
Abstract
Mesenchymal stem cells (MSCs) are important components of the tumor microenvironment, which play an important role in tumor development. Exosomes derived from tumor cells can affect the biological characteristics of MSCs. Our study examined the effects of exosomes derived from gastric cancer cells on MSC immunomodulatory functions. Exosomes were extracted from gastric cancer cell line AGS (AGS-Exos) and cultured with MSCs. MSCs were then cocultured with both human peripheral blood mononuclear cells and macrophages [phorbol-12-myristate-13-acetate (PMA)-stimulated THP1 cells]. The activation levels of T cells and macrophages were detected by flow cytometry and real-time quantitative polymerase chain reaction (RT-PCR). Changes in the MSC signaling pathway after AGS-Exos stimulation were studied using RNA Chip, and the molecular mechanisms of functional change in MSCs were studied by inhibiting the signaling pathway. MSCs treated with AGS-Exos could promote macrophage phagocytosis and upregulate the secretion of proinflammatory factor, and promote the activation of CD69 and CD25 on the surface of T cells. RNA Chip results indicated the abnormal activation of the NF-kB signaling pathway in MSCs after AGS-Exos stimulation, and this was verified by the identification of key proteins in the pathway using western blot analysis. After NF-kB signaling pathway inhibition, the effect of MSCs stimulated by AGS-Exos on T cells and macrophages was markedly weakened. Therefore, AGS-Exos affected the immunomodulation function of MSCs through the NF-kB signaling pathway, which enhanced the ability of MSCs to activate immune cells, maintain the inflammatory environment, and support tumor growth.
Introduction
Gastric cancer is a common global malignancy, and its incidence and mortality rate are among the highest worldwide [1]. The early diagnosis rate of gastric cancer in China is relatively low, and the vast majority of cases are at an advanced stage at the initial visit [2,3]. Therefore, several advances must be made to improve the diagnosis and treatment of gastric cancer.
More studies have shown that the behavior of tumors is not completely determined by the tumor cells themselves, and a large number of nontumor cells are homed to the tumor site; therefore, the tumor microenvironment influences tumor progression [4].
The mesenchymal stem cells (MSCs) in normal tissues are homed to the tumor site, and they become tumor-enhanced MSCs based on the action of tumors and the inflammatory microenvironment, thus playing an important role in promoting tumor development, invasion, and metastasis [5]. Tumor cells have been found to have two ways for the education of MSCs. One is the action of soluble inflammatory factors secreted by tumor cells on MSCs, and the other is the direct exchange of exosome with MSCs by tumor cells [6]. Previous studies found that lung cancer exosomes can induce normal MSC immune functions to transform into MSC phenotypes that promote tumor progression and the abnormal activation of the NF-kB signaling pathway [7].
In addition, it has been reported that exosomes derived from breast cancer [8], ovarian cancer [9], cholangiocarcinoma [10], and prostate cancer cell lines [11] were reported to induce MSC differentiation into carcinoma-associated fibroblasts, which can promote tumor proliferation and migration.
However, the methods and mechanisms for promotion differ. On the one hand, reprogrammed MSCs promote the development of tumors by secreting cytokines acting directly on tumor cells [12]. On the other hand, reprogrammed MSCs support tumor growth by interacting with neighboring cells in the tumor microenvironment [13]. For example, reprogrammed MSCs can act on endothelial cells to promote angiogenesis [14], and a coculture of tumor cells with MSCs or subcutaneous injections of an MSC–tumor cell mixture in nude mice can promote tumor vasodilation and tumor growth [15]. Immunohistochemical results also showed more macrophages around tumor tissue in the coculture group. Thus, the effects of reprogrammed MSCs on immune cells in the tumor microenvironment also support tumor growth.
We know that MSCs are immunomodulatory cells [16] that have the ability to regulate immune effector cells (including T and B lymphocytes, NK cells, monocyte macrophages, and dendritic cells) [16,17], and a large number of studies have revealed the immunosuppressive effects of MSCs. For instance, it was reported that MSCs have a significant inhibitory effect on the proliferation of T cells [18,19], and they cause the cell cycle arrest of T cells [20]. Studies have also shown that MSCs can reduce the inflammatory response induced by macrophages [21 –23].
However, because of differences in experimental conditions and experimental protocols, researchers sometimes arrive at the opposite conclusion. For example, reports suggest that when MSCs are cocultured with T cells in smaller proportions, they can promote the proliferation of T cells, thus indicating that MSCs have enhanced immunity effects [24]. However, based on our knowledge of tumor exosomes, how is MSC regulating the function of immune cells? Studies have shown that tumor cell-induced MSCs recruit macrophages [25], and they can support tumor growth [26 –28]. However, related research is still relatively limited, especially regarding the study of gastric cancer.
Therefore, in this study, exosomes derived from gastric cancer cells were used to induce MSCs to study the effects of altered MSCs on related immune cells and associated mechanisms. This study explores new mechanisms of MSC immune regulatory functions, and it also provides possibilities and new strategies that can be used to identify targets for the prevention and treatment of gastric cancer.
Materials and Methods
Cell culture
Isolation and culture of human adipose-derived MSCs
This study has been performed in accordance with the Declaration of Helsinki and had been approved by the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences (Project No. 022-2015). All patients were informed about the use of their tissue samples for biological research. Adult fat samples were obtained from orthopedic hospitals, and all samples were used with informed consent from donors.
Adipose tissue was washed twice with D-Hanks' solution containing antibiotics (penicillin and streptomycin) and centrifuged at 800 rpm for 3 min. The lower fluid was aspirated with a pipette, and adipose tissue was transferred to a new 50-mL centrifuge tube. Samples were digested with 0.2% collagenase P, and shaken at 37°C for 30 min. An appropriate amount of D-Hanks' solution was added, and digested adipose tissue was filtered with a 100-μm cell strainer to remove undigested tissue. Samples were centrifuged at 1,500 rpm for 10 min, and the upper liquid was aspirated with a pipette. The supernatant was discarded, cells were resuspended in D-Hanks' solution, washed once, and centrifuged at 1,500 rpm for 10 min. The supernatant was then discarded. The cell pellet was resuspended in the working solution, and was inoculated into T75 flasks with 2 × 106 cells. Cells were incubated at 37°C in a 5% CO2 humidified incubator.
After the primary cells were inoculated for 48 h, the upper layer without adhered cells was discarded. Cell culture continued, and cultures were replaced every 2–3 days. When the cells reached 80% confluence, they were passaged or frozen for conservation.
AGS gastric cells [purchased from the Cell Resource Center; Public Union Medical College (PUMC)] were cultured in Dulbecco's modified Eagle medium (DMEM)/F12 with 10% fetal calf serum, and were then incubated at 37°C in a 5% CO2 humidified incubator. THP1 cells [purchased from Cell Resource Center (PUMC)] were cultured in 1640 (modified) culture medium with 10% fetal calf serum, and were then incubated at 37°C in a 5% CO2 humidified incubator.
Isolation and culture of peripheral blood mononuclear cells
Peripheral blood was collected from healthy individuals, and equal volumes of human peripheral blood and phosphate-buffered saline (PBS) were mixed. First, 5-mL of Ficoll solution was added to a 15-mL centrifuge tube, and an equal volume of PBS was slowly added for dilution. Peripheral blood samples from humans were centrifuged at 2,000 rpm for 20 min. The peripheral blood mononuclear cell (PBMC) layer was white, so cells in the layer were pipetted into another clean 15-mL centrifuge tube. About 10–15 mL of PBS was added, and the sample was centrifuged at 1,500 rpm for 10 min. After centrifugation, the supernatant was removed, and the medium was added to a 15-mL dish. Samples were then incubated in RPMI 1640 cell culture medium with 10% fetal calf serum at 37°C in a 5% CO2 humidified incubator.
Animal experiments
All nude mice (female) were purchased from the Laboratory Animal Center of the Chinese Academy of Medical Sciences (Beijing, China), and were bred and maintained under specific pathogen-free conditions. Animal use and experimental procedures were approved by the Animal Care and Use Committee of the Chinese Academy of Medical Sciences.
All nude mice received a subcutaneous injection of 5 × 106 AGS cells. The first group received a subcutaneous injection of 1 × 106 MSCs stimulated by AGS-Exos. The second group received a subcutaneous injection of 1 × 106 normal MSCs. The last group only received an injection of AGS cells. We used the method of coinjection of MSCs and AGS cells, with 10 mice in each group. The tumor volume was measured after 3 weeks. The tumor tissues were fixed with 10% PFA for immunohistochemical analysis. Each group was treated with Ki67 and F4/80 staining. Ki67 antibody was purchased from Servicebio (GB11027). F4/80 antibody was purchased from Servicebio (GB13030-2).
Identification of human adipose-derived MSCs
Regarding human adipose-derived MSCs (hAD-MSCs) with third-generation cells at 70%–80% confluence, cells were washed twice with D-Hanks' solution, and 0.25% trypsin (containing 0.01% EDTA) was added to digest cells. Cells were counted using a cell counter, and were then centrifuged at 1,200 rpm for 5 min. The supernatant was discarded, and the cell pellet (2 × 105/mL) was resuspended in the appropriate amount of PBS. Samples were aliquoted into 1.5-mL microcentrifuge tubes (1 mL/tube), centrifuged at 1,200 rpm for 5 min, and resuspended after the supernatant was discarded. A total of 50 μL of a 1:100 dilution of mouse antihuman primary antibodies (CD106, CD34, CD73, CD105, CD29, CD90, and HLA-DR) were added to each tube and mixed well.
Samples were incubated at 4°C for 30 min. PBS (1 mL) was added, and the samples were centrifuged at 1,200 rpm for 5 min before being washed twice. The supernatant was discarded, and the cells were resuspended in 50 μL of a 1:50 dilution of antihuman primary antibody in test and control tubes, and samples were incubated for 30 min at 4°C. PBS (1 mL) was added, and samples were centrifuged at 1,200 rpm for 5 min before being washed twice. The supernatant was discarded, and 200 μL of 4% paraformaldehyde was added at room temperature. Cells were analyzed using the BD Accuri C6.
Extraction and identification of AGS-Exos
Twenty-four hours before exosome extraction, the AGS medium was replaced with fetal bovine serum-free DMEM/F-12 medium. Cells were cultured for 24 h, and culture supernatants were collected. Supernatants were centrifuged at 1,500 rpm for 10 min to remove viable and dead cells. The medium was filtered through a 0.22-μm microporous filter, and the remaining cell debris and large vesicles were removed by filtration.
The filtered supernatant was transferred to a 100,000 Mw molecular weight ultrafiltration membrane, and an ultrafiltration method was applied at 4°C until the supernatant was completely centrifuged. The appropriate amount of D-Hanks' solution was used to wash the sample twice, and the resulting liquid was colorless and transparent. Approximately 1 mL of the remaining liquid was repeatedly pipetted and transferred to a 1.5-mL EP tube and filtrated with a 0.2-μm Millipore filter, and 200 dL of the sample was dispensed into a 1.5-mL sterile EP tube and stored at −80°C.
Exosomes identified by transmission electron microscopy
The purified exosomes were diluted in multiples and precipitated on a copper grid for 5 min. The filter paper, which absorbed excess liquid, was drained of excess liquid, dried, and diluted with 3% aqueous phosphotungstic acid for 2 min. Exosomes were analyzed and photographed using a transmission electron microscope. Nanosight (Zetasizer Nano ZS90) was used to identify the uniformity and size of exosomes extracted from gastric cancer cell line AGS (AGS-Exos). AGS-Exos were diluted, the appropriate amount was added to the transparent tube, and particle size was measured on the machine.
Exosome labeling and uptake experiments
1′-Dioctadecyl-3,3,3′,3-tetramethylindocarbocyanine perchlorate (Dil) is a lipophilic carbayanine dye that binds lipoproteins in a manner similar to phospholipids, and it is embedded in a biofilm. Diffusion exercises can be used to observe cell binding or the endocytosis of lipoproteins under fluorescence microscopy, and the exercise can be used to perform semiquantitative analyses.
The purified exosomes were added to 1 μM Dil, stained for 10 min, and washed through ultra-high-speed centrifugation (700,000 g at 4°C for 40 min). The process was repeated, and the supernatant was discarded. Labeled exosomes were resuspended in DMEM and coincubated with MSCs. The dishes were placed in Live cell Imaging System (BioTek), and the setup procedure was conducted at 30-min intervals for 24 h to observe MSC uptake by exosomes. After 24 h, the medium was discarded, washed three times with PBS, fixed in 4% paraformaldehyde for 10 min, stained with Hoechst 33342 (1:1,000 dilution), washed at room temperature for 5 min, washed with PBS three times, and observed under a fluorescence microscope.
Western blot
The protein concentration was measured by the bicinchoninic acid (BCA) assay. The cells were discarded from each well and washed twice with prechilled PBS. The PBS was then aspirated, neutral Radio Immunoprecipitation Assay lysate containing 1 mM phenylmethyl sulfonylfluoride was added, and the cells were lysed on ice for 10 min. The cells were then scraped off and collected in a 1.5-mL EP tube and centrifuged at 12,000 rpm for 30 min. The pellet was discarded, and the supernatant was transferred to a new EP tube. The cells were divided into two parts: one was frozen at −20°C; the other was used for concentration detection.
A BCA protein concentration measurement kit (P0011; Beyotime) was used to measure protein concentrations. Regarding protein gel electrophoresis, 5 × protein electrophoresis loading buffer was added to the sample mix at a 1:4 ratio, and the mixture was boiled in a boiling water bath (5–10 min).
Vertical gel electrophoresis and protein loading were conducted, and 20 μg samples per lane were loaded onto an SDS-PAGE (sodium dodecyl sulfate–polyacrylamide gel electrophoresis) protein gel. One lane contained a prestained marker that was used as a molecular weight standard. The voltage was adjusted to 200 mA until the dye reached the bottom of the gel. After the completion of electrophoresis, the gel was carefully removed, and excess parts were removed with a surgical blade.
A wet transfer membrane was used, blocking was performed, and the antibody was subjected to a hybridization reaction. The primary antibody was incubated at 4°C overnight, and the Tris-buffered saline with Tween 20 (TBST) was washed five times for 5 min each time. Horseradish peroxidase-conjugated secondary antibodies were then incubated for 1 h at room temperature, and TBST was washed five times for 5 min each time. Electrochemiluminescence analysis data were then obtained.
RNA extraction
The cell culture solution was discarded, and 1 mL of Trizol was added to 1 × 106 cells for 5 min at room temperature. The reaction was repeated with a nonenzymatic pipette to completely lyse the cells. The lysate was then transferred to an enzyme-free EP tube, and was stored at −20°C or directly subjected to RNA extraction.
Samples were transferred to an ultraclean bench, and 200 μL of chloroform was added to each tube. Tubes were shaken vigorously for 15 s, mixed for 5 min at room temperature, and centrifuged at 12,000 rpm for 15 min at 4°C. The upper aqueous phase was aspirated into a new enzyme-free EP tube (protein aspiration was avoided). An equal volume of isopropanol was added, the tube was inverted gently, and the sample was incubated for 10 min at room temperature.
After incubation, the sample was centrifuged at 12,000 rpm for 15 min at 4°C. After centrifugation, 1 mL of prechilled 75% ethanol [dehydrated ethanol plus enzyme-free diethyl pyrocarbonate (DEPC) water] was added to each tube, the solution was inverted, the pellet was washed (without dispersing the pellet), and the sample was centrifuged at 7,500 rpm for 5 min at 4°C. This process was then repeated once. The supernatant was discarded, and the remaining liquid was removed. The sample was air-dried on a clean bench until the precipitate was clear. An appropriate amount of enzyme-free DEPC water was added to dissolve the RNA precipitate, and a Nanodrop (Thermo Scientific NanoDrop 2000/2000C) was used to measure RNA concentrations.
Real-time polymerase chain reaction
Complementary DNA was reverse transcribed (using a 40-μL system), and experiments were performed according to the method recommended by the TaKaRa M-MLV reverse transcriptase product specifications. Experiments were then performed according to the recommended methods provided by the manufacturer of the TaKaRa SYBR® Premix Ex Taq™ kit (Takara Bio, Inc.). All quantitative real-time polymerase chain reactions (qRT-PCRs) were carried out in duplicate and normalized to GAPDH.
Coculture
MSCs were cocultured with PBMCs. The AGS-Exos-stimulated MSCs were digested, centrifuged, resuspended, and plated in 24-well plates. The number of cells was ∼1 × 105 cells per well. After the cells were adherent, the medium was discarded and washed with PBS, and the process was repeated once. Polyhydroxyalkanoates (PHA)-stimulated PBMCs were plated (MSC:PBMC = 1:5), and three replicates per sample were examined. Samples were measured at three different time points (12, 24, and 48 h), and specific PBMC activation markers were labeled at each time point for flow detection.
The MSCs were cocultured with THPI cells. The AGS-Exos-stimulated MSCs were digested, centrifuged, resuspended, and plated in a six-well transwell chamber with ∼5 × 105 cells per well, and the upper chamber was stimulated with lipopolysaccharide for 6 h. The THP1 cells were washed twice with PBS and allowed to stand for 24 h (MSC:THP1 = 1:5) before coculture with MSCs. After 24 h of coculture, the expression levels of inflammatory factors were determined.
The MSCs were cocultured with macrophages. THP1 cells were plated in a transwell lower chamber of a six-well plate with 5 × 105 cells per well, and samples were stimulated with 100 ng/mL of phorbol-12-myristate-13-acetate (PMA) for 24 h. After the macrophages were adherent, the cells were washed with PBS. The new medium was changed and allowed to stand for 24 h, and the upper chamber was paved with AGS-Exos-stimulated MSCs. After 24 h of cocultivation, the upper chamber of the transwell was discarded, and 3 μL of fluorescent microspheres (3-μm pore size) added to each well of the lower chamber. The cells were incubated for 6 h in the cell incubator, and were washed three times with PBS to collect the cells. Flow cytometry was performed to detect macrophages for the uptake of fluorescent microspheres.
Confocal
The MSCs were digested, centrifuged, resuspended, and plated in confocal wells. After the cells were adherent, the medium was discarded, and the cells were washed twice with PBS. The PBS was discarded, and the cells were fixed with 4% paraformaldehyde for 10 min. Cells were then washed again using a membrane and ice-cold acetone for 10 min. Cells were washed three times with PBS, incubated for 1 h at room temperature, and washed three more times with PBS. Samples were incubated for 1 h at room temperature, and were washed three times with PBS. A confocal microscope was used for detection and photography.
Statistical analysis
All data are expressed as mean ± standard deviation, and two-tailed t-tests and one-way ANOVA (analysis of variance) were performed. P < 0.05 was considered significant. Each experiment was repeated at least three times to obtain a P value and to control for systematic errors.
Results
Phenotypes of hAD-MSCs
Three passages of hAD-MSCs, which were 70%–80% confluent, were assessed for their differentiation ability. Oil red O staining, Alizarin red staining, and alkaline phosphatase staining were used to detect the ability of lipid formation, osteogenesis, and cartilage differentiation. As shown in Fig. 1A–D, hAD-MSCs are capable of adipogenic osteogenic differentiation. Further analysis of the immunophenotype of stem cells indicated that CD90, CD29, CD105, and CD73 were positive (positive rate >95%), and HLA-DR, CD 34, and CD 106 were negative (positive rate <5%; Fig. 1E).
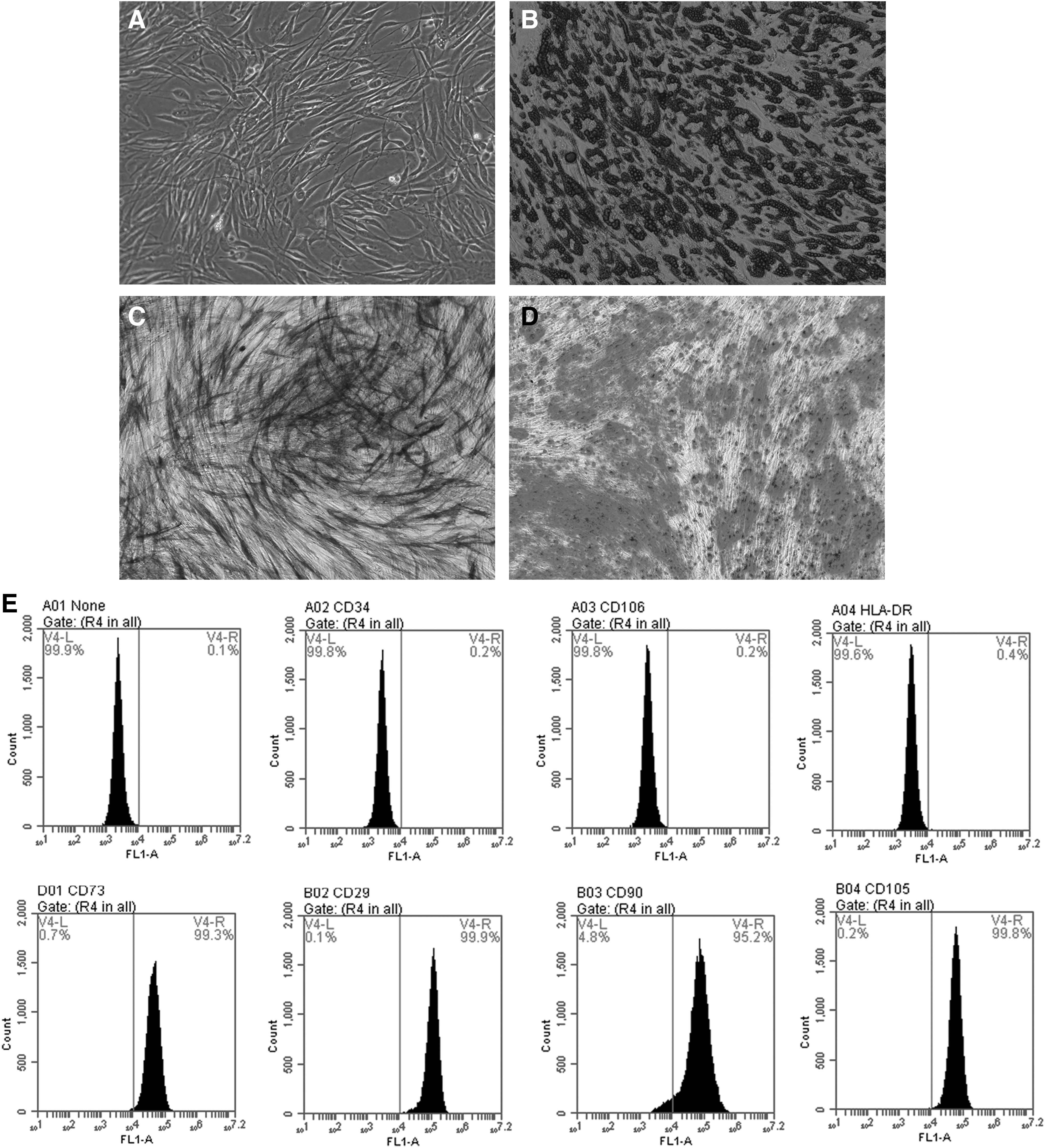
Differentiation potential and immunophenotype identification of hAD-MSCs.
Separation and identification of AGS-Exos
To study the effects of AGS-Exos on MSCs, AGS-Exos was isolated and purified using ultrafiltration. Electron microscope results showed that the exosome was a vesicle with a double-layer membrane structure (∼80 nm in diameter; Fig. 2A). Western blot results indicated that AGS-Exos expressed HSP70, HSP90, and CD63 (Fig. 2B). The Nanosight Particle Size Analyzer showed that the extracted AGS-Exos sample was stable in size (30–100 nm; Fig. 2C).
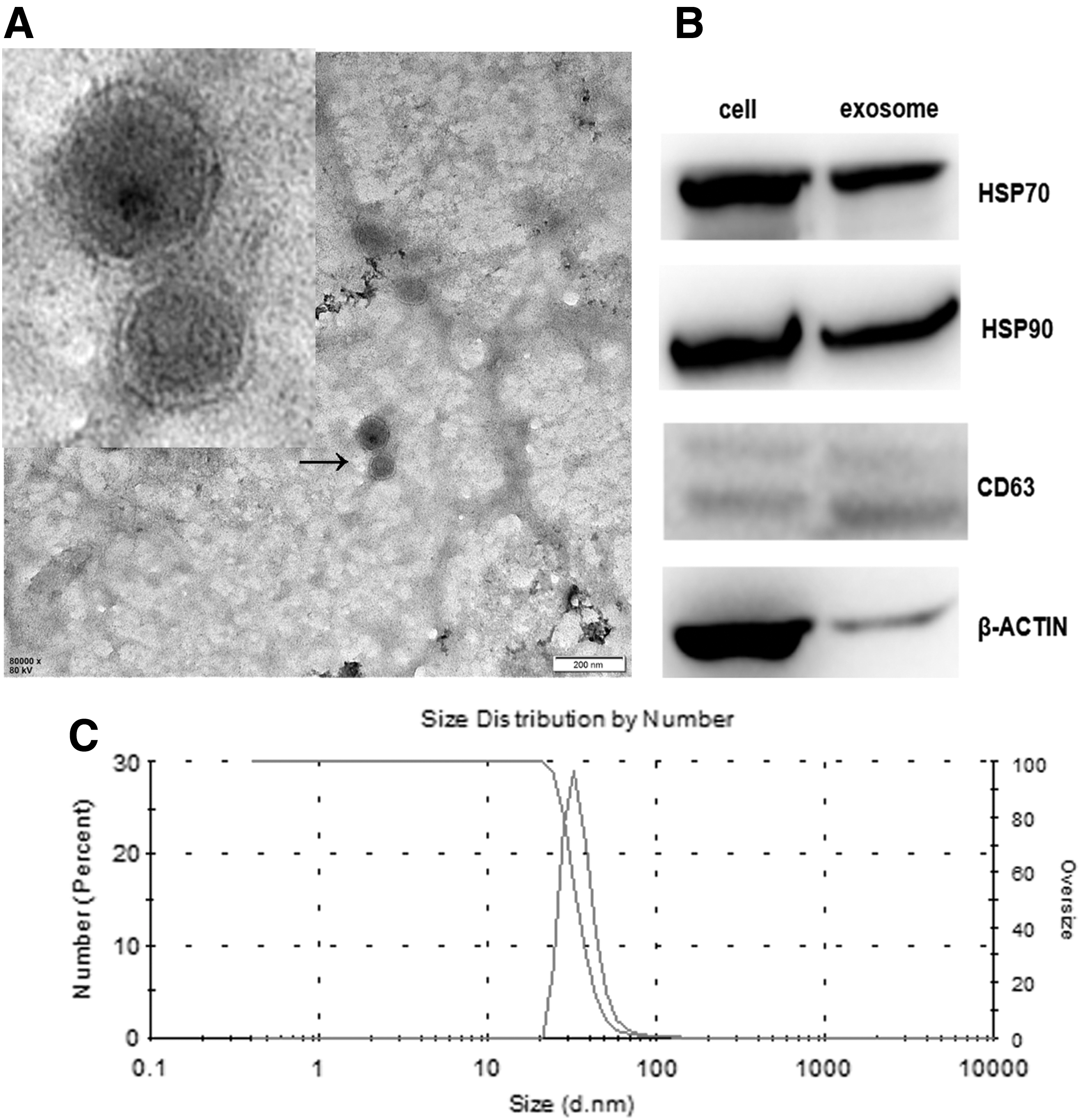
Identification of exosomes derived from hAD-MSCs.
Uptake of AGS-Exos by MSCs
To detect whether AGS-Exos can be ingested by MSCs, AGS-Exos was labeled with Dil and cocultured with MSCs. The uptake was observed in real time using a living cell imager. As shown in Fig. 3A, the uptake of AGS-Exos at different time points in the same visual field was observed. Fluorescence signals were observed in the field of vision for ∼2 h, indicating that MSCs began to ingest AGS-Exos. Moreover, the strength of the fluorescence signal increased over time. After 24 h of uptake, MSCs were photographed after DAPI staining (Fig. 3B), and AGS-Exos were located in the cytoplasm and near the nucleus.
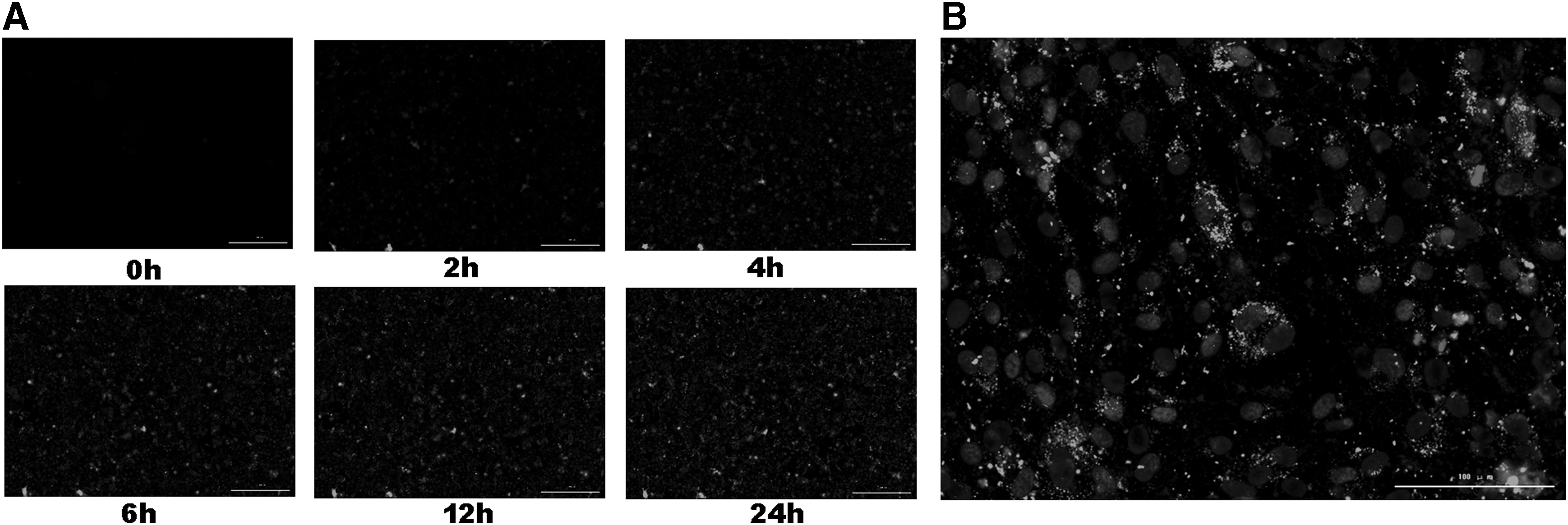
Dil-tagged AGS-Exos transported to hAD-MSCs.
Effects of AGS-Exos on the biological characteristics of MSCs
To detect the changes in biological characteristics of MSCs after uptake of AGS-Exos, the proliferation, cycle, and migration of MSCs were examined. The results indicated that after the fourth day of AGS-Exos stimulation, the proliferation rate of MSCs was significantly lower than that of the control group (P < 0.05), thus suggesting that AGS-Exos could inhibit the proliferation of MSCs (Fig. 4A). The results of cell cycle analyses showed that the proportion of the G0/G1 phase increased in MSCs treated with AGS-Exos, and the G0/G1 phase ratios of MSCs treated with AGS-Exos and MSCs in the control group were 59.9% ± 4.1% and 35.5% ± 1.9%, respectively (P < 0.05). AGS-Exos significantly blocked the cell cycle of MSCs (Fig. 4B). Moreover, the mobility of MSCs in the AGS-Exos-stimulated group was significantly higher than that of the control group, and the migration rate of MSCs increased with increasing AGS-Exos concentrations, which were dose dependent (Fig. 4C).
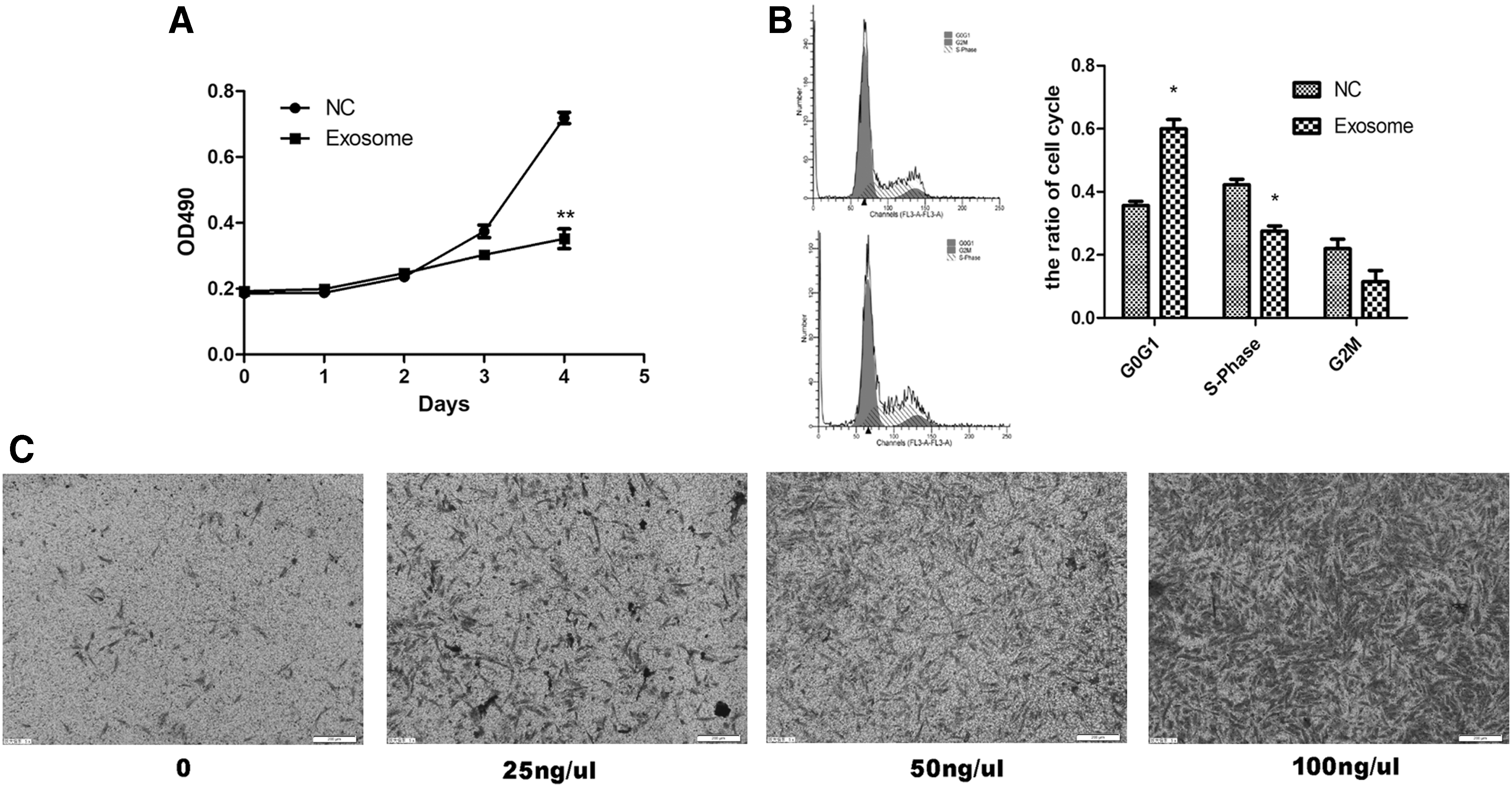
Effects of AGS-Exos on MSC proliferation, cell cycles, and migration.
MSCs stimulated by AGS-Exos promote tumor growth in vivo
To detect the effects of MSCs treated with AGS-Exos in the development of tumor growth, we subcutaneously injected AGS cells with MSCs stimulated by AGS-Exos, normal MSCs, or PBS.
As shown in Fig. 5A, the size of tumors significantly increased in the presence of MSCs treated with AGS-Exos in respect to tumor alone or tumor coinjected with normal MSCs. Such stimulation in tumor growth was confirmed by measurement of tumor diameter (Fig. 3B). Since macrophage recruitment was relevant with tumor growth, the tumors were excised for examination of immune cell infiltration, and we observed that F4/80 macrophages were much more abundant in the first group than in the other two (Fig. 3C, D). In addition, Ki67 staining showed elevated cell proliferation rate in the first group (Fig. 3E, F). Thus, MSCs educated by AGS exosomes gained an enhanced capacity in recruiting macrophages and in promoting tumor growth in vivo.
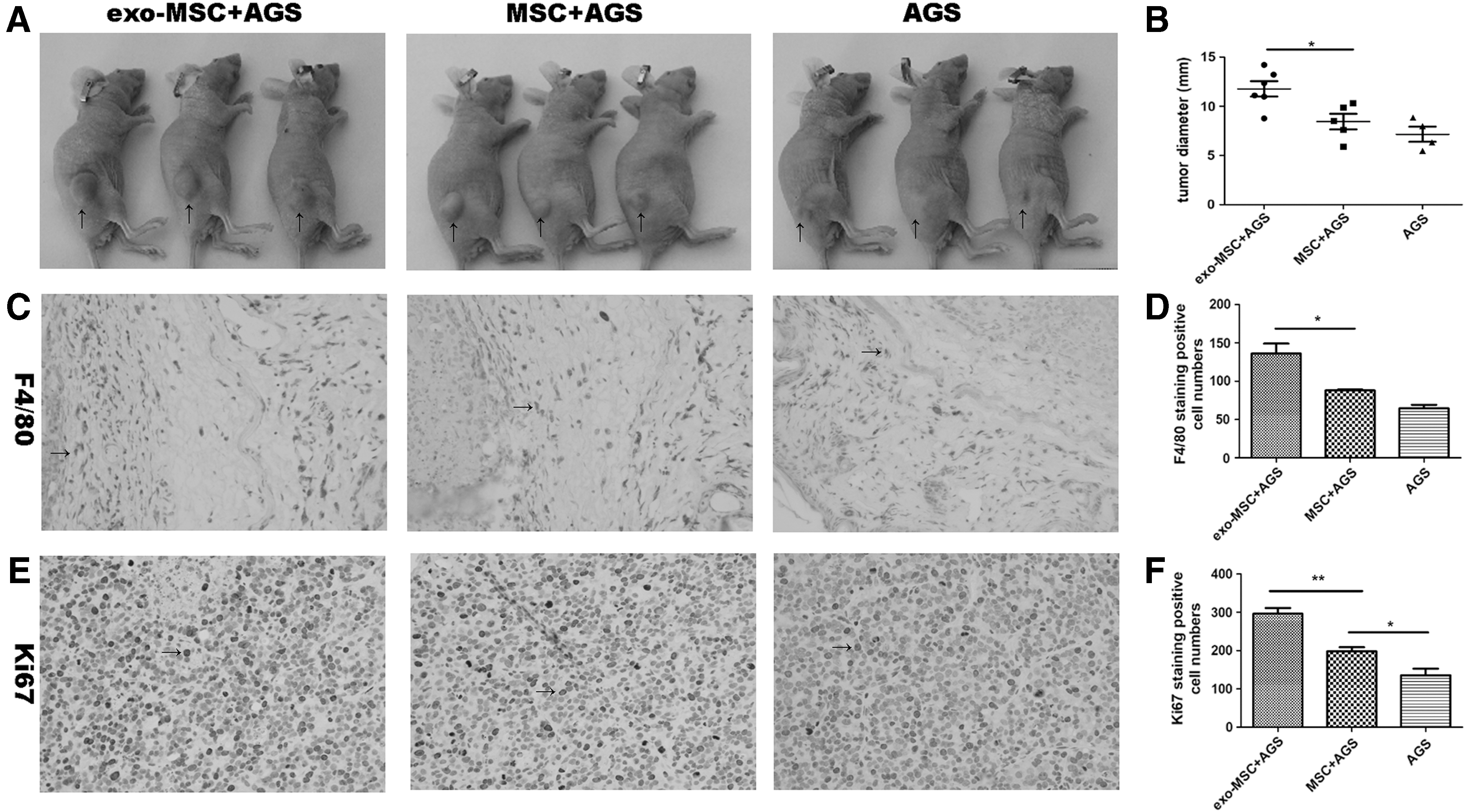
MSCs stimulated by AGS-Exos promote tumor growth in vivo.
Effects of MSCs on T cell activation after 24 h of AGS-Exos treatment
To detect the effects of MSCs treated with AGS-Exos for 24 h on T cell activation, PBMCs with PHA-stimulated activation were cocultured with normal MSCs and MSCs stimulated by AGS-Exos, respectively. The T cell activation level was detected by flow cytometry. Results showed that the double-positive T cell rates of CD4/CD69 in the PBMC group, normal MSCs group, and AGS-Exos-stimulated MSCs group were 4.9% ± 0.5%, 5.4% ± 0.31%, and 16.1% ± 2.7%, respectively. There was a significant difference between the latter two groups (P < 0.05; Fig. 6A, B) in which normal MSCs had no effect on the activation of CD4/CD69 double-positive T cells, while AGS-Exos-stimulated MSCs significantly increased the activation of CD4/CD69 double-positive T cells.
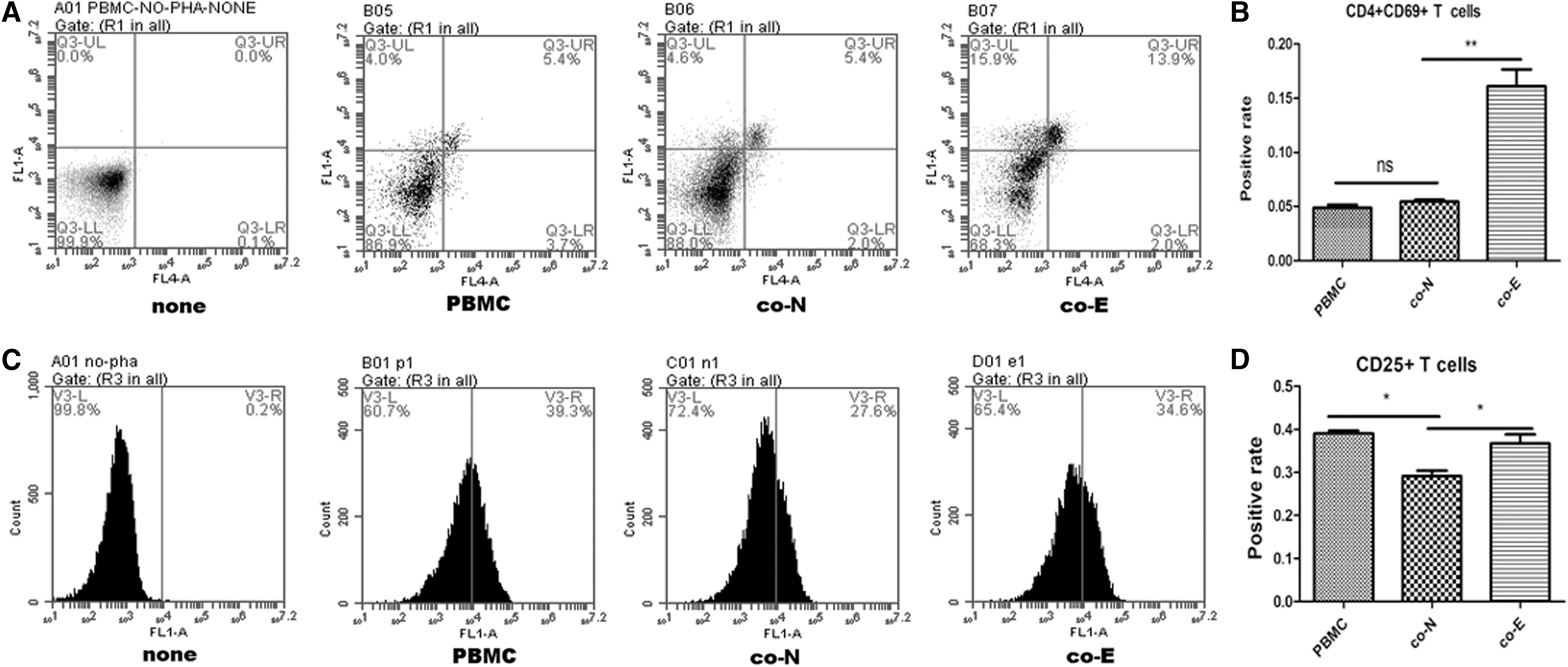
Effects of MSCs treated with AGS-Exos on T cell activation.
The same results were obtained regarding the activation of CD25-positive T cells (the proportions of CD25-positive T cells in the three groups were 39.0% ± 1.2%, 29.1% ± 2.1%, and 36.7% ± 3.5%, respectively; Fig. 5C, D). The results suggest that AGS-Exos stimulation can enhance the ability of MSCs to promote T cell activation. CD69 is an early marker of T cell activation, which is associated with both regulatory T cell (Treg) and memory T cell, and it is also a coactivating molecule of CD25. CD25 is considered to be a characteristic marker molecule on the surface of regulatory T cells, which are beneficial to maintain the microenvironment of tumor immunosuppression.
Effects of MSCs on macrophage activation after 24 h of AGS-Exos treatment
To detect the effects of MSCs treated with AGS-Exos for 24 h on the phagocytosis of macrophages, THP1 cells were induced into adherent macrophages by PMA, and were then cocultured with MSCs. The phagocytosis of fluorescent microspheres was detected by flow cytometry. The results showed that the average fluorescence intensity of phagocytosis of macrophages cocultured with MSCs in the AGS-Exos group was significantly higher than that in normal MSCs group (P < 0.05), suggesting that MSCs in the AGS-Exos treatment group could promote the phagocytosis of macrophages (Fig. 7A–C).
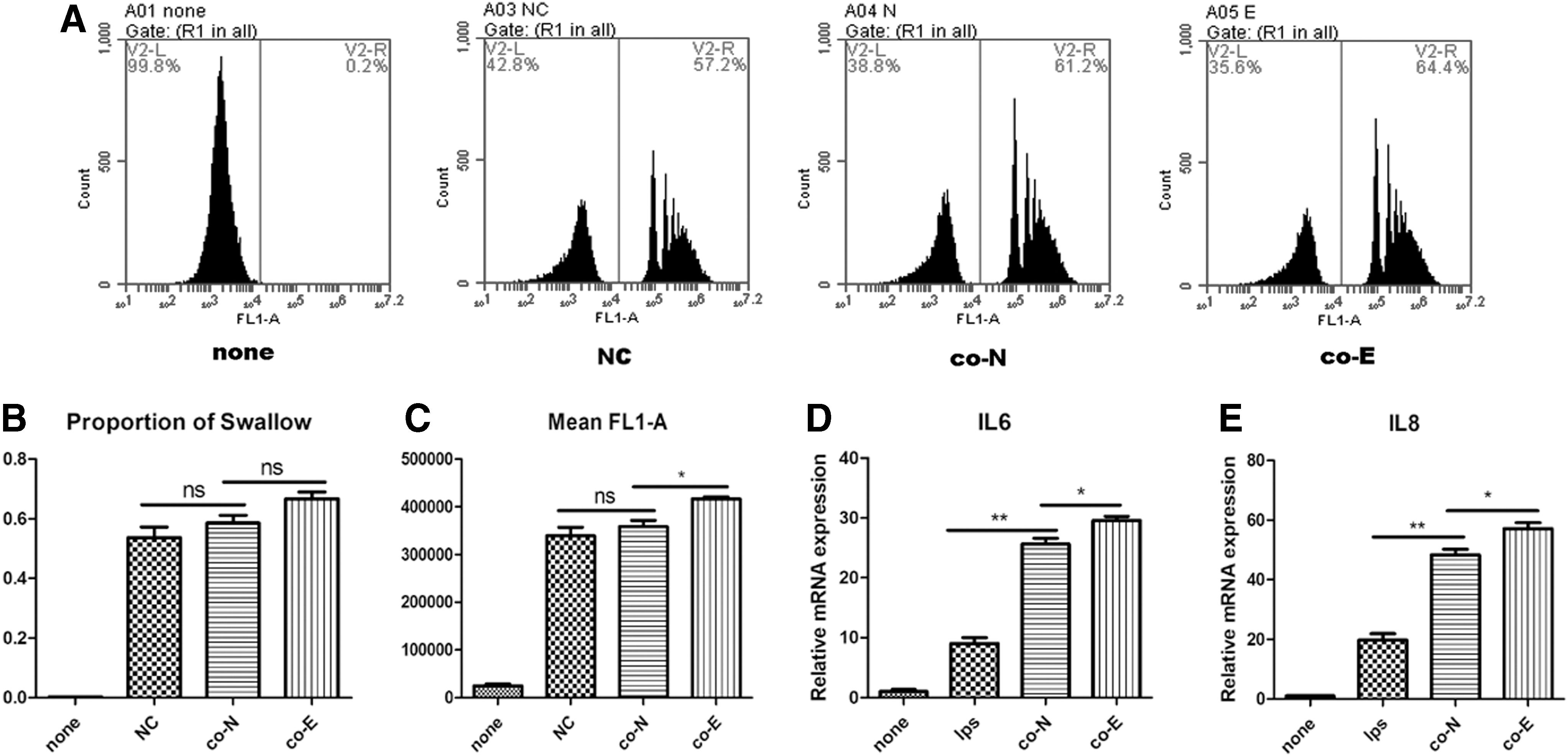
Effects of MSCs treated with AGS-Exos on macrophage activation.
Furthermore, the level of inflammatory cytokines secreted by macrophages after coculture was detected by quantitative PCR. It was found that the level of IL6 and IL8 cytokines secreted by macrophages cocultured with MSCs treated by AGS-Exos was significantly higher than that observed in the normal MSCs group (P < 0.05; Fig. 7D). The results suggest that MSCs treated with AGS-Exos can promote the activation of macrophages.
Analysis of RNA expression profiles based on RNA Chip and bioinformatic analyses before and after MSC treatment by AGS-Exos
Based on the above results, we concluded that MSCs treated with AGS-Exos were capable of activating immune cells, but the mechanism was unknown. Therefore, RNA microarray analyses were used to detect changes in the gene expression of MSCs before and after AGS-Exos treatment. We found that the number and type of genes that changed were relatively large (Fig. 8A–C), including CircRNA, LncRNA, and mRNA (Fig. 8D). Moreover, the enrichment of signal pathways was analyzed, and the results indicated that infection- and immune system-related signal pathways increased most significantly (Fig. 8E, F), including the NF-kB signaling pathway (Fig. 8G).
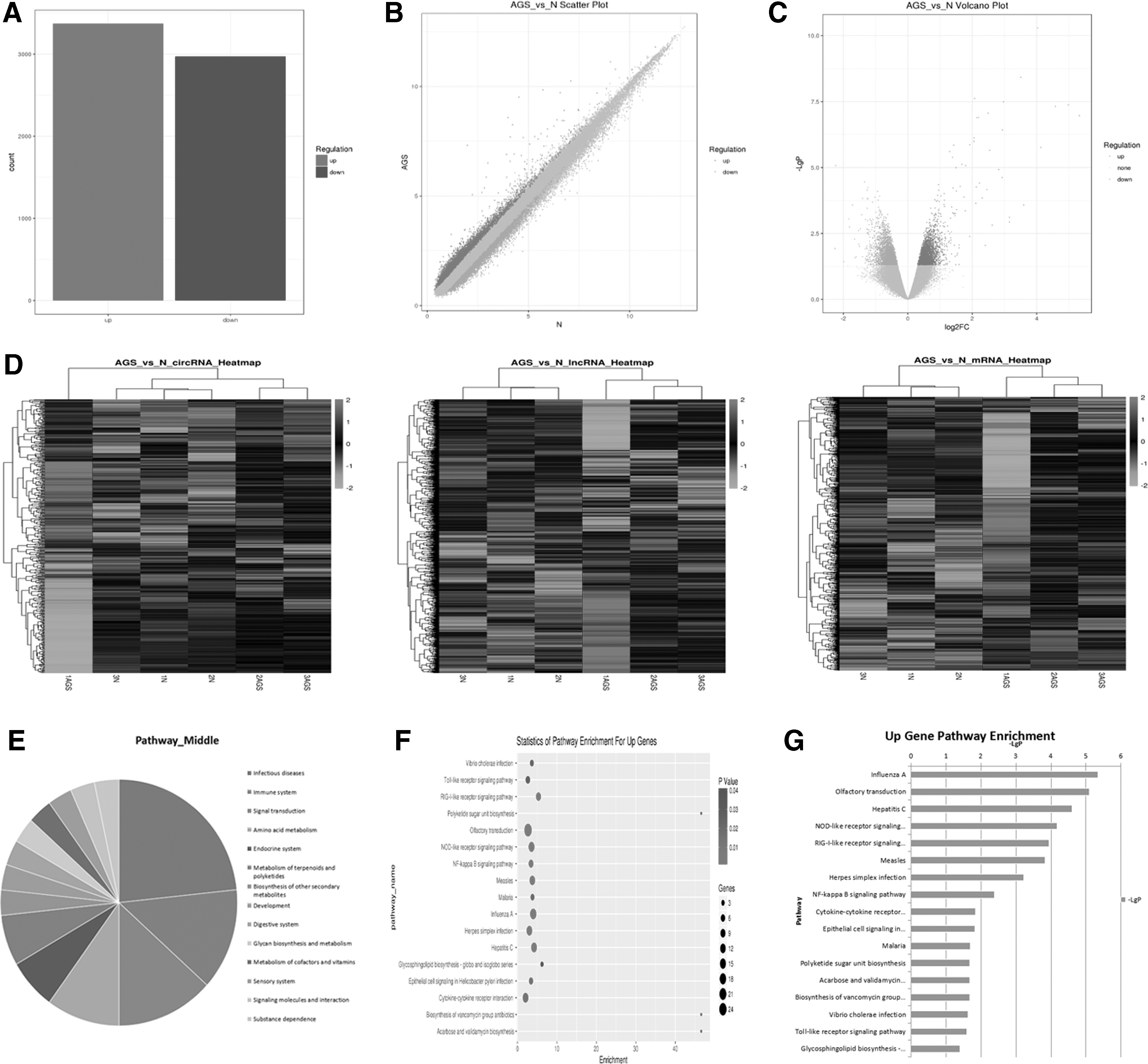
Analyses of RNA expression profiles before and after MSC treatment by AGS-Exos.
AGS-Exos changed MSCs immunomodulatory functions mainly by activating the NF-kB signaling pathway
It has been reported that exosomes from other tumor cells can activate the NF-kB signaling pathway of MSCs [7], and this is consistent with the microarray prediction results of this study. Therefore, we first verified through RNA Chip analysis that the AGS-Exos treatment activated the NF-kB signaling pathway of MSCs. The western blot result of the key protein of the NF-kB signaling pathway was verified at different time points after adding AGS-Exos. The results showed that the phosphorylated p65 protein began to increase 10 min after the addition of the exosome, peaked after 20 min, and then decreased gradually (Fig. 9A). This result indicated that the activation level of the NF-kB signaling pathway was altered, and that p65 phosphorylation was caused by the degradation of IKBa, thus activating the NF-kB signaling pathway (Fig. 8A). Accordingly, we also carried out immunofluorescence experiments to detect the level of P-p65, and found that it was consistent with the results of the western blot (Fig. 9B).
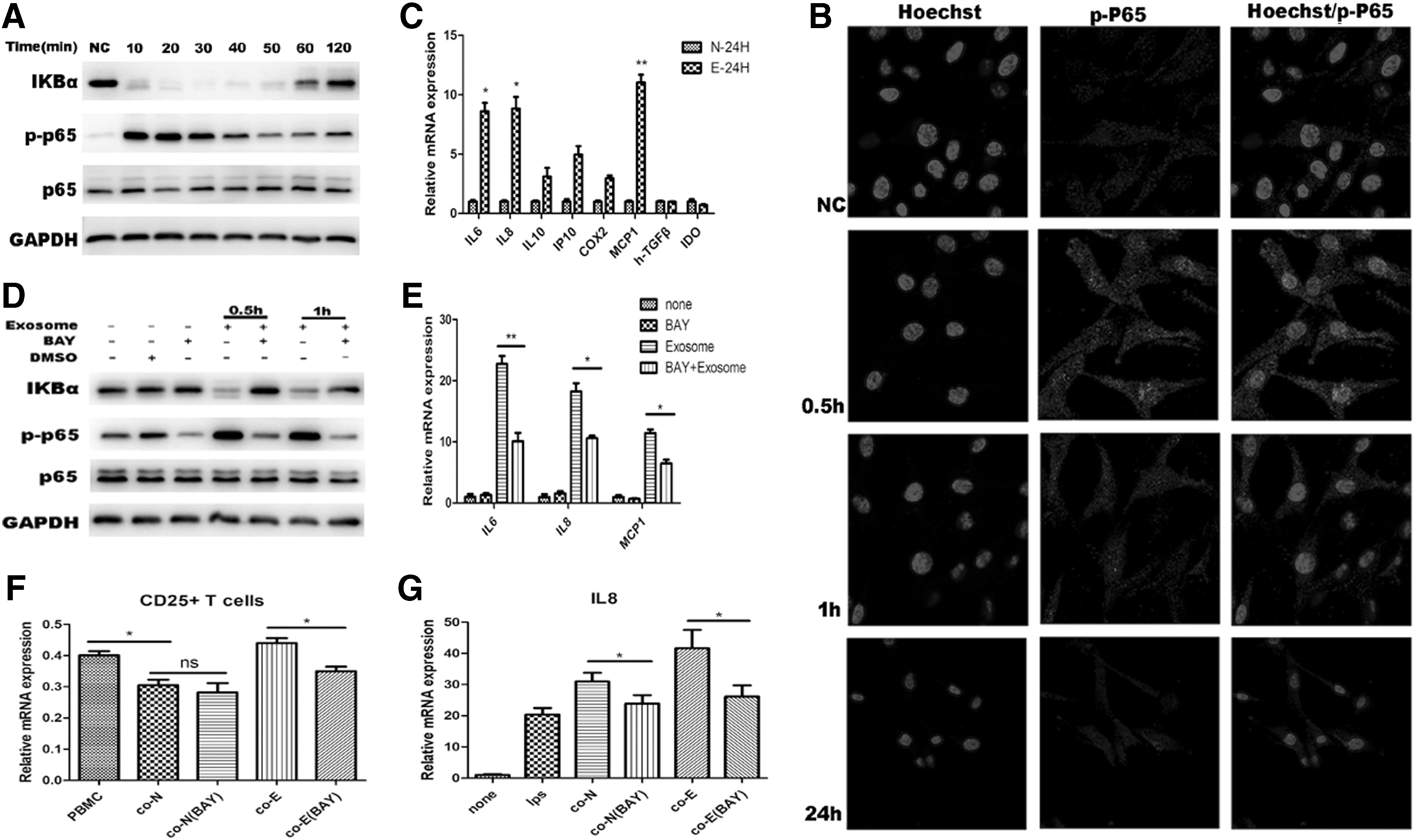
AGS-Exos changed MSCs immunomodulatory functions mainly by activating the NF-kB signaling pathway.
In addition, we detected the expression of some inflammatory factors downstream of the NF-kB signaling pathway, and found that the expression of proinflammatory factors IL6, IL8, and MCP1 increased (Fig. 9C), thus suggesting that the alteration of the MSC immune regulatory function is probably caused by NF-kB signaling pathway activation.
The NF-kB signaling pathway was further inhibited to observe whether the changes in MSC immunomodulatory functions were inhibited by AGS-Exos. The inhibitor Bay 11-7082 can inhibit the phosphorylation of IKBa and inhibit the components of the ubiquitin system. Based on pre-experimental results, we selected a concentration of 4 μM to act for 6 h before conducting subsequent experiments.
First, western blot tests showed that p65 phosphorylation was inhibited (Fig. 9D). Furthermore, the levels of the inflammatory cytokines IL6 and IL8 and MCP1 in MSCs decreased significantly (P < 0.05), but the levels were still detectable (Fig. 9E). The immunomodulatory function of MSCs on T cells and THP1 cells was further observed, and it was determined that after the inhibition of the NF-kB signaling pathway, MSCs treated with AGS-Exos could weakly promote the activation of T cells (Fig. 9F). Moreover, the level of inflammatory cytokines secreted by THP1 cells was significantly decreased (Fig. 8G). These results suggest that NF-kB is an important factor associated with the influence of exosome bodies on the immune regulatory function of MSCs.
Discussion
MSCs are immunomodulatory cells [16], and their immunomodulatory effects may have a direct effect on the tumor microenvironment. However, a large number of studies have shown that the biological characteristics of tumors have undergone major changes [25,29,30]. Compared with normal MSCs, the proliferation, migration, and tumor support functions of MSCs were significantly enhanced [31 –34]. Therefore, whether there is a difference in the immunoregulatory effects of MSCs between local tumor and normal MSCs is important for our understanding of the relationship between MSCs and the tumor inflammatory microenvironment. In this study, we found that MSCs stimulated by AGS-Exos can significantly promote T cell activation markers CD69 and CD25, but MSCs derived from normal adipose tissue have little effect on T cells.
This study found that AGS-Exos-stimulated MSCs significantly promoted the early activation of T cell markers CD69 and CD25, while the role of normal adipose tissue-derived MSCs in T cells was not obvious. A large number of studies have shown that MSCs derived from bone marrow can inhibit the proliferation of PBMCs, thus inhibiting the function of T cells [18 –20], but there are few reports on the detection of activation markers. Furthermore, studies of healthy human bone marrow-derived MSCs showed no significant changes in the early activation of T cell marker CD69 [20], and this is consistent with the results of this experiment.
In this experiment, the normal fat-derived MSCs were cocultured with PBMCs, and there was no significant change in the activation of T cells. In addition, with respect to the study of the immunoregulatory properties of PBMCs by MSCs, most studies reported that bone marrow-derived MSCs were used, and the reference data for AD-MSCs are still relatively limited.
Studies have found differences in the inhibition of PBMC proliferation between fat, umbilical cord, and bone marrow-derived MSCs, and the results showed that bone marrow-derived MSCs had the strongest immunosuppressive effects, followed by umbilical cords and fat-derived MSCs (the weakest immunosuppressive effects) [35]. These results suggest that there are differences in immunomodulatory functions among the three groups, and this may explain the instability of MSCs derived from adipose tissue in early T cell activation. However, the results also indicated that MSCs stimulated by AGS-Exos promoted T cell activation markers.
Furthermore, consistent with this result, MSCs stimulated by AGS-Exos significantly enhanced the ability of THP1 cells to secrete proinflammatory cytokines (Fig. 7D, E), and the promotion of phagocytosis in macrophages was also more pronounced (Fig. 7A, C), thus suggesting that MSCs stimulated by tumor exosomes can promote the activation of THP1 cells and macrophages. It has been reported that tumor growth depends on the recruitment of macrophages by MSCs [25], and the upregulation of macrophage proinflammatory factors may contribute to the proliferation and migration of tumors [36]; therefore, indirectly confirming the significance of our findings.
According to the results of RNA microarray and bioinformatic analyses, it was suggested that the possible reason is that the NF-kB signaling pathway of MSCs activated by AGS-Exos and proinflammatory cytokines increased, resulting in MSCs that promoted T cells and macrophages. After inhibiting the NF-kB signaling pathway of MSCs, the secretion of inflammatory cytokines of MSCs was significantly reduced, but was still detectable. Furthermore, the promotion of the early activation T cell markers and the secretion of inflammatory cytokines in THP1 cells were weakened, indicating that the NF-kB signaling pathway was activated.
In conclusion, our study found that exosomes secreted by gastric cancer cells affect the immune regulatory functions of MSCs through the NF-kB signaling pathway, strengthen the activation ability of MSCs for immune cells, maintain the inflammatory environment, and support the growth of tumors. This study has explored new mechanisms for the immune regulatory function of MSCs, and also provided possibilities and new strategies for finding targets for the prevention and treatment of gastric cancer.
Footnotes
Acknowledgments
This study was supported by grants from National Natural Science Foundation of China (81672313, 81473450); the National Key Research and Development Program of China (2016YFA0101000, 2016YFA0101003); CAMS Innovation Fund for Medical Sciences (2017-I2M-3-007); and Beijing Key Laboratory of New Drug Development and Clinical Trial of Stem Cell Therapy (BZ0381).
Author Disclosure Statement
No competing financial interests exist.