
Abstract
Cerebral infarction is one of the major causes of severe morbidity and mortality, and thus, research has focused on developing treatment options for this condition. Zinc (Zn) is an essential element in the central nervous system and has several neuroprotective effects in the brain. In this study, we examined the neuroprotective effects of Zn on neural stem cells (NSCs) exposed to hypoxia. After treatment with several concentrations of Zn, the viability of NSCs under hypoxic conditions was measured by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, Trypan blue staining, and a lactate dehydrogenase assay. To evaluate the effect of Zn on the proliferation of NSCs, bromodeoxyuridine/5-bromo-2′-deoxyuridine (BrdU) labeling and colony formation assays were performed. Apoptosis was also examined in NSCs exposed to hypoxia with and without Zn treatment. In addition, a western blot analysis was performed to evaluate the effect of Zn on intracellular signaling proteins. NSC viability and proliferation were decreased under hypoxic conditions, but treatment with sublethal doses of Zn restored viability and proliferation. Sublethal doses of Zn reduced apoptosis caused by hypoxia, increased the expression levels of proteins related to the phosphatidylinositol-3 kinase (PI3K) pathway, and decreased the expression levels of proteins associated with neuronal cell death. These findings confirm that in vivo, sublethal doses of Zn protect NSCs against hypoxia through the activation of the PI3K pathway. Thus, Zn could be employed as a therapeutic option to protect NSCs in ischemic stroke.
Introduction
Hypoxia is one of the main pathogenic mechanisms of neuronal cell death by ischemic stroke [1 –3], and it is known to produce reactive oxygen species (ROS), including free radicals and peroxides, which are closely associated with apoptosis and necrosis in ischemic stroke [4]. Despite unsatisfactory results in clinical studies, numerous studies have tried to identify the neuroprotective effects of promising drugs, such as antioxidant, antiapoptotic, and anti-inflammatory agents, for ischemic stroke for many years [5].
Neural stem cells (NSCs) are multipotent stem cells that can differentiate into neurons, astrocytes, and oligodendrocytes. In recent years, NSCs have been studied as a promising option for developing neuroprotective strategies for the treatment of cerebral infarction [6]. NSCs are known to have a major role in endogenous neurogenesis following ischemic insult. Once activated by ischemic injury, NSCs migrate toward the involved lesions where they assist in their recovery [7]. However, NSCs can also be damaged by ischemic stroke. Therefore, it is essential to prevent NSC damage following ischemic insult to reduce brain injury and promote the restoration of neurological deficits.
Zinc (Zn) is an essential element that has vital functions in the central nervous system [8]. Although the exact physiological mechanisms of Zn remain to be elucidated, several studies suggest that in certain pathological states, high doses of Zn could induce neuronal damage. Previous studies have demonstrated that excessive Zn plays an important role in acute neural injury caused by epilepsy, traumatic brain injury, and stroke [9 –12]. However, several recent studies have shown that Zn also has several neuroprotective effects in the brain [13 –16]. It is essential for neurogenesis and influences cell division and proliferation.
Therefore, it is unsurprising that Zn deficiency induces abnormal brain development [17]. Despite numerous investigations, the mechanisms behind its neurotoxic and neuroprotective effects remain unclear. In addition, the effect of Zn on NSCs in cerebral ischemia has not been fully investigated. In this study, we aimed to investigate the neuroprotective effects of sublethal doses of Zn on NSCs under hypoxic conditions.
Methods
Materials
To create hypoxic conditions, we used an anaerobic chamber (Forma Anaerobic System Model 1025; Thermo Forma, Marietta, OH) and N2 medium (Gibco, NY). Zinc chloride was provided as a generous gift from Il-Dong (Korea). Before use, we dissolved zinc chloride in methyl chloride to 500 mM, diluted it with dimethyl sulfoxide (DMSO) to 100 mM, and then further diluted it in culture medium to the desired concentration (0.1–100 μM).
Culture of NSCs under hypoxic conditions and Zn treatment
All procedures using animals were performed in accordance with Hanyang University guidelines for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee (IACUC) of Hanyang University. Every effort was made to minimize the number and suffering of animals.
NSCs were isolated from rodent embryonic brains, cultured, and increased. The NSC culture protocol was used as previously described in previous studies [18 –20]. Briefly, rat embryos were decapitated at embryonic day 13 (E13), and the brains were rapidly removed and placed in a petri dish filled halfway with ice-cold Hank's balanced salt solution [HBSS; 137 mM NaCl, 5.4 mM KCl, 0.3 mM Na2HPO4, 0.4 mM KH2PO4, 5.6 mM glucose, and 2.5 mM HEPES (Gibco BRL, NY)].
Single cells were dissociated from the whole cerebral cortexes, lateral ganglionic eminences, and ventral midbrains of the fetal rats. The cells were plated at 2 × 104 cells/cm2 on culture dishes precoated with poly-
All hypoxia experiments were conducted in an anaerobic chamber. A gas mixture that contained CO2 (5 mol %), O2 (0.2 mol %), and N2 (94.8 mol %) was flushed through the chamber for 0–24 h. This process preserved an environment of nonfluctuating hypoxia below 1 mol % O2 [21] with several experimental concentrations of Zn (0.1–100 μM) in N2 medium.
To evaluate whether NSCs were affected by hypoxia conditions delivered by this process, we counted the surviving cells using a Trypan blue and lactate dehydrogenase (LDH) assay. To observe the effect of Zn itself, NSCs were treated with several concentrations of Zn in both normal and anaerobic conditions for 8 h. Cell viability was assessed by Trypan blue and LDH assays. Based on our preliminary data, NSCs were treated with varying concentrations of Zn (0, 0.1, 0.5, 1, 5, 10, 20, 50, and 100 μM) for 8 h under hypoxic conditions. The cells were then gently washed, and cell viability was evaluated.
Cell viability was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and LDH assays. Finally, to examine the role of the phosphatidylinositol-3 kinase (PI3K) pathway in the neuroprotective effects of Zn, we also treated NSCs with 10 μM of the PI3K inhibitor LY294002 60 min before treatment with Zn under hypoxic conditions.
NSCs were separated into seven groups as follows: control (group 1); hypoxia for 8 h (group 2); hypoxia +1 μM Zn for 8 h (group 3); 10 mM of the PI3K inhibitor LY294002 for 9 h, hypoxia, and 1 μM Zn for 8 h (group 4); 10 mM LY294002 for 9 h+hypoxia for 8 h (group 5); 10 mM LY294002 for 9 h+1 μM Zn for 8 h (group 6); and 10 mM LY294002 alone for 9 h (group 7). Cell viability was assessed by MTT and LDH assays.
MTT assay, Trypan blue staining, and LDH assay
Cells were plated in 96-well plates at a density of 1 × 104 cells/well in 200 μL of medium. An aliquot (220 μL) of the resulting solution was removed from each well, followed by the addition of 150 μL DMSO. The precipitate from each well was resuspended on a microplate mixer for 10 min, and optical densities (OD) at 540 nm were measured using a plate reader. All results were subtracted by OD values measured from an identically treated well without cultured cells.
For Trypan blue staining, 10 μL of Trypan blue solution (BMS, Seoul, Korea) was incubated with 10 μL of dissociated cells from each sample for 2 min. Unstained live cells were counted using a hemocytometer [19]. A colorimetric assay kit (Roche Boehringer–Mannheim, IN) was used to quantify LDH released from cultured NSCs according to the manufacturer's instructions. Cell cytotoxicity was assessed using an ELISA plate reader (Synergy H1 Hybrid reader; Bio-Tek, Winooski) by measuring the absorbance at 490 nm at a reference wavelength of 690 nm. All results were subtracted from the OD of an identical well without cells.
Immunostaining for nestin and Ki-67
NSCs were seeded (1 × 105 cells) on chamber well plates, and the cells were treated with different concentrations of Zn (0, 0.1, 1, or 10 μM) for 8 h under hypoxic conditions. The cells were fixed in 2% paraformaldehyde in Dulbecco's PBS (DPBS) for 15 min and were permeabilized with 0.5% Triton X-100 in DPBS for an additional 5 min. The endogenous peroxidase activity was blocked using 3% H2O2 in DPBS for 20 min and cells were washed several times with DPBS.
The cells were incubated in 5% normal serum in DPBS for 1 h. The cells were incubated overnight in 2% normal serum in DPBS that contained the primary antibodies, mouse anti-nestin (1:100; Abcam) and rabbit anti-Ki-67 (1:100; Abcam). The next day, the cells were incubated for 60 min in 2% normal serum in PBS that contained the secondary antibodies tetramethylrhodamine goat anti-rabbit lgG (HþL Life Technologies) and goat anti-mouse Alexa Fluor 488 (Life Technologies), washed several times with PBS, and mounted on glass slides using Moviol 4-88 solution. The cells were visualized with a fluorescence microscope (eclipse Ti; Nikon) to estimate the percentage of anti-nestin- and anti-Ki-67-positive cells [22].
BrdU cell proliferation assay and colony-forming unit assay
The proliferation of NSCs was calculated using a 5-bromo-2′-deoxyuridine (BrdU) cell proliferation assay and the colony-forming unit (CFU) assay. Approximately 1 × 104 cells were seeded in a 60-mm grid plate and treated with Zn (0, 0.1, 1, and 10 μM) under hypoxic conditions for 8 h. The cells were incubated in BrdU labeling medium (10 μM BrdU) for 6 h, and the cell proliferation assay was performed using a BrdU Labeling and Detection Kit (Roche Boehringer–Mannheim) according to the manufacturer's instructions.
Cell proliferation was assessed using an ELISA plate reader at 370 nm with a reference wavelength of 492 nm. All results were subtracted by the OD of an identical well without cells [22]. For colony-forming assays, the cells were washed with DPBS, and the culture media were changed. After 14 days, the cells were washed again with DPBS and stained with 0.5% crystal violet (Sigma) in methanol for 30 min at room temperature. After staining, the plates were washed with DPBS and allowed to dry. The colony count was performed using a dissecting microscope. Colonies that were less than 2 mm in diameter or faintly stained were excluded [22].
DAPI and TUNEL staining to evaluate apoptosis
NSCs were seeded on collagen-coated 13-mm diameter glass coverslips and treated with Zn (0. 0.1, 1, and 10 μM) under hypoxic conditions for 8 h. The cells were then rinsed twice with PBS, air dried, and fixed with 4% paraformaldehyde in PBS for 60 min at room temperature. Apoptotic cell death was identified by terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL; Roche Boehringer–Mannheim). To monitor intact, condensed, and fragmented nuclei, TUNEL-stained cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, 100 μg/mL in PBS; Sigma) for 20 min, washed several times with PBS, and mounted on glass slides with Moviol 4-88 solution. The cells were observed under an Olympus fluorescence microscope with the appropriate excitation wavelengths for TUNEL and DAPI staining [23].
Western blot analysis
Levels of nestin, Ki67, p85a PI3K, phosphorylated Akt (pAkt) (Ser473), phosphorylated glycogen synthase kinase-3β (pGSK-3β) (Ser9), Bcl-2, Bax, cleaved caspase 9, cleaved caspase 3, and cytosolic cytochrome C were analyzed using western blotting. Briefly, 5 × 106 cells treated under several conditions were washed twice in cold PBS and incubated in lysis buffer [50 mM Tris (pH 8.0), 150 mM NaCl, 0.02% sodium azide, 0.2% sodium dodecyl sulfate (SDS), 100 mg/mL phenylmethylsulfonylfluoride (PMSF), 50 mL/mL aprotinin, 1% IGEPAL 630, 100 mM NaF, 0.5% sodium deoxycholate, 0.5 mM EDTA, and 0.1 mM EGTA] for 10 min on ice.
Cell lysates were centrifuged at 10,000g and evaluated for levels of nestin, Ki67, p85a PI3K, pAkt, pGSK-3β, Bcl-2, Bax, cleaved caspase 9, cleaved caspase 3, and cytosolic cytochrome C. Protein concentrations of cell lysates and postmitochondrial fractions were determined using a Bio-Rad protein assay kit (Bio-Rad, Hercules, CA). Samples containing equal amounts (20 mg) of protein were resolved by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes (Whatman, Maidstone, United Kingdom). Membranes were blocked using 2% skim milk before incubation with specific primary antibodies against nestin (1:100; Abcam), Ki67 (1:100; Abcam), p85a PI3K (1:1000; Cell Signaling, Beverly, MA), pAkt (1:100; Cell Signaling), pGSK-3β (1:100; Cell Signaling), Bcl-2 (1:200; Cell Signaling), Bax (1:500; Cell Signaling), cytosolic cytochrome C (1:200; Cell Signaling), cleaved caspase 9 (1:500; Cell Signaling), and cleaved caspase 3 (1:500; Cell Signaling). Membranes were washed with Tris-buffered saline containing 0.05% Tween 20 and then processed using a horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibody (Jackson Immunoresearch Laboratories, Bar Harbor, ME) followed by the West-Q Chemiluminescent Substrate Plus kit (GenDEPOT, Barker, TX). The western blotting results were quantified using an image analyzer (Quantity One-4,2,0; Bio-Rad).
Reverse transcriptase PCR
Total RNA was extracted from the NSCs using the TRIzol (Invitrogen). cDNA was synthesized using the amfiRivert cDNA Synthesis Platinum Master Mix (GenDEPOT) according to the manufacturer's protocol. Reverse transcriptase polymerase chain reaction (RT-PCR) was performed using amfiEco Taq DNA polymerase (GenDEPOT) and the conditions were as follows; 94°C for 2 min, 58–60°C for 2 min, and 72°C for 1 min (40 cycles). GAPDH was used as internal control. The PCR products were confirmed by using 1%–2% agarose gel electrophoresis and quantified using an image analyzer (ImageQuant LAS 4000; GE Healthcare, Little Chalfont, United Kingdom) [24 –26].
Statistical analyses
All data are presented as the mean ± standard deviation from five independent experiments. Comparisons between the different treatment groups of viability, cytotoxicity, apoptosis rates, proliferation rates, immunocytochemistry, free radical production, and western blotting results were conducted using a one- or two-way analysis of variance (ANOVA) (depending on the data) followed by Tukey's post hoc test. P values less than 0.05 were considered statistically significant. All statistical analyses were performed using SPSS 17.0 software package for Windows (SPSS, Seoul, Korea).
Results
Effect of hypoxia and Zn on the viability of NSCs
To confirm the effect of hypoxia on NSC viability, NSCs were incubated in an anaerobic chamber for different amounts of time (between 0 and 24 h). Cell viability was measured using Trypan blue staining (TBS) and LDH assays. Under hypoxic conditions, cell viability was significantly reduced in a time-dependent manner (Fig. 1A, B, E). After 8 h of exposure to hypoxic conditions, cell viability was 60%–70%; therefore, it was chosen as an optimal exposure time for further experiments. To evaluate the effect of Zn itself on NSCs, we treated NSCs with several concentrations of Zn (0, 0.1, 0.5, 1, 5, 10, 20, 50, and 100 μM) under normal conditions. We found Zn to be cytotoxic at concentrations of over 5 μM (Fig. 1C, D, F).
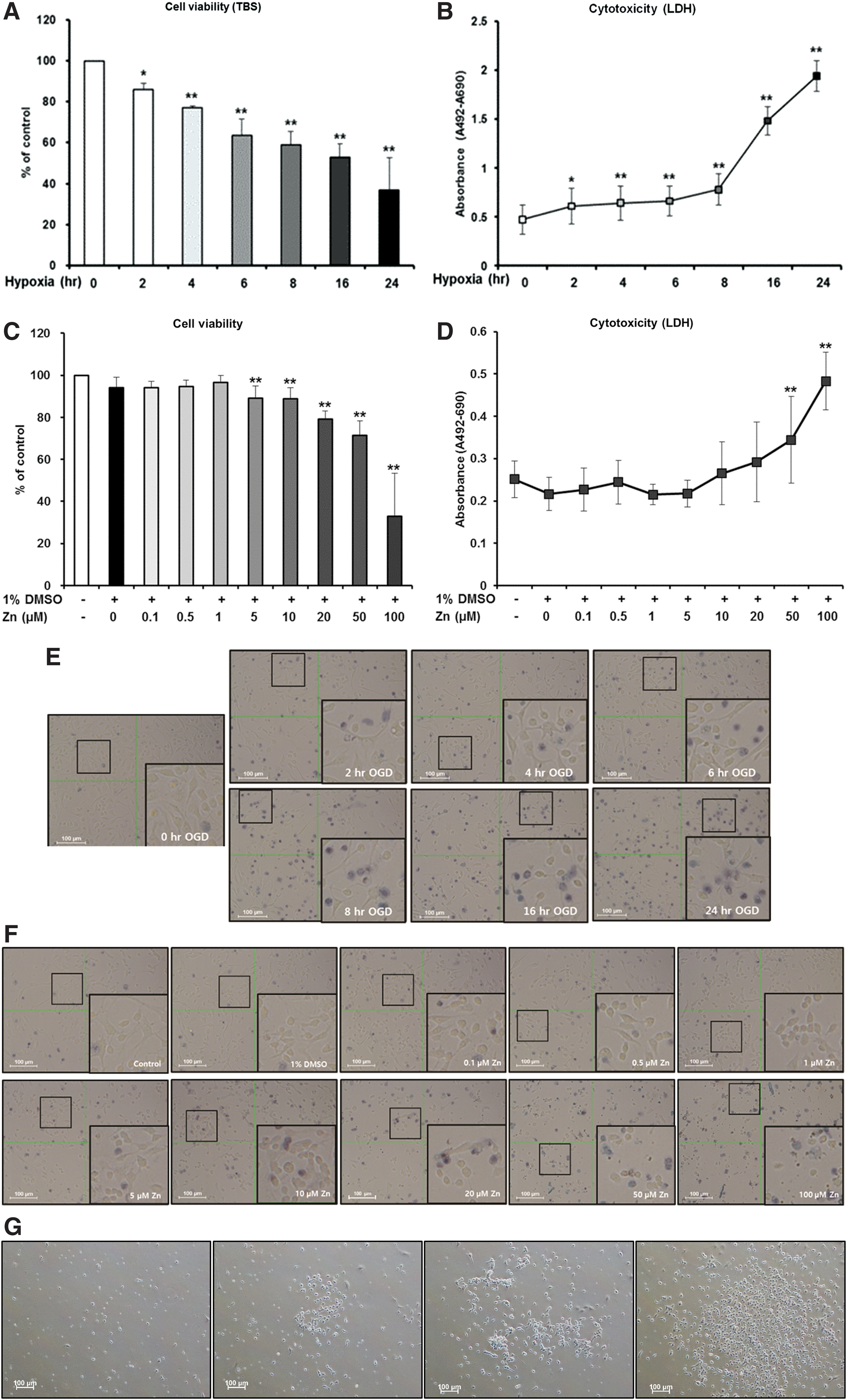
Effect of hypoxia or Zn on NSCs. Trypan blue staining was performed to check the viability of NSCs and an LDH assay was performed to evaluate the damage of NSCs. The viability and damage of cells exposed to hypoxia for different exposure times were compared to controls using a one-way ANOVA and Tukey's post hoc test. *P < 0.05 (vs. control group)
To evaluate the effects of Zn on NSCs exposed to hypoxia, NSCs were treated with several concentrations of Zn (0, 0.1, 1, or 10 μM) under hypoxic conditions for 8 h. Compared to the NSCs exposed to hypoxia without Zn, NSC viability significantly increased when they were simultaneously treated with 1 μM Zn (Fig. 2A, B, E). To investigate whether the PI3K pathway, which is known to be a very important signaling pathway for NSCs, is implicated in cell viability, NSCs were separated into seven groups as Methods section.
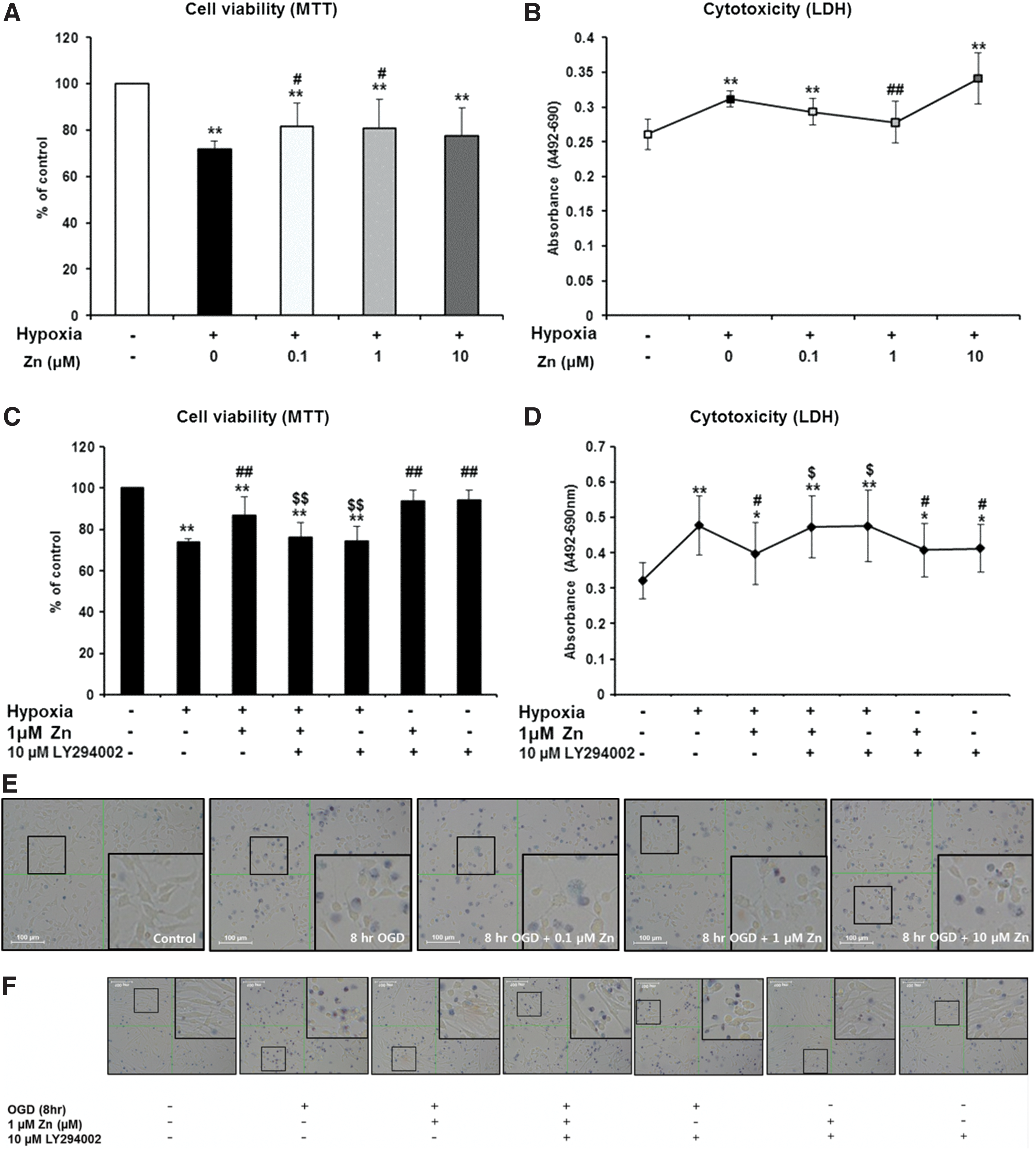
Effect of Zn on the viability of NSCs injured by hypoxia. An MTT assay was performed to measure the viability of NSCs and an LDH assay was performed to evaluate NSC damage and to confirm the role of the PI3K pathway in the neuroprotective effects of Zn against hypoxia in NSCs. Cells were exposed to hypoxia for 8 h and were simultaneously treated with Zn (1 μM) and/or LY294002 (10 μM). All data were compared using a two-way ANOVA and Tukey's post hoc test. *P < 0.05 (vs. control group); **P < 0.01 (vs. control group); #
P < 0.05 (vs. hypoxia alone for 8 h); ##
P < 0.01 (vs. hypoxia alone for 8 h)
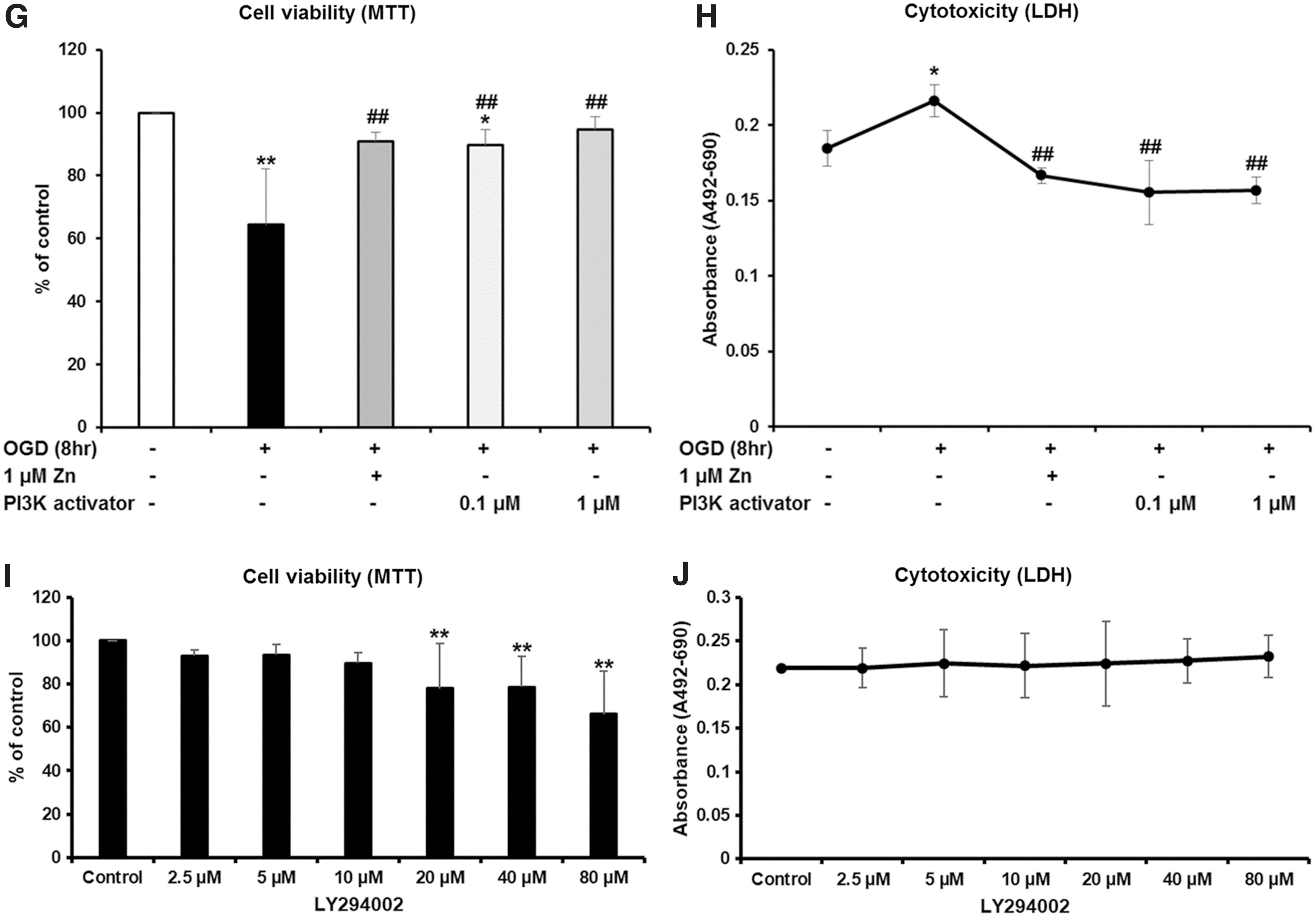
In this study, pretreatment with the PI3K inhibitor (group 4) blocked the protective effect of Zn on NSCs under hypoxic conditions (Fig. 2C, D, F). Treatment with PI3K activator-740Y-P was also analyzed for NSCs treated with Zn under hypoxic conditions. It showed similar results to the PI3K inhibitor (Fig. 2G, H). To evaluate the effect of PI3K inhibitor LY294002 itself on NSCs, we treated LY294002 with several concentrations (0, 2.5, 5, 10, 20, 40, and 80 μM) under normal conditions. We found LY294002 to be cytotoxic at concentrations of over 10 μM, and chose the dose of PI3K inhibitor LY294002 (10 μM) (Fig. 2I, J).
Effect of hypoxia and Zn on the proliferation of NSCs
Cell proliferation was assessed using BrdU labeling, a colony formation assay, and immunostaining for Ki-67 and nestin, which are NSC markers and reflect proliferative activity. Compared to NSCs exposed to hypoxia for 8 h, cells cotreated with Zn exhibited significantly higher cell proliferation activity (Fig. 3A). BrdU labeling and colony formation assays also showed that Zn treatment markedly increased cell proliferation (Fig. 3B–D).

The effects of Zn on NSC proliferation under hypoxic conditions. NSCs were simultaneously exposed to hypoxia for 8 h and different concentrations of Zn (0, 0.1, 1, or 10 μM). NSCs were immunoreactive for the proliferation capacity marker Ki67 and the NSC marker nestin
Antiapoptotic effects of Zn on NSCs under hypoxic conditions
DAPI and TUNEL staining were performed to examine apoptosis rates. There were significantly more apoptotic NSCs under hypoxic conditions, but this number significantly decreased with Zn treatment (0.1 and 1 μM) (Fig. 4A, B). Treating NSCs with 1 μM Zn resulted in the greatest decrease in the percentage of apoptotic cells.

Antiapoptotic effects of Zn on NSCs under hypoxic conditions. NSCs were stained with DAPI and TUNEL. The data are presented as the percentage of TUNEL-positive cells ± SD. Each treatment group was compared to all other groups using a two-way ANOVA followed by Tukey's post hoc test. **P < 0.01 compared to the control group; # P < 0.05 compared to NSCs under hypoxic conditions alone; ## P < 0.01 compared to NSCs under hypoxic conditions alone. DAPI, 4′,6-diamidino-2-phenylindole; TUNEL, terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling. Color images are available online.
Effects of Zn on intracellular signaling protein levels
To investigate the effects of Zn on several intracellular signaling proteins, we performed western blot analyses to measure the expression of nestin, Ki67, p85a PI3K, pAkt (Ser473), pGSK-3β (Ser9), Bcl-2, Bax, cytosolic cytochrome C, cleaved caspase 9, and cleaved caspase 3. Following exposure to hypoxic conditions, the expression of nestin, Ki67, p85a PI3K, pAkt (Ser473), pGSK-3β (Ser9), and Bcl-2 significantly increased in NSCs treated with Zn compared to those without treatment. In contrast, Zn treatment significantly decreased the expression of Bax, cytosolic cytochrome C, cleaved caspase 9, and cleaved caspase 3 in NSCs exposed to hypoxia for 8 h (Fig. 5A–J).
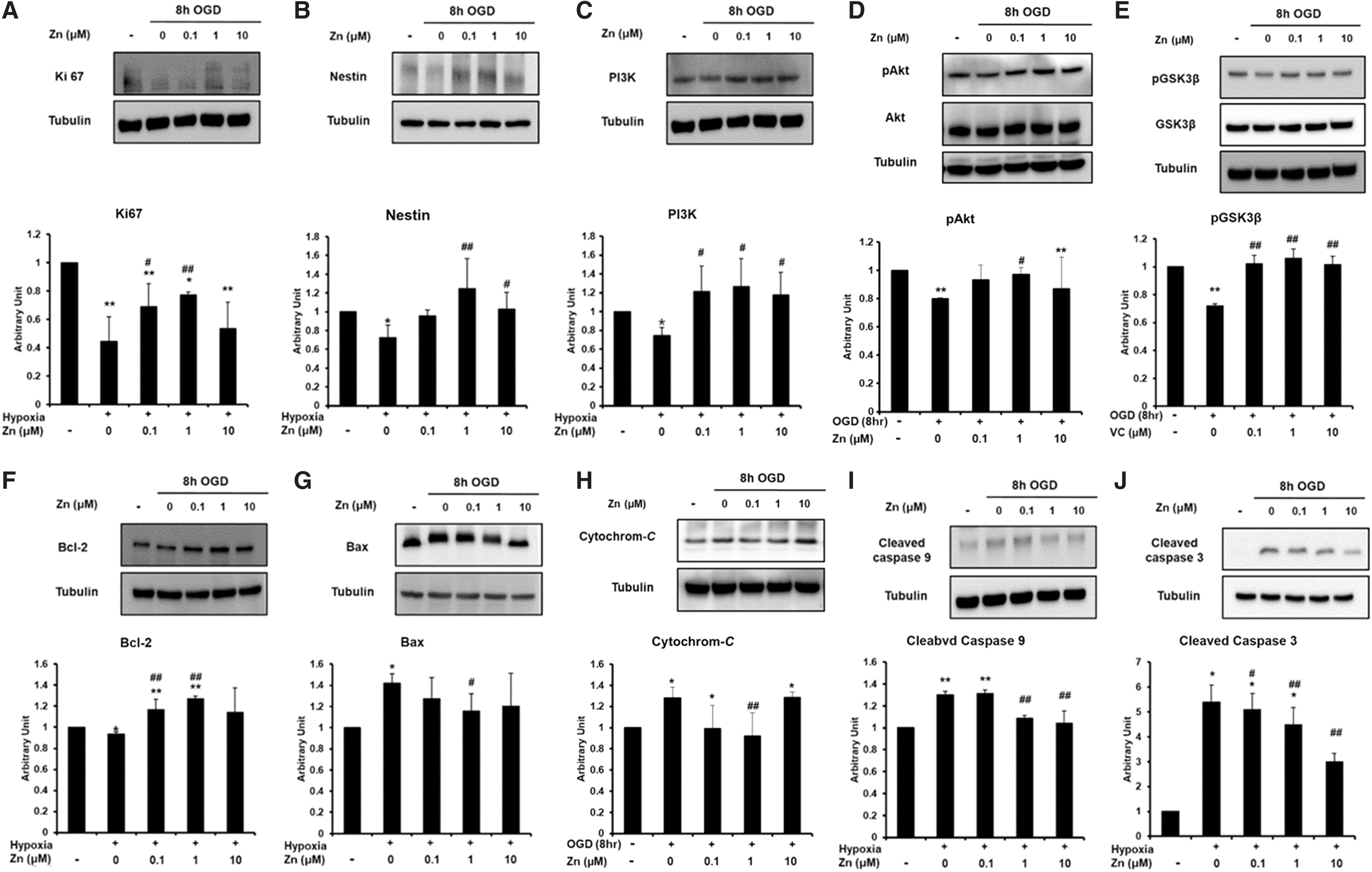
Effect of Zn on intracellular signaling protein in NSCs under hypoxic conditions. Each treatment group was compared to all other groups using a two-way ANOVA followed by Tukey's post hoc test. The immunoreactivity of nestin, Ki67, p85a PI3K, phosphorylated Akt (pAkt) (Ser473), phosphorylated GSK-3β (pGSK-3β) (Ser9), Bcl-2, Bax, cytosolic cytochrome C, cleaved caspase 9, and cleaved caspase 3 in neural stem cells was assessed by western blot analysis
RT-PCR was performed as previously described [23 –25]. Expression profiles of NSCs were compared with human embryonic kidney 293 cells. NSCs showed the expression of some typical undifferentiated stem cell marker, such as Nestin and sex-determining region Y (SRY)-box2 (SOX2) (Fig. 6A). We confirmed the effect of Zn on viability of NSCs injured by hypoxia, by RT-PCR detection of cell cycle signaling genes such as PI3K, Akt, and GSK3β. Following exposure to hypoxic conditions, the mRNA expression of PI3K, Akt, significantly increased in NSCs treated with Zn compared to those without treatment (Fig. 6B). The predicted size of each PCR product is shown (Table 1).

Effect of Zn on mRNA expression of NSCs injured by hypoxia. Expression profiles of NSCs compared with HEK 293 cells
Primer Sequences for Reverse Transcriptase Polymerase Chain Reaction
Discussion
Ischemic stroke provokes neuronal cell death by energy failure, metabolism imbalance, free radical production, and several homeostasis impairments. All of these pathogeneses induce necrotic or apoptotic cell death and inflammation, and contribute to cerebral damage after ischemia [27,28]. Neurogenesis by endogenous or transplanted NSCs could be crucial for the recovery of brain damage induced by ischemic stroke. However, NSCs can be damaged during ischemic injury and transplantation. Consistent with this, our results demonstrated that NSCs are vulnerable to hypoxia. In a previous study, we showed that the proliferation of NSCs can be inhibited by ischemic injury [29,30], and our current results confirm this. This study found that NSC damage caused by hypoxia is associated with an increase in apoptosis.
Zn is an essential factor of numerous enzymes and is important for the regulation of various cellular processes, including cell division and DNA synthesis [31]. Zn is also known to be an essential element for brain function [32 –34]. Recent studies have demonstrated that Zn is involved not only in cell division, proliferation, migration, and development but also in neurogenesis and cognitive functions [35,36]. However, excess Zn increases excitotoxic neuronal injury. Zn has an important role in oxidative and nitrosative damage in animal models [37]. It has been reported that exposure to zinc increases levels of super oxides and lipoperoxides [38]. Furthermore, abnormal mitochondrial function, the major cellular source of ROS production, might be due to Zn [39].
In contrast, several studies have demonstrated that Zn decreases oxidative injury. Zn could be associated with increasing metallothionein expression [40,41]. Metallothioneins are a group of intracellular, cysteine-rich, metal-binding proteins involved in diverse intracellular functions, including distributing Zn2+ to lipids and protecting proteins damaged by oxidative stress [40]. They are also known to be ROS scavengers. Zn could also promote Cu/Zn superoxide dismutase (SOD1) activity, an enzyme that removes ROS [42]. In this study, we found that Zn reduced cell apoptosis in NSCs under hypoxic conditions. To explain these results, we hypothesized that at low concentrations (sublethal doses), Zn has more neuroprotective than neurotoxic effects.
The cell death pathways induced by hypoxia are very complex and involve various protein families. For example, hypoxia inhibited PI3K, but activated GSK-3β in NSCs, similar to other cells [43]. Several studies have demonstrated the cardioprotective effects of Zn through the activation of PI3K/Akt signaling [44 –46]. Thus, there must be a specific condition in which the antiapoptotic effects of PI3K/Akt signaling activation are greater than the known toxicity of Zn. In this study, we found that Zn reduces cell apoptosis in NSCs under hypoxic conditions.
The PI3K/Akt pathway is a major pathway for cell survival. Activated PI3K phosphorylates its downstream target to mediate several biochemical cascades [6,47 –49]. Phosphorylated Akt directly influences GSK-3β, the Bcl-2-associated apoptosis promoter (BAD)/Bcl-2, caspase-9, the inhibitor of nuclear factor κB (IκB) kinase, and forkhead-related transcription factor 1 [6,47]. Therefore, the inactivation of GSK-3β by pAkt is important for neuronal cell survival. In this study, we demonstrate that diverse survival-related proteins, such as pAkt (Ser-473), pGSK-3β (Ser-9), and Bcl-2, increased, while several death-related proteins, such as Bax, cytosolic cytochrome C, cleaved caspase-9, and cleaved caspase-3, decreased when NSCs exposed to hypoxia were treated with Zn. We also found that the mRNA expression of PI3K, Akt, significantly increased in NSCs treated with Zn by RT-PCR. These findings suggest that Zn can increase survival-related proteins and decrease apoptosis-related proteins in NSCs; thus, Zn contributes to the protection of NSCs against hypoxia. Based on our western blot data, we hypothesized that the PI3K pathway is involved in the neuroprotective effects of Zn against hypoxia in NSCs. To confirm this, we treated NSCs with a specific PI3K inhibitor and activator 60 min before NSCs were exposed to both Zn and hypoxia and found that this was sufficient to block a majority of the protective effects of Zn against hypoxia in NSCs. This means that activation of the PI3K pathway by Zn is critical for its neuroprotective effects against hypoxia in NSCs.
There were some limitations to this study. First, because this study was performed under in vitro conditions, these results might be different from those under in vivo conditions or in clinical settings, where more complex factors are involved. Second, this study was performed in embryonic NSCs. Because oxidative injury in stroke and neurodegenerative diseases is common in elderly individuals, it may be more appropriate to perform these studies using adult NSCs; however, we could not prepare sufficient adult NSCs in this study, so we used embryonic NSCs instead. Therefore, future studies must confirm the precise effects of oxidative stress on adult NSCs.
In conclusion, this study demonstrated that sublethal doses of Zn are neuroprotective against hypoxia in NSCs and that this mechanism is associated with the restoration of proliferation, inhibition of apoptosis, and activation of the PI3K pathway. The neuroprotection of NSCs as well as neurons and glial cells is important in ischemic stroke. The observed neuroprotective effects of Zn on NSCs might provide an experimental basis for future clinical treatment of ischemic stroke.
Footnotes
Acknowledgments
This research was supported by the Basic Science Research Program of the National Research Foundation of Korea, which is funded by the Ministry of Science, ICT and Future Planning (2018R1A2A2A15023219 and 2018R1C1B5044530), by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI17C2160 and HI18C1254), and by the Medical Research Center (2017R1A5A2015395).
Author Disclosure Statement
No competing financial interests exist.