells were plated in a 96-well plate (4 × 103 cells/well) and incubated for 1, 2, 3, 4, 5, 6, and 7 days, respectively. At each time point, the medium was changed to fresh BCM supplemented with 10% CCK-8 solution and then incubated for an additional 2 h before measuring the absorbance at 450 nm wavelength using a microplate reader (SPECTROstar Nano; BMG Labtech, Germany).
For EdU labeling assay, cells were seeded in 24-well plates and incubated for 72 h; 50 μM EdU labeling medium was added to each well at 37°C for 2 h, and the cells were fixed and then stained by Apollo®567 and Hoechst33342. The stained cells were examined with fluorescence microscope (Olympus, Japan) and photographed with camera. The proliferation rate of cells was assessed with the proportion of EdU-positive nucleus (red) to blue fluorescent nucleus by counting six microscopic fields randomly for each well.
Cell apoptosis assay
Approximately 1 × 105 cells were cultured in six-well plates for apoptosis detection. At 80%–90% confluency, the cells were treated with 250 μM H2O2 for 4 h. The percentage of apoptotic cells was detected by Annexin V-FITC/PI staining kit (Sungene Biotech, China). After treatment, the cells were harvested, resuspended in 200 μL of binding buffer containing 5 μL Annexin V-FITC for 15 min, and then 300 μL binding buffer containing 5 μL propidium iodide was mixed for 5 min in darkness at room temperature. Cell apoptosis was then detected by flow cytometer (BD Accuri TM) and analyzed by BD Accuri TM C6 Plus Software.
ALP staining and ALP activity assay
ALP activity was measured using ALP activity assay kit (Nanjing Jiancheng Bioengineering Institute, China) according to manufacturer's instructions and the absorbance was measured at 520 nm wavelength with a microplate reader (SPECTROstar Nano; BMG Labtech). ALP staining was implemented using an NBT/BCIP staining kit (Beyotime, China). Briefly, the cells were rinsed with PBS and fixed in 4% paraformaldehyde and then stained with NBT/BCIP solution evades light.
Protein extraction and Western blot analysis
Cells were washed with ice-cold PBS and lysed using RIPA lysis buffer (Solarbio, China) containing 1% phenylmethylsulfonyl fluoride (PMSF) (Solarbio, China) and 1% phosphatase inhibitor (Solarbio, China). After ultrasonic lysis and centrifugation at 12,000 rpm at 4°C for 15 min, the supernatant lysate with proteins was collected and the concentration was measured by BCA Protein Assay Kit (Solarbio, China). Then proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membranes. Membranes were blocked with nonfat dry milk for 1 h, incubated with primary antibodies overnight at 4°C, and incubated with secondary antibodies for 1 h at room temperature.
The following primary antibodies were used: COL1A1 (1:1,000, #84336; Cell Signaling Technology), ALP (1:20,000, ab108337; Abcam), osteocalcin (OCN) (1:500, ab93876; Abcam), p-AKT (ser473) (1:1,000, #4060; Cell Signaling Technology), AKT (pan) (1:1,000, #4691; Cell Signaling Technology), p-ERK (1:1,000, #4370; Cell Signaling Technology), ERK (1:1,000, #4695; Cell Signaling Technology), p-p38 (1:1,000, #4511; Cell Signaling Technology), p38 (1:1,000, #8690, Cell Signaling Technology), Caspase-9 (1:1,000, #9508; Cell Signaling Technology), BAX (1:1,000, #5023; Cell Signaling Technology), Bcl-2 (1:1,000, wl01556; Wanleibio), c-IAP1 (1:500, wl01680; Wanleibio), c-IAP2 (1:1,000, wl01254; Wanleibio), Survivin (1:500, wl01684; Wanleibio), and GAPDH (1:20,000, HRP-60004; Proteintech). The protein bands were detected by enhanced chemiluminescence (Millipore) under Amersham Imager 600.
Statistical analysis
All experiments were performed independently at least thrice and all data were presented as mean ± standard deviation. One-way analysis of variance (ANOVA) was used to determine statistical differences between groups. The statistical analyses were conducted by SPSS 19.0 and differences at P < 0.05 were considered to be statistically significant.
Results
MSC morphology and identification
Plastic-adherent cells were successfully isolated from all the types of tissues and exhibited a homogeneous fibroblast-like morphology with abundant cytoplasm and large nuclei. After the second passage, no obvious differences in morphology were noted among the seven populations (Fig. 1A). The mesenchymal cell-specific surface markers, CD73, CD90, CD105 and CD44, were positively expressed in all cell types with the percentage of >95% by flow cytometry analysis. Meanwhile, the cells were negative for hematopoietic or endothelial-specific antigens CD34, CD11b, CD19, CD45, and HLA-DR with the percentage of <5%. The results confirmed that cells isolated in this study were all from mesenchymal origins (Fig. 1B).

Characterization of the morphology and immunophenotype of MSCs derived from seven tissues.
To identify the multilineage differentiation potential of the MSCs derived from various tissues, they were cultured in osteogenic, adipogenic, and chondrogenic induction medium for 4 weeks and detected with their own specific staining methods, respectively. The results confirmed that MSCs from the seven kinds of tissues could differentiate into osteoblasts (Fig. 2A), adipocytes (Fig. 2B), and chondrocytes (Fig. 2C). However, differences in staining strength of cells can be observed, which reflected the varying capability of differentiation between cell sources.
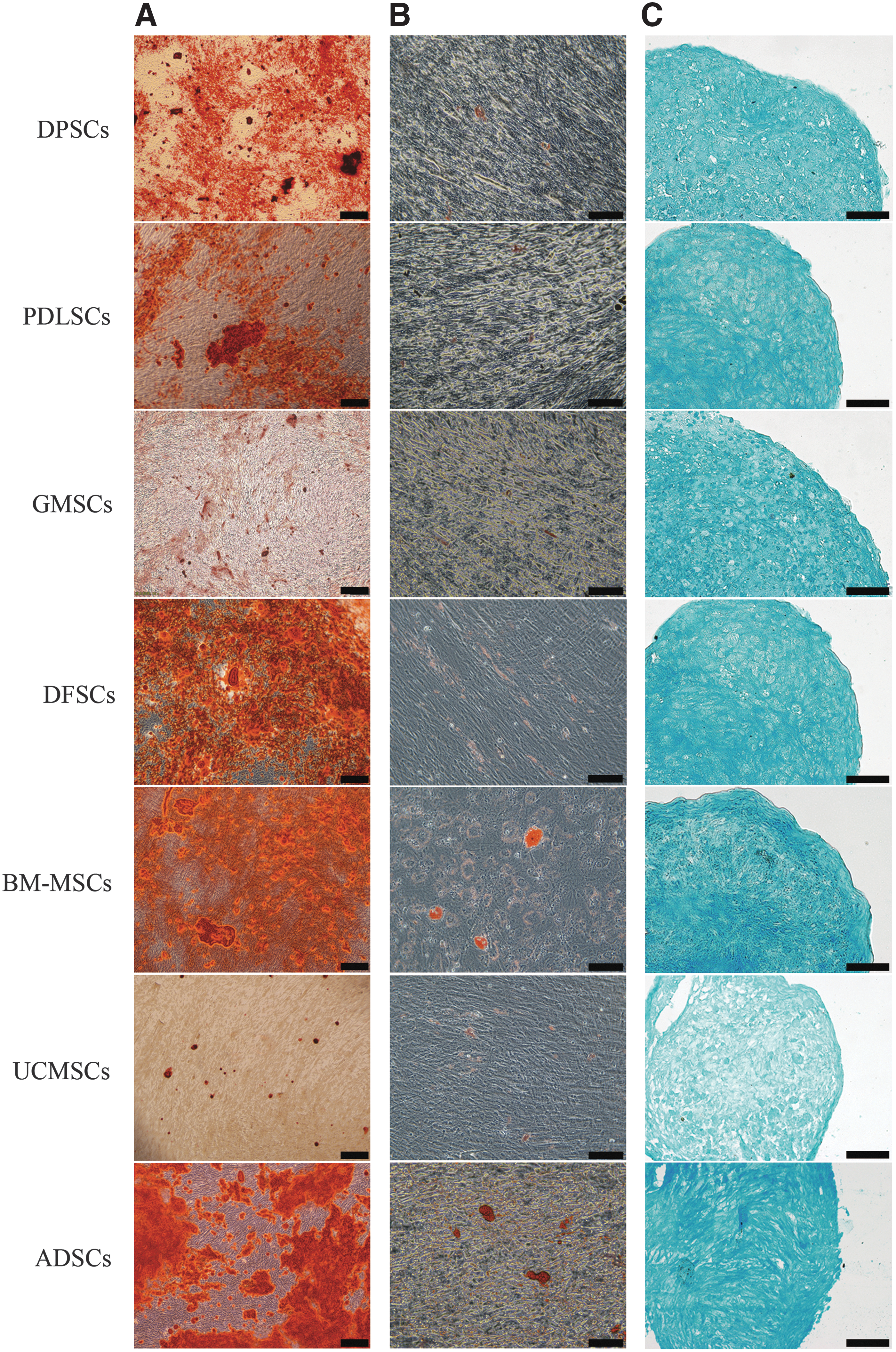
Multilineage differentiation ability of seven kinds of cells.
Proliferation ability of the MSCs
The growth curves were drawn according to the results of the CCK-8 analysis in a row of 7 days of proliferation and the percentage of cells undergoing DNA replication was measured by EdU assay after 72 h of plating. The CCK-8 results demonstrated that MSCs proliferated slowly during the first 2 days and then entered the subsequent logarithmic phase. The four dental MSCs and UCMSCs seemed to have comparable proliferation rate (P > 0.05) and proliferated significantly faster than BM-MSCs and ADSCs from the fifth and third day, respectively (P < 0.05) (Fig. 3A). Parallel results were revealed by EdU labeling analysis. A higher ratio of EdU-positive cells was observed in DPSCs, PDLSCs, GMSCs, DFSCs, and UCMSCs than in BM-MSCs and ADSCs (Fig. 3B, C), suggested greater proliferation ability of the former five MSCs.
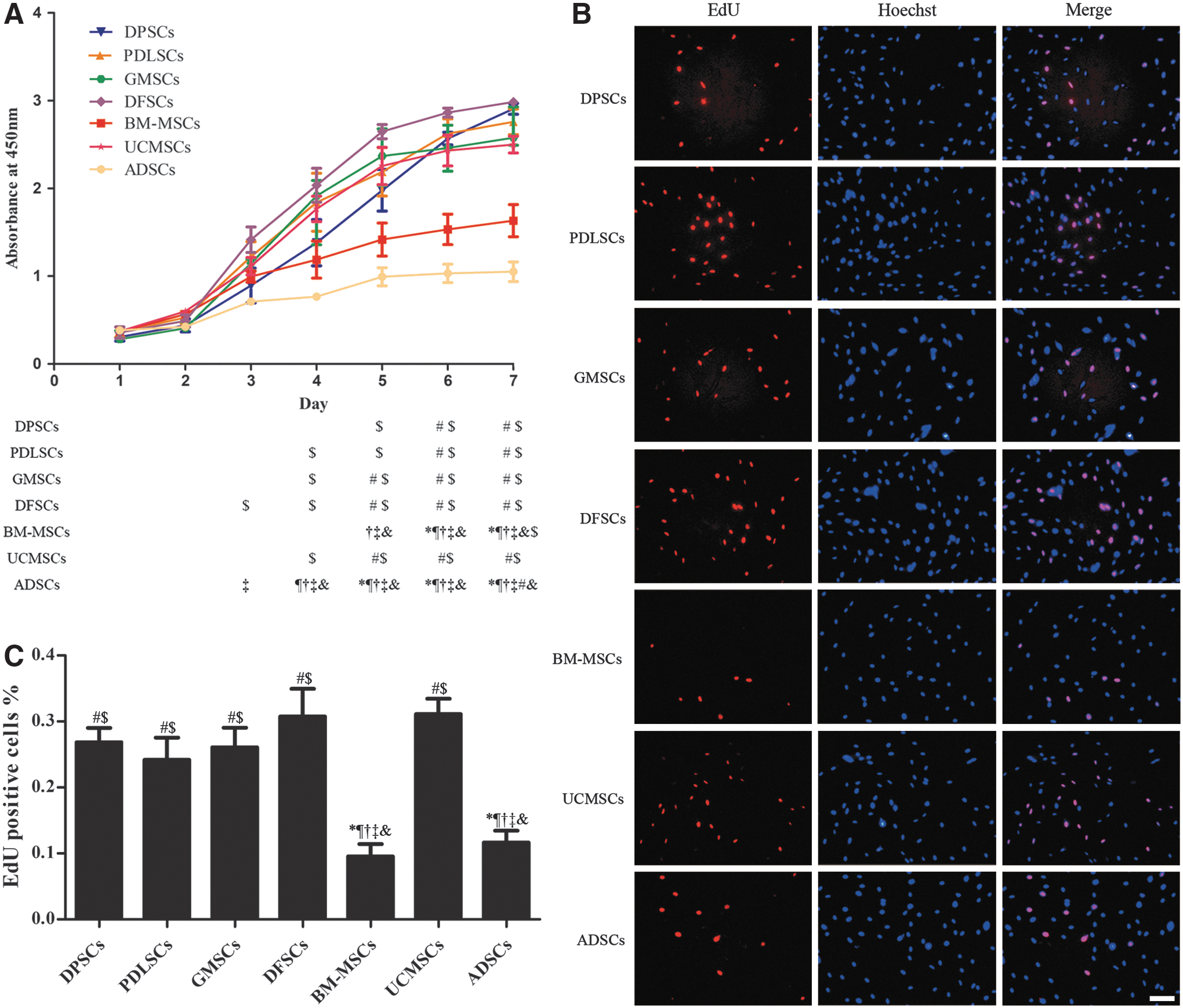
Proliferation ability of different MSCs.
Antiapoptotic ability of the MSCs
Cell apoptosis was induced by the H2O2-created oxidative stress combined with serum deprivation. The results showed that the apoptosis rates of DPSCs and PDLSCs were lower than GMSCs, DFSCs, BM-MSCs, and UCMSCs (P < 0.05). The highest rate of apoptosis occurred in DFSCs and UCMSCs compared with all other MSCs (P < 0.05). The apoptosis rate of ADSCs was not statistically different when compared to DPSCs, PDLSCs, GMSCs, and BM-MSCs (P > 0.05) (Fig. 4A, B).

Antiapoptosis ability of the MSCs.
The expression levels of some proapoptotic and antiapoptotic proteins of untreated cells were detected to provide some clues for why these cells have different antiapoptotic capabilities. The results showed that PDLSCs expressed less Caspase-9 and BAX. GMSCs and UCMSCs expressed a higher Caspase-9 level. Even though the Caspase-9 level was low in DFSCs, the BAX/Bcl-2 ratio was high. Furthermore, DFSCs behaved the lowest c-IAP2 and Survivin level (Fig. 4C).
Comparison of osteogenic differentiation capacity
To confirm the osteogenesis capacity of MSCs derived from different tissues for bone tissue engineering, ALP activity, mineralized nodules and osteogenic differentiation-related proteins were detected at different time points. The ALP staining was deeper in PDLSCs, DFSCs, BM-MSCs, and ADSCs followed by DPSCs, while GMSCs and UCMSCs were less stained on the 14th day (Fig. 5A). A similar tendency was observed by the detection of ALP activity (Fig. 5B). The protein expressions of COL1 and ALP were consistent with the ALP activity (Fig. 5E).
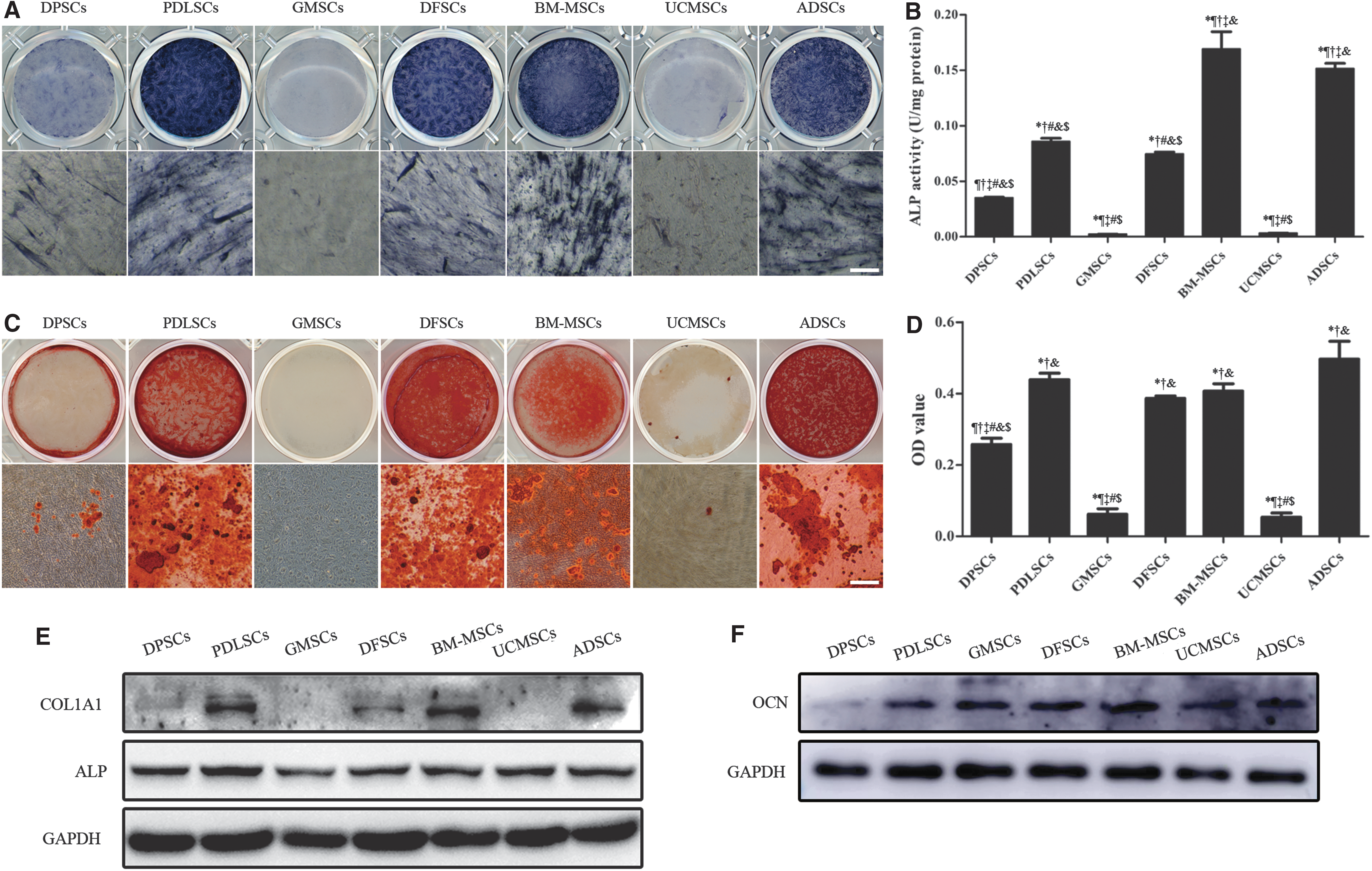
Comparison of osteogenic differentiation capacity of the MSCs.
After 28 days of induction, Alizarin Red staining and quantitative analysis demonstrated that BM-MSCs and ADSCs formed the most mineralized nodules followed by PDLSCs and DFSCs. GMSCs and UCMSCs formed the least mineralized nodules (Fig. 5C, D). The protein level of OCN on day 28 was almost the same with the ability of mineralization, except for GMSCs (Fig. 5F).
Changes of canonical signaling pathways in MSCs after osteogenic induction
To observe changes in the activation of several canonical cascade signaling pathways during the osteogenic differentiation of MSCs from different tissue origins, the proteins of the cells after 7 days of osteogenic induction were examined. Results showed that the phosphorylation of AKT at serine 473 was significantly decreased in DFSCs, but remained unchanged in other MSCs. The MAPK/ERK signaling pathway was greatly activated only in PDLSCs. The p38 MAPK signaling pathway had complex changes among these cells. A significant downregulation of phosphorylated p38 was observed in BM-MSCs and ADSCs, while upregulated in UCMSCs. DPSCs, PDLSCs, GMSCs, and DFSCs had little changes in the phosphorylation of p38 (Fig. 6).

Western blot analysis of the changes of canonical intracellular protein kinase-based cascade signaling pathways on day 7 of induction. (n = 3) OI, osteogenic induction; NC, negative control.
Discussion
A solid understanding of MSCs is crucial for tissue engineering and regenerative medicine. Since the discovery and characterization of BM-MSCs, MSC-like populations from other tissues have now been characterized based on the “gold standard” criteria established for BM-MSCs. There are many studies evaluating the biology of MSCs derived from other tissues versus bone marrow [21 –26]. However, when comparison occurs among MSCs original from different tissues that are nonbone marrow derived, fewer studies can be consulted.
Conditions for cell culture varied in every independent experiment, so comparison between different studies was difficult. This study compared the biological properties of different MSCs in a completely uniform culture condition so that the results of comparison were convictive, even though the culture condition in our study may not be the best for all seven kinds of MSCs. Since culture conditions affect the properties of MSCs [27], the results in this study are supposed to understand and apply in this certain culture condition.
Assessment of the optimal seed cell for tissue engineering includes many aspects. Besides the consideration of cell properties in proliferation and differentiation toward a specific lineage, the determination of the cell source is also of great importance. For example, one important factor is that tissue samples should be easily accessible and can be obtained in a minimally invasive way. Our study demonstrated that the MSCs could be isolated from different types of tissue with a similar protocol and condition.
However, compared with the two somatic MSCs (BM-MSCs and ADSCs), dental MSCs (DPSCs, PDLSCs, GMSCs, and DFSCs) and perinatal MSCs (UCMSCs) can be obtained from medical waste such as wisdom teeth and umbilical cord, which is very convenient and brings up less ethical arguments. Meanwhile, these cells can be stored for later use when needed. As for BM-MSCs, the extra invasive operation might bring serious complications to donors such as infection. Furthermore, low cell numbers upon harvest was a shortcoming, which was frequently encountered in obtaining BM-MSCs. ADSCs, although can be obtained by suction-assisted lipectomy or caesarean section, it is still invasive [21,28]. In addition, the process of pregnancy might affect the status of ADSCs.
In view of the above, dental and perinatal tissue-derived stem cells perform better than somatic stem cells for their easy availability. However, obtaining cells from wisdom tooth extracted with pericoronitis and infection should be avoided and no prophylactic use of antibiotics before extraction should be considered.
The proliferation capacity of cells is important with regard to their application in cell therapy and tissue engineering, requiring enormous amounts of cells. Our results showed that the somatic tissue-derived MSCs, BM-MSCs and ADSCs, exhibited lower proportion of replication compared with other cells. Instead, the dental tissue-derived MSCs, as well as UCMSCs, performed better in cell proliferation under identical culture conditions. Previous studies compared MSCs isolated from bone marrow, adipose tissue, and umbilical cord and found that BM-MSCs had the lowest proliferative capacity, while umbilical cord-derived MSCs showed the highest proliferation potency [29,30]. Our results once again proved these findings, but we did not find a significant difference between BM-MSCs and ADSCs.
As dental tissue-derived MSCs have been used for tissue engineering studies in large animals to assess their potential in preclinical applications [31], four kinds of dental MSCs (DPSCs, PDLSCs, GMSCs, and DFSCs) were evaluated synchronously in our study. It turned out that these dental MSCs proliferated as fast as UCMSCs, which demonstrated their virtue in quantity production. This result is also in accordance with the former study that MSCs isolated from dental tissues possessed greater proliferative potential than BM-MSCs [24].
The antiapoptotic capacity of cells is also an important aspect when considering therapeutic applications. It has been reported that ∼99% of transplanted MSCs are lost during the first 24 h after transplantation and apoptosis is thought to be a main influencing factor [32,33]. Multiple mechanisms could contribute to the apoptosis of grafted cells, including nutrient deprivation and inflammatory environment [34]. The use of H2O2 and serum deprivation condition partially simulated the harsh microenvironment of the transplant sites and can successfully induce MSC apoptosis [35].
Our results demonstrated that DPSCs and PDLSCs possess the strongest antiapoptotic capacity and could lead to significant advantages in clinical application. The low level of proapoptotic protein Caspase-9 in these two kinds of cells might be a possible reason. On the contrary, UCMSCs had the highest Caspase-9 expression and as a result, the highest apoptosis rate. Similarly, DPSCs have been described by another report as having lower cell apoptosis compared with UCMSCs [20].
On the other hand, the antiapoptotic proteins, c-IAP and Survivin, are inhibitors of Caspase-9 and apoptosis [36]. The low level of c-IAP and Survivin in DFSCs may contribute to their increased level of apoptosis. Previous study reported that the ratio of Bcl-2 to Bax determines survival or death under apoptotic stimuli [37]. The lower ratio of BAX/Bcl-2 in PDLSCs and higher one in DFSCs could also affect their sensitivities to apoptosis.
The expected plasticity of MSCs is paramount for upcoming therapeutic strategies. Osteogenic differentiation was further explored on account of the imperative requirement in bone regeneration. As confirmed by ALP staining, ALP activity assay, and mineralized nodules with Alizarin Red stain, our results indicated that BM-MSCs and ADSCs are most sensitive toward the osteogenic differentiation, whereas UCMSCs and GMSCs rarely differentiated toward osteoblasts under the common differentiation protocols. Moreover, PDLSCs and DFSCs also had great osteogenic capabilities, which were slightly weaker compared with BM-MSCs and ADSCs and better than DPSCs.
A similar result was observed in a study that demonstrated the osteogenic differentiation of ADSCs was greater compared with DPSCs [38]. Another research that revealed DPSCs may have significant advantages for osteogenic differentiation compared with UCMSCs is consistent with ours [20]. BM-MSCs showed higher osteogenesis than DPSCs, was also reported by other scholars [39]. However, there was a study that showed UCMSCs had the greatest potential in osteogenesis than ADSCs and BM-MSCs [30]. The reason might be that the Wharton's jelly was obtained from individuals who were much younger than the individuals from whom the adipose tissue and bone marrow were obtained in the previous study.
Nevertheless, the donors were between 15 and 25 years of age in our study, which might be more comparable to the comparative study because the effect of donor age on the self-renewal and differentiation potential is presented [40]. In this study, we demonstrated MSCs derived from other potential sources exert as good osteogenic potential as BM-MSCs, which can be helpful in choosing alternative cell sources for bone regeneration and tissue engineering.
Intracellular protein kinase-based cascade signaling pathways such as the PI3K/AKT and MAPK/ERK play important roles in the physiology and pathophysiology of many types of cells [41]. In this study, a unique suppression effect on AKT was detected in DFSCs during osteogenic induction. This result is conflicting with a previous study that reported AKT signaling pathway was activated in DFCs after the induction of osteogenic differentiation [42]. The reason for the inverse results might be that the DFCs were induced by BMP2 in the previous research, while the dexamethasone system was used in this study. Another study reported that uncarboxylated osteocalcin stimulates osteoblastic differentiation by inhibiting the activation of PI3K/AKT in MC3T3-E1 cells [43], suggesting that different stimuli may lead to different changes of signaling pathway and the exact mechanism remains to be elucidated.
The MAPK/ERK pathway, which was only significantly activated in PDLSCs, indicated that under the osteogenic induction of dexamethasone and ascorbic acid, only PDLSCs responded through MAPK/ERK signaling pathway among the seven MSCs studied. Although numerous reports demonstrated that the ERK signaling pathway was induced in other cell types [44 –46], different agents were used such as FGF2, Rap1A protein, and chlormadinone acetate.
The p38 MAPK signaling pathway remained unchanged in the dental MSCs, significantly inhibited in somatic MSCs, and activated in perinatal MSCs, and demonstrated that this pathway may have a distinct sensibility and function in cells from different tissue origins. A lot of studies indicated that the osteogenic differentiation of BM-MSCs was promoted with the increase in p-p38, which is contradictory to our result [47 –49]. This may be because these studies have additional stimuli other than osteogenic induction. Additional stimulation activates p38, which is itself reduced after osteogenic induction, and promotes osteogenic differentiation.
Despite the fact that the p38 pathway may not be involved in the osteogenic differentiation of UCMSCs, raised by some scholars [50], the significant increase of p38 in UCMSCs after osteogenic induction in our study may be an important reason for the poor osteogenic ability of UCMSCs. This completely opposite change between somatic and perinatal MSCs once again proves that, facing the same stimulus, different tissue-derived cells have different molecular responses and regulatory mechanisms. Meanwhile, the inactivity of GMSCs in the protein kinase pathway might be one of the reasons for their poor osteogenesis.
Collectively, MSCs can be successfully obtained from various kinds of tissues, including dental, somatic, and perinatal origins, with a similar approach. They all have self-renewal and multidirectional differentiation ability under the same culture condition. In a comprehensive consideration of the source accessibility, proliferation, antiapoptosis, and osteogenic differentiation capacity, PDLSCs are considered a good potential alternative to BM-MSCs in bone tissue engineering. Simultaneously, the canonical intracellular protein kinase-based cascade signaling pathways function in a cell type-specific manner. Nevertheless, the molecular mechanisms regulating osteogenic differentiation of different MSCs have to be intensively studied and further experiments are still needed to assess their regeneration potentials in vivo.
Footnotes
Acknowledgments
We thank Dr. Weiting Gu from Qilu hospital for the collection of umbilical cords. This work was supported by grants from Shandong Provincial key research and development program (2016GSF201115, 2016GSF201220, and 2017GSF18117), Shandong Provincial Natural Science Foundation (ZR2018MH018 and ZR2017MH031), Young Scholars Program of Shandong University (2015WLJH53), and the Construction Engineering Special Fund of Taishan Scholars (ts201511106).
Author Disclosure Statement
No competing financial interests exist.