
Abstract
Osteogenic differentiation is a complex and still poorly understood biological process regulated by intrinsic cellular signals and extrinsic microenvironmental cues. Following appropriate stimuli, mesenchymal stem cells (MSCs) differentiate into osteoblasts through a tightly regulated multistep process driven by several transcription factors and characterized by the expression of a number of bone-specific proteins. In this study, we describe a novel transcription factor that we named osteoblast inducer (ObI)-1, involved in MSC differentiation toward the osteogenic lineage. ObI-1 encodes for a nuclear protein subjected to proteasomal degradation and expressed during osteoblast differentiation both in a murine multipotent mesenchymal cell line (W20-17) and in primary murine MSCs. RNA interference-mediated knockdown of ObI-1 expression significantly impairs osteoblast differentiation and matrix mineralization with reduced expression of the osteogenic markers, Runt-related transcription factor 2 (Runx2) and osteopontin. Conversely, ObI-1 overexpression enhances osteogenic differentiation and bone-specific markers expression. ObI-1 stimulates bone morphogenetic protein (BMP)-4 expression and the consequent activation of the Smad pathway; treatment with a BMP receptor type I antagonist completely abolishes ObI-1-mediated stimulation of osteogenic differentiation. Collectively, our findings suggest that ObI-1 modulates osteogenic differentiation, at least in part, through the BMP signaling pathway, increasing Runx2 activation and leading to osteoblast commitment and maturation.
Introduction
Osteogenic differentiation is a sophisticated and tightly regulated biological process. In response to specific stimuli, mesenchymal stem cells (MSCs) differentiate into osteoblasts, the “bone-forming” cells, through a multistep process characterized by four major phases: commitment toward the osteogenic lineage, osteoprogenitor cells proliferation, osteoblast maturation, and bone matrix mineralization. Induction of the differentiation process and progression through the different steps are driven by a complex network of cytokines, hormones, and growth factors that modulates expression and activity of transcription factors and other bone-related proteins [1,2].
Among growth factors involved in osteogenic regulation, bone morphogenetic proteins (BMPs) play a role of paramount relevance in inducing bone and cartilage development [3,4]. BMPs belong to the transforming growth factor-beta superfamily and signal through two types of transmembrane serine–threonine kinase receptors, type I and type II [BMP receptor type I (BMPR-I) and BMP receptor type II BMPR-II], which, upon ligand binding, activate downstream signaling pathways such as Smad1/5/8 and MAPK pathways [5]. BMPs locally synthesized accumulate in the extracellular matrix and play a critical role in both inducing commitment of MSCs toward the osteogenic lineage and enhancing the activity of mature osteoblasts [6 –8].
Bone morphogenetic proteins induce the expression of several genes, including transcription factors that control the genetic programs required for commitment and differentiation toward the osteoblastic pathway [5]. Among them, Runt-related transcription factor 2 (Runx2), although expressed also in other cell types, is considered as one of the most important transcriptional regulators of osteogenic differentiation. Indeed, Runx2 is the earliest and most specific marker of osteogenesis, and its expression is absolutely required for osteoblast differentiation; Runx2 ablation in mice results in a skeleton constituted mainly by cartilage with lack of bone due to the absence of osteoblasts [9 –11].
In addition, Runx2 haploinsufficiency in humans causes a condition known as cleidocranial dysplasia, characterized by dental anomalies and delayed skeletal development [11 –13]. Runx2 is not only necessary for MSC commitment toward the osteogenic lineage but it also regulates osteoblast maturation and functions, such as matrix formation and mineralization, inducing expression of several osteoblast-specific genes, including alkaline phosphatase (ALP), type I collagen, osteopontin (OPN), and osteocalcin [14,15]. Therefore, due to its prominent role throughout osteogenic differentiation, Runx2 is regarded as the master gene of bone formation. BMPs induce Runx2 expression and stimulate its function, and both Smad-dependent and -independent signaling pathways activated by these factors converge to Runx2 activation to modulate osteoblast differentiation [16,17].
However, despite the efforts to elucidate the molecular mechanisms underlying osteogenic differentiation, our knowledge of this complex biological process is still incomplete. Indeed, a detailed understanding of the molecular processes that drive differentiation of MSCs toward the osteogenic lineage is essential to identify new molecular targets and develop novel and more effective therapeutic strategies for the treatment of bone-related pathologies, such as osteopenia/osteoporosis, bone fractures, and osteosarcoma.
Moreover, by virtue of their potential to differentiate into several mesenchymal lineages and their favorable immune-modulatory and anti-inflammatory properties, MSCs have emerged as a promising cell source for a wide range of regenerative medicine and tissue engineering applications and are largely used in preclinical and clinical studies to treat a broad spectrum of conditions [18]. Therefore, a thorough evaluation and comprehension of the molecular mechanisms regulating MSC self-renewal and differentiation toward a specific cell lineage is of paramount importance to optimize the use and fully exploit the potential of MSCs in clinical applications.
To identify genes involved in osteoblast differentiation, we have previously performed a high-throughput screening based on an RNA-interference (RNAi) approach. The murine bone marrow-derived stromal cell line W20-17 was used as cellular model and differentiated into osteoblasts, and mineral matrix deposition was chosen as read-out [19].
The present report focuses on the characterization of a previously undescribed transcription factor involved in osteogenic regulation, identified during this screening, that we named osteoblast inducer (ObI)-1. Subsequently to our screening, Jin et al. showed that the same gene is expressed in mouse gonads and the encoded protein is localized in the nucleus [20]. We also observed nuclear localization in primary murine MSCs (mMSCs) and showed that ObI-1 expression increases during osteogenic differentiation in W20-17 cells and in mMSCs, and its knockdown impairs this biological process. In addition, we observed that ObI-1 acts, at least in part, by stimulating BMP signaling and Runx2 expression. These findings identify a novel player of the BMP–Runx2 axis regulating MSCs osteogenic differentiation.
Materials and Methods
Cell cultures
W20-17 cell line was obtained from American Type Culture Collection (Cat. No. CRL-2623/LGC standards; Milan, Italy). Primary mMSCs have been previously isolated from bone marrow of C57Bl/6 mice and characterized [21]. Human MSCs (hMSCs) were isolated from bone marrow of healthy adult donors after informed consent according to the procedure established by the local Bioethics Institutional Committee [22]. mMSCs and W20-17 cells were seeded at a density of 6,000 cells/cm2 in regular medium consisting of D-MEM (COD. ECM0749L Euroclone, Siziano, Italy) supplemented with 10% fetal bovine serum (FBS; HyClone, Northumberland, United Kingdom) and 4 mM
For adipogenic differentiation of W20-17 cells, cells were grown until they reached 80%–90% confluence and then cultured in D-MEM with 10% FBS, 4 mM
Small hairpin RNA-mediated screening
The high-throughput screening procedure used to identify novel genes potentially relevant for osteoblast differentiation has been previously described [19]. In brief, W20-17 cells were plated in 96-well plates and transfected with different small hairpin RNA (shRNA)-expressing plasmids (Open Biosystem, Huntsville, AL) using Lipofectamine 2000 (Invitrogen, Paisley, United Kingdom) according to manufacturer's instructions. Cells were then cultured in osteogenic differentiation medium for 21 days and mineral deposition was evaluated with alizarin red staining. Genes that, when silenced, impaired the ability of W20-17 cells to produce a mineralized matrix were considered potential candidate genes and further analyzed.
Alizarin red and ALP staining
For alizarin red staining, cells were washed with phosphate-buffered saline (PBS) and fixed in 10% formaldehyde (Sigma-Aldrich) for 1 h; after rinsing with distilled water, they were incubated with 2% alizarin red S (Sigma-Aldrich) at pH 4.1 for 10 min. Excess staining was removed using water. For W20-17 cell cultures, after visual examination the mineralized deposit-bound dye was extracted adding 100 μL of 4 M guanidine–hydrochloric acid (Sigma-Aldrich) and incubating them over night at room temperature. A semi-quantitative assay was used to determine the amount of cell-bound dye assessing absorbance at 490 nm of a 10-times dilution of the resulting supernatant. For ALP staining, cells were washed with PBS and fixed in 10% cold neutral formalin buffer (10% formalin, 0.1 M Na2HPO4, 0.029 M NaH2PO4 (Sigma-Aldrich). Cells were then rinsed with distilled water and stained with a substrate solution containing 0.24 mM naphthol AS MX-PO4 (Sigma-Aldrich), 0.4% N,N-dimethylformamide (Sigma-Aldrich), and 1.6 μM red violet LB salt (Sigma-Aldrich) in 0.2 M Tris-HCl pH 8.3 (Carlo Erba Reagenti, Milano, Italy) for 45 min at room temperature. Excess staining was removed washing twice with distilled water and ALP-positive cells visualized under microscope.
shRNA transfection
Murine MSCs or W20-17 cells were grown until they reached 90% confluence and then transfected with shRNAs from the same library used in the screening (Open Biosystem, Huntsville, AL) against ObI-1 gene or a scrambled shRNA [non-silencing shRNA (shNS)] as a control using Lipofectamine 2000, according to manufacturer's instructions. Two days after transfection, cells were plated in selective media containing 2 μg/mL puromycin (Sigma-Aldrich), and the medium was replaced every 3 days. A plate of untransfected cells was used as a control for the selection.
Construction of ObI-1 Flag-expressing plasmid and stable transfection
RNA was extracted from mouse lungs and retro-transcribed in cDNA as described below.
The following primers were used to amplify ObI-1 (≈2 KB): Forward (BamHI restriction site-containing) 5′-GGCCACGGGATCCAGCCATGGTGAGAAGACG-3′ and Reverse (NotI restriction site-containing) 5′-GGGCTCGCGGCCGCCAAATCCCAGATAGGGTTTG-3′. We used for PCR amplification Phusion Taq polymerase (New England Biolabs, EuroClone, Pero, Italy) with the following protocol: 98°C for 3 min, 15 cycles at 98°C for 1 min, 49°C for 20 s, and 72°C for 2 min, 5 cycles at 98°C for 1 min, 60°C for 20 s, and 72°C for 2 min, 15 cycles at 98°C for 1 min and 72°C for 2 min, and a final extension at 72°C for 10 min.
The amplified product was resolved by agarose gel electrophoresis (1% agarose with 0.5 μg/mL ethidium bromide; Sigma-Aldrich); the band was purified using Wizard SV Gel and PCR Cleanup system (Promega Italia Srl, Milano, Italy). PCR product and pcDNA3.1 vector (Invitrogen) were digested for 2 h at 37°C with BamHI and NotI (New England Biolabs, EuroClone), resolved by agarose gel electrophoresis and extracted.
Vector dephosphorilation and ligation reactions were performed using the Rapid Dephosphorilation and Ligation kit (Roche, Milano, Italy) according to manufacturer's instructions. The ligation mixture was transformed into calcium chloride competent DH5α cells (Invitrogen). Plasmids that after BamHI-NotI digestion showed that the presence of a 2 KB insert were validated by sequencing (Ceinge Sequencing Service). mMSCs were grown until they reached 90% confluence and then transfected with either ObI-1 Flag-expressing plasmid or pcDNA3.1 empty vector using Lipofectamine 2000 and, after 2 days, plated in selective media containing 400 μg/mL G418 (Gibco); the medium was replaced every 3 days. A plate of untransfected cells was used as a control for the selection.
Mice experiments
C57Bl/6 mice were obtained from Jackson Laboratory (Bar Harbor, ME). Experiments were performed in conformity with protocols approved by the veterinary department of the Italian Ministry of Health and in accordance with the ethical and safety rules and guidelines for the use of animals in biomedical research provided by the relevant Italian laws and European Union's directives (no. 86/609/EC). All efforts were made to minimize the animals' suffering. Mice were euthanized with carbon dioxide vapors; organs and tissues were immediately collected in ice and homogenized in Tri Reagent (Sigma-Aldrich).
RNA extraction and real-time PCR
Total RNA was extracted from cells or murine tissues and organs using Tri Reagent (Sigma-Aldrich) according to manufacturer's instructions. cDNA was amplified from 2 μg of RNA using M-MuLV reverse transcriptase (New England Biolabs, EuroClone). The following protocol was performed: RNA was incubated with dNTPs (Amersham Biosciences) and Random Examers (Promega Italia Srl) at 70°C for 10 min and in ice for 1 min; then, after the addition of M-MuLV and the specific buffer, the incubation was performed at 42°C for 1 h and at 90°C for 10 min. Real-time PCR was performed using the SYBR Green PCR master mixture in an ABI PRISM 7500 apparatus (Applera, Foster City, CA). Levels of target genes were quantified using specific oligonucleotide primers and normalized for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or ribosomal protein L13A (RPL13A) expression. Primer sequences are available upon request.
Immunofluorescence analysis
Murine MSCs were grown until they reached 60%–70% confluence and then transfected with either pcDNA3.1 expressing ObI-1 Flag or an empty vector using Lipofectamine 2000. After 24 h, cells were fixed in 4% paraformaldehyde (Sigma-Aldrich), permeabilized with 0.2% Triton X-100 (Sigma-Aldrich) and blocked in 10% bovine serum albumin (BSA) in PBS for 30 min at room temperature. The samples were incubated with primary anti-Flag antibody (mouse monoclonal, Cat. No. F1804, 1:250 dilution; Sigma-Aldrich) and then with an anti-mouse immunoglobulin G (IgG) conjugated with Alexa Fluor 488 (Molecular Probes, Eugene, OR; Cat. No. A-21202, 1:500 dilution). Both antibodies were diluted in 3% BSA/PBS. Cells nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) diluted 1:5,000 in PBS. Images were captured using a confocal microscope (LSM 510 Meta; Zeiss).
Bromo-2′-deoxyuridine incorporation assay
Murine MSCs were plated at 50% confluence and the following day transfected with either stealth small interfering RNAs (siRNAs) against ObI-1 or scramble negative control siRNA (20 nM, NS# 462001, 1# 291188A08, 2# 291188A10; Invitrogen) using Lipofectamine 2000 (Invitrogen) according to manufacturer's instructions. Twenty-four hours after transfection, cells were cultured in regular media or complete osteogenic medium for the next 24 h. Cell proliferation was then assessed performing a bromo-2′-deoxyuridine (BrdU) incorporation assay (BrdU labeling and detection kit I; Roche) according to manufacturer's instructions; cells were incubated in the BrdU labeling medium for 40 min at 37°C. Cells nuclei were counterstained with DAPI diluted 1:5,000 in PBS and the percentage of BrdU-positive nuclei was determined by counting 30 fields (10 × magnification).
Proteasomal inhibition
Murine MSCs, stably transfected with either pcDNA3.1 empty vector or the ObI-1 Flag-expressing plasmid, were seeded at a density of 19,000 cells/cm2 and the following day treated with either dimethyl sulfoxide (DMSO, control) or proteasome inhibitor MG 132 (Sigma-Aldrich) at 5 or 10 μM concentration. After 6 h, cells were lysed and protein collected for further analyses.
Cycloheximide chase assay
Murine MSCs were grown until they reached 70%–80% confluence and then transfected with either pcDNA3.1 expressing ObI-1 Flag or the empty vector using Lipofectamine 2000. After 24 h, cells were treated with either DMSO (control) or 10 μg/mL cycloheximide (Sigma-Aldrich) and proteins collected at different time points and subjected to western blot analysis.
BMP-4 pathway stimulation
Murine MSCs, stably transfected with either pcDNA3.1 empty vector or ObI-1 Flag-encoding plasmid, were seeded at a density of 19,000 cells/cm2 and starved overnight in D-MEM supplemented with 2% FBS to reduce cell proliferation. The following day, cells were treated with 40 ng/mL recombinant BMP-4 (Sigma-Aldrich) and proteins collected at different time points and subjected to western blot analysis. Unstimulated cells were used as control. Optimization experiments have been performed to select the BMP-4 concentration able to induce a robust and reproducible activation of the Smad pathway (Smad1/5/8 phosphorylation) in our cell system (data not shown).
BMPR-I inhibition
Murine MSCs, stably transfected with either pcDNA3.1 empty vector or, ObI-1 Flag-encoding plasmid, were grown until they reached 80%–90% confluence and then were cultured in regular medium, osteogenic medium alone, or supplemented with the BMPR-I antagonist LDN193189 (Sigma-Aldrich) at a concentration of 100 nM, chosen on the basis of previous reports [23,24]. LDN193189 was added to the culture medium every 48 h during osteogenic differentiation. After 14 days of differentiation, alizarin red staining was performed to evaluate mineral matrix deposition.
Western blot assays
Total cell lysates were obtained by treatment with lysis buffer (1 mM ethylenediaminetetraaceticacid, 50 mM Tris-HCl, pH 7.5, 70 mM NaCl, 1% Triton X-100), protease inhibitors (Complete Protease Inhibitor Cocktail; Roche), and phosphatase inhibitors sodium fluoride (50 mM) and sodium orthovanadate (1 mM; Sigma-Aldrich). Proteins were quantified with the Bradford solution according to manufacturer's instructions (AppliChem, Darmstadt, Germany).
Protein extracts were electrophoresed on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels, transferred on Immuno-Blot polyvinylidene fluoride membranes (Bio-Rad, Hercules, CA) and analyzed using the following antibodies: anti-Flag (mouse monoclonal, Cat. No. F1804, 1:1,000 dilution; Sigma-Aldrich), anti-GAPDH (mouse monoclonal, Cat. No. sc-32233, 1:1,000 dilution; Santa Cruz Biotechnology), anti-Ubiquitin (Ub, rabbit polyclonal, Cat. No. sc-9133, 1:700 dilution; Santa Cruz Biotechnology), anti-p53 (rabbit polyclonal, Cat. No. sc-6243, 1:1,000 dilution; Santa Cruz Biotechnology), anti-pSmad1/5/8 (rabbit monoclonal, 1:1,000 dilution, Cat. No. 9516; Cell Signaling, Danvers, MA), anti-Smad1 (rabbit polyclonal, Cat. No. 9743, 1:1,000 dilution; Cell Signaling), anti-HA (mouse monoclonal, Cat. No. sc-7392, 1:1,000 dilution; Santa Cruz Biotechnology), and secondary horseradish peroxidase-conjugated antibody (Amsterdam Bioscience, Uppsala, Sweden; 1:5,000 dilution). Proteins were detected by chemiluminescence (ECL or ECL plus; GE Healthcare, United Kingdom). Quantitative analysis was performed using ChemiDoc (Bio-Rad) according to manufacturer's instructions. pSmad1/5/8 levels were normalized to Smad1 levels and results are expressed as fold change of normalized pSmad1/5/8 levels in BMP4-treated cells at different time points versus untreated cells.
Immunoprecipitation
Murine MSCs, stably transfected with the ObI-1 Flag expressing vector or pcDNA3.1 empty vector, were grown until they reached 90% confluence and then transiently transfected with HA-Ub-encoding pcDNA3 plasmid (HA-Ub). HA-Ub (Addgene plasmid No. 18712) was a gift from Yeh [25]. Twenty-four hours after transfection, cells were treated with a 5 μM concentration of MG 132. Six hours after treatment, cell extracts were collected and subjected to western blot analysis. HA-Ub-untransfected mMSCs and mMSCs treated with either DMSO or MG 132 were included as controls.
Protein lysate (1.5 mg) was subjected to immunoprecipitation (IP). A preclearing step with an irrelevant antibody (mouse IgG1 isotype control, Cat. No. ab81032; Abcam) was performed to reduce nonspecific binding. Cell lysates were then incubated with 3.75 μg of anti-Flag antibody (Sigma-Aldrich) for 3 h at 4°C and with protein A/G-agarose (Santa Cruz Biotechnology; Cat. No. sc-2003) overnight at 4°C. The following day, samples were washed three times and analyzed by western blot as previously described.
Statistical analysis
Data are reported as mean ± standard deviation of three independent experiments. Two-tailed Student's t-test was performed using GraphPad Prism 5.0 software (San Diego, CA). A P-value <0.05 was considered statistically significant.
Results
ObI-1 is a putative transcription factor with increasing expression during osteoblast differentiation
To identify genes that regulate osteoblast differentiation, we have previously used single clones from a library of shRNA-expressing plasmids able to silence specific murine genes in the multipotent murine cell line W20-17, as previously published [19]. We also used the same library to successfully identify genes involved in other biological processes, such as murine embryonic stem cell self-renewal or differentiation [26,27]. With this approach, we identified 650 candidate genes that, when silenced, impair the ability of W20-17 cells to differentiate in osteoblasts and produce mineralized matrix [19].
Among the identified genes, we focused on the characterization of a previously undescribed gene (that we named ObI-1), and its role in osteogenic differentiation. ObI-1 (A430033k04Rik) belongs to the Riken family of transcripts and is predicted to encode for two short and two long isoforms originating from alternative splicing (Supplementary Fig. S1A). The shortest isoforms are predicted to be ∼100 aa in length (Supplementary Fig. S1B, C) and contain a Kruppel-associated box (KRAB) domain usually associated to transcriptional repression [28]. The long isoforms contain, in addition to the amino-terminal KRAB domain, several zinc-finger domains at the C-terminus (Supplementary Fig. S1D, E). The presence of both an effector and a DNA-binding domain suggests that ObI-1 long isoforms could play a role as transcriptional regulators.
According to the expressed sequence tag (EST) profiles on UniGene database of NCBI, ObI-1-matching ESTs are present in several murine adult tissues, including bone marrow, as well as during embryonic development (Supplementary Fig. S2).
Subsequently, we investigated ObI-1 expression during osteogenic differentiation in vitro. Real-time PCR analysis in W20-17 cells indicates that the ObI-1 long isoform (A430033K04Rik-002) transcript levels increase from the first days of differentiation and remain high during the whole process. On the contrary, both the short forms and the other long isoform (A430033K04Rik-001) were almost undetectable, indicating that they may represent only a bioinformatic prediction or they are tissue-specific isoforms not expressed in MSCs nor involved in osteogenic differentiation (Fig. 1A and data not shown). Therefore, these isoforms were not further investigated.
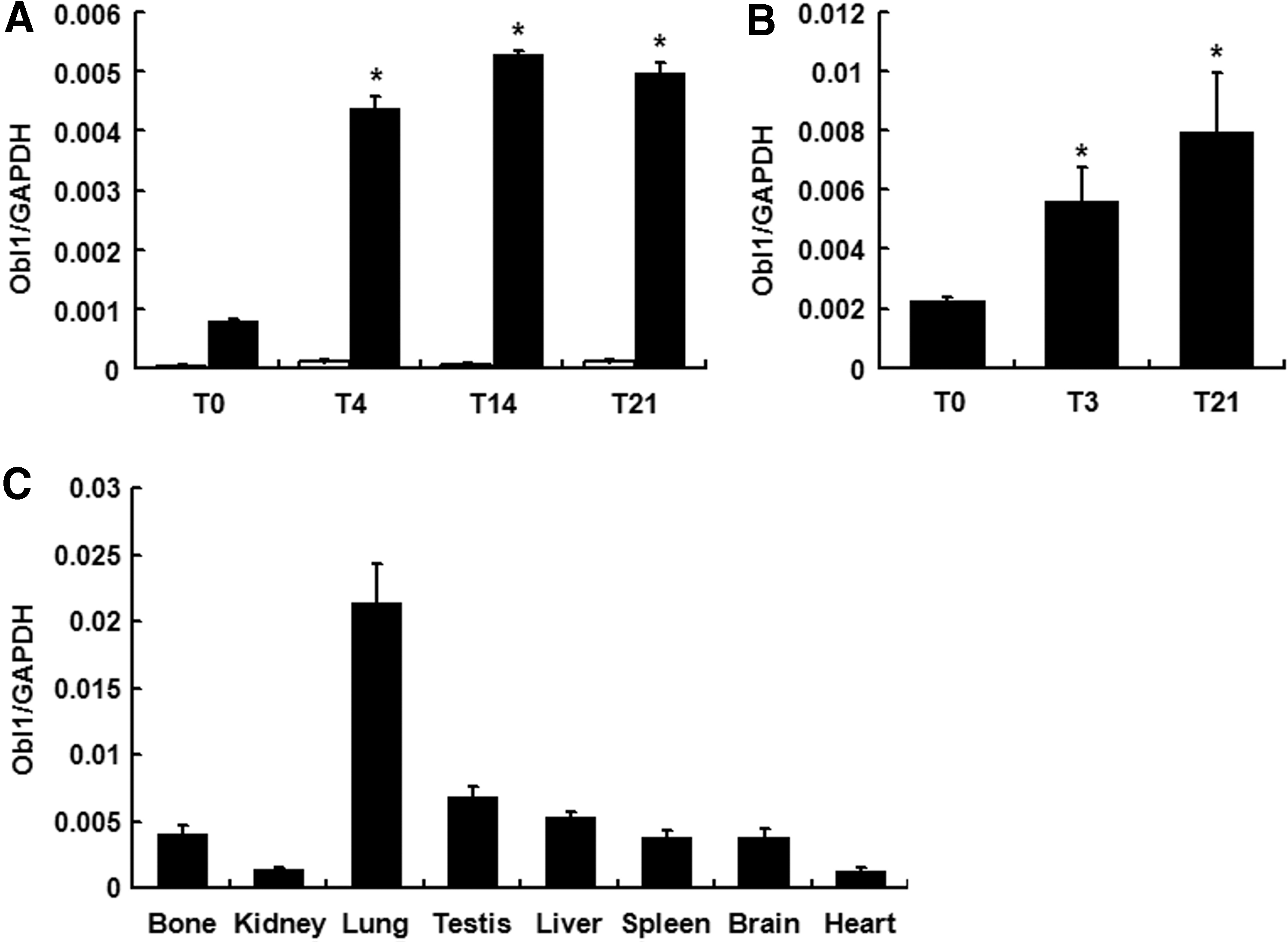
ObI-1 is expressed during osteogenic differentiation and in several murine tissues. Expression profiles of ObI-1 transcripts during osteogenic differentiation of
ObI-1 long form (A430033K04Rik-002) expression profile during osteogenic differentiation of primary mMSCs, isolated from bone marrow, is comparable to what observed in W20-17, with transcript levels significantly increasing during the first days of differentiation (Fig. 1B). This finding further supports a role for ObI-1 gene in osteogenic differentiation. Moreover, ObI-1 transcription appears to be selectively up-regulated during osteogenic differentiation of MSCs compared to other differentiation fates, as its transcript levels decrease during adipogenic differentiation of W20-17 cells (Supplementary Fig. S3).
Recently, studies have shown that some of the most popular housekeeping genes, used as internal controls to normalize the expression of genes of interest in real time (RT)-quantitative polymerase chain reaction experiments, do not have a stable expression during MSC differentiation, although it is still unclear which housekeeping gene is the most appropriate and reliable reference gene [29,30]. Therefore, we decided to analyze ObI-1 expression profile during osteogenic differentiation of mMSCs using as a reference gene the ribosomal protein RPL13A in addition to GAPDH. We chose RPL13A among other housekeeping genes since it has been previously shown to have a stable expression in bone marrow tissues and during differentiation of MSCs toward the osteogenic lineage [30,31]. Our results confirmed that ObI-1 transcript levels increase during osteogenic differentiation, regardless of the reference gene used for the normalization (Supplementary Fig. S4A). Indeed, the expression levels of ObI-1 as well as the osteogenic markers Runx2 and OPN normalized to RPL13A or GAPDH were very similar (Supplementary Fig. S4B, C).
Moreover, we observed that ObI-1 expression is not restricted to osteoblasts; Real-time PCR analysis in several murine tissues showed a broad expression pattern since ObI-1 transcript could be detected at variable levels in all the tissues examined. Surprisingly, we observed the highest levels of expression in lung (Fig. 1C). This finding suggests that ObI-1 could play a role in different biological processes in a number of tissues.
We performed a phylogenetic analysis of the murine ObI-1 gene to identify potential orthologous genes in other species using MetaPhOrs, a public repository of phylogeny-based orthology and paralogy predictions computed using resources available in seven different homology prediction services [32]. Potential ObI-1 orthologs were identified in several species, including Human (Homo sapiens), Macaque (Macaca mulatta), Orangutan (Pongo abelii), and Rat (Rattus Norvegicus). In particular, the predicted human ortholog corresponds to the zinc-finger protein-717 (ZNF717) gene; this gene, similarly to ObI-1, contains KRAB and zinc-finger domains. Interestingly, ZNF717 expression also increases during osteoblast differentiation of hMSCs from day 3 (Supplementary Fig. S5); this observation supports the predicted orthology and, therefore, suggests a possible role of ZNF717 in human osteoblastogenesis.
ObI-1 is a nuclear protein subjected to proteasomal degradation
As previously described by Jin et al. [20], we confirmed ObI-1 nuclear subcellular localization in mMSCs: immunofluorescence analysis in mMSCs transiently transfected with a vector expressing ObI-1 fused to a Flag tag (ObI-1 Flag) showed that the protein is exclusively localized into the nucleus, thereby supporting a possible role in transcriptional regulation (Fig. 2).

ObI-1 is localized in the nucleus.
We then attempted to generate mMSCs stably expressing the ObI-1 Flag protein. However, despite a robust expression of the ObI-1 transcript (Fig. 3A), we were not able to detect the corresponding protein through western blot analysis (data not shown and Fig. 3B). This result suggested that ObI-1, as most transcriptional factors, could be subjected to posttranslational modifications that regulate its stability and/or function [33].
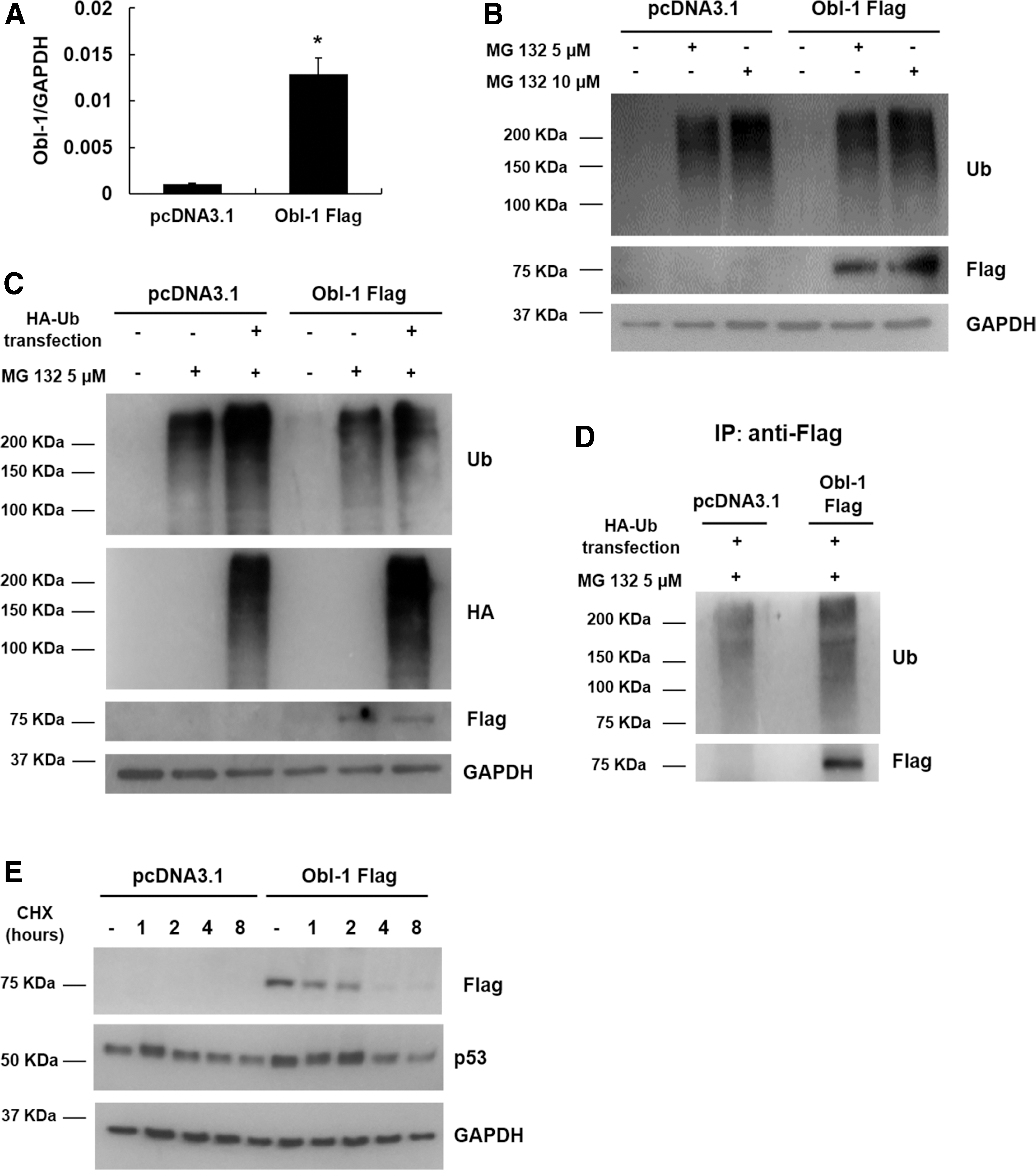
ObI-1 protein is subjected to proteasomal degradation.
To assess whether mechanisms such as Ub-mediated proteasomal degradation may be involved, we evaluated the presence of the ObI-1 Flag protein after inhibition of proteasomal function. Consistently with our hypothesis, treatment with the proteasome inhibitor MG 132 results in ObI-1 Flag protein accumulation in mMSCs (Fig. 3B). We then performed an IP analysis in mMSCs stably expressing ObI-1 Flag and transiently transfected with a vector expressing HA-Ub to increase the cellular pool of free Ub. Efficiency of both HA-Ub transfection and MG 132 proteasomal inhibition were confirmed by western blot analysis (Fig. 3C). The same cell lysates were subjected to IP with an anti-Flag antibody, and western blot analysis confirmed that the ObI-1 protein, stabilized following MG 132 treatment, is polyubiquitylated (Fig. 3D).
These results indicate that ObI-1 protein levels, and therefore its function, are finely regulated by Ub-mediated proteasomal degradation, a mechanism commonly used by eukaryotic cells to tightly control the activity of transcription factors and other chromatin-associated proteins [34].
To further confirm this finding, we then performed a cycloheximide chase experiment in mMSCs transiently transfected with ObI-1 Flag or the empty vector to evaluate ObI-1 stability when translation, and therefore de novo protein synthesis, is inhibited. Consistently, we showed that ObI-1 encodes for a highly unstable, rapidly degraded protein (Fig. 3E).
ObI-1 is necessary for osteoblast differentiation of mMSCs
Following ObI-1 identification, we confirmed the relevance of this gene for proper osteoblast maturation with additional silencing experiments, to ensure the specificity of the results and minimize the possibility of RNAi off-target effects. Therefore, we stably transfected W20-17 cells with three independent shRNAs that recognize different region of the ObI-1 transcript, and observed an almost complete absence of matrix mineralization, as assessed by alizarin red staining after 21 days of osteogenic differentiation (Fig. 4A–E). Staining quantification showed a significant reduction of mineralized deposits in ObI-1 silenced cells compared with untransfected cells and W20-17 cells transfected with shNS, used as controls (Fig. 4F). Effective reduction of ObI-1 levels was confirmed by RT-polymerase chain reaction (RT-PCR) (Fig. 4G).
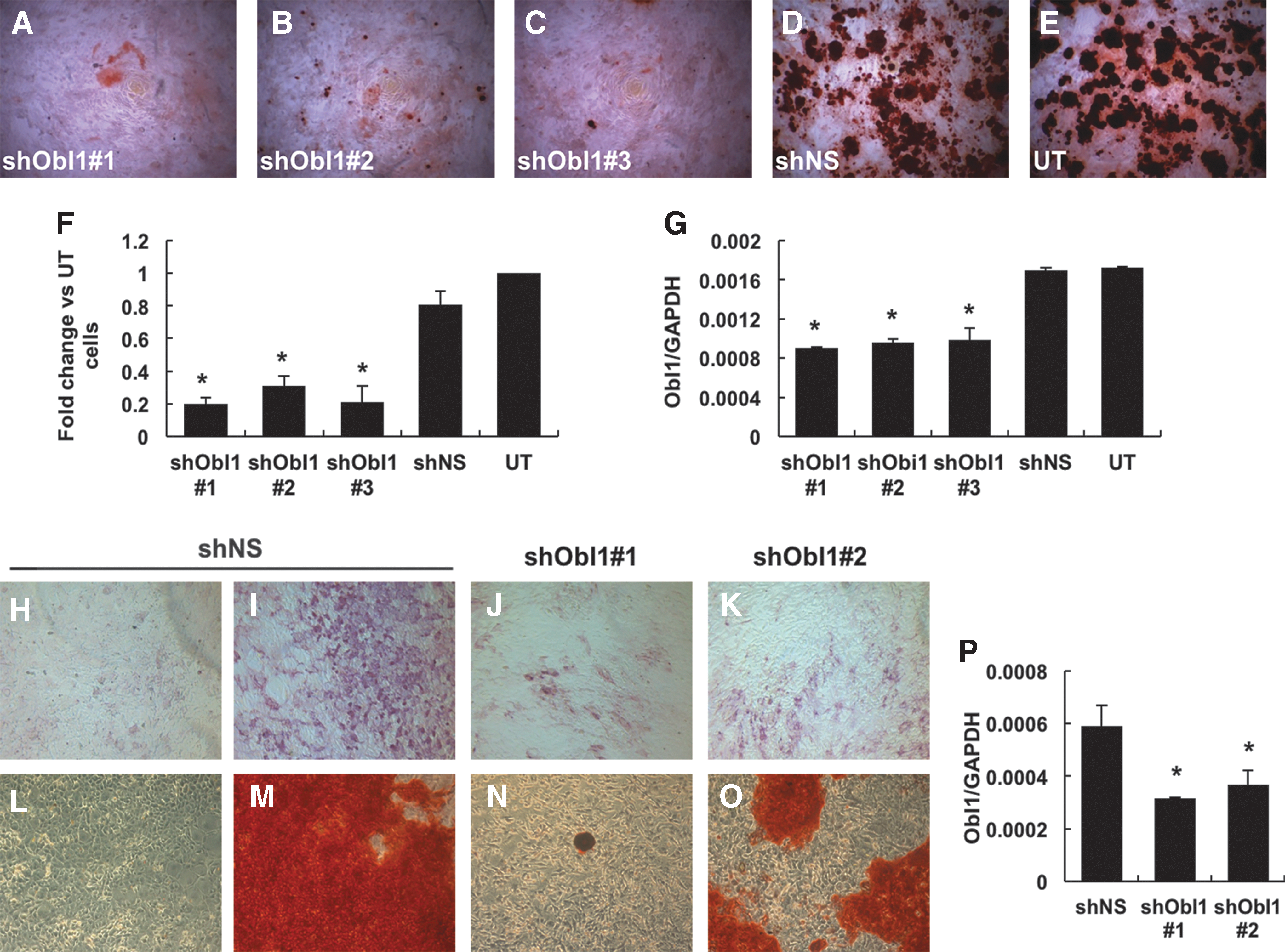
ObI-1 gene silencing impairs osteoblast differentiation. W20-17 cells transfected with three different shRNAs against ObI-1 transcript,
We evaluated ObI-1 involvement in osteogenic regulation also in primary mMSCs. As observed in W20-17 cells, ObI-1 silencing significantly impairs the ability of cells to produce mineralized nodules 18 days after osteogenic induction (Fig. 4L–O and Supplementary Fig. S6). RT-PCR confirmed silencing efficiency (Fig. 4P).
To characterize the role of ObI-1 in osteoblast differentiation, we decided to investigate the phase of the differentiation process, in which this gene is required. We observed that ObI-1 silencing results in a reduced expression of ALP, a protein expressed by mature osteoblasts that promotes extracellular matrix mineralization and considered an intermediate marker of osteogenic differentiation, after 12 days of differentiation (Fig. 4H–K) [35].
This finding indicates that ObI-1 is not involved only in the mineralization phase, but its function is required in earlier steps of osteogenic differentiation. Furthermore, this result is also in agreement with the observation that ObI-1 is expressed early during osteogenic differentiation of both W20-17 and mMSCs (Fig. 1A, B).
The hypothesis that ObI-1 could play a role during an early step of osteoblastogenesis is further corroborated by the observation that its silencing results in reduced levels of Runx2 and OPN during W20-17 cell differentiation (Fig. 5A, B). Indeed, the transcription factor Runx2 is an early marker of osteogenesis, and it is absolutely required for the commitment of MSCs toward the osteogenic lineage as well as for the function of mature osteoblasts, whereas OPN is a bone matrix sialoprotein expressed throughout osteogenic differentiation, from the first day of the process, reaching a peak during the mineralization phase [9,10,36]. As the transcript levels of both markers were significantly reduced in ObI-1 silenced cells, ObI-1 is likely to be involved in an early phase of osteogenic commitment, acting probably upstream of Runx2.
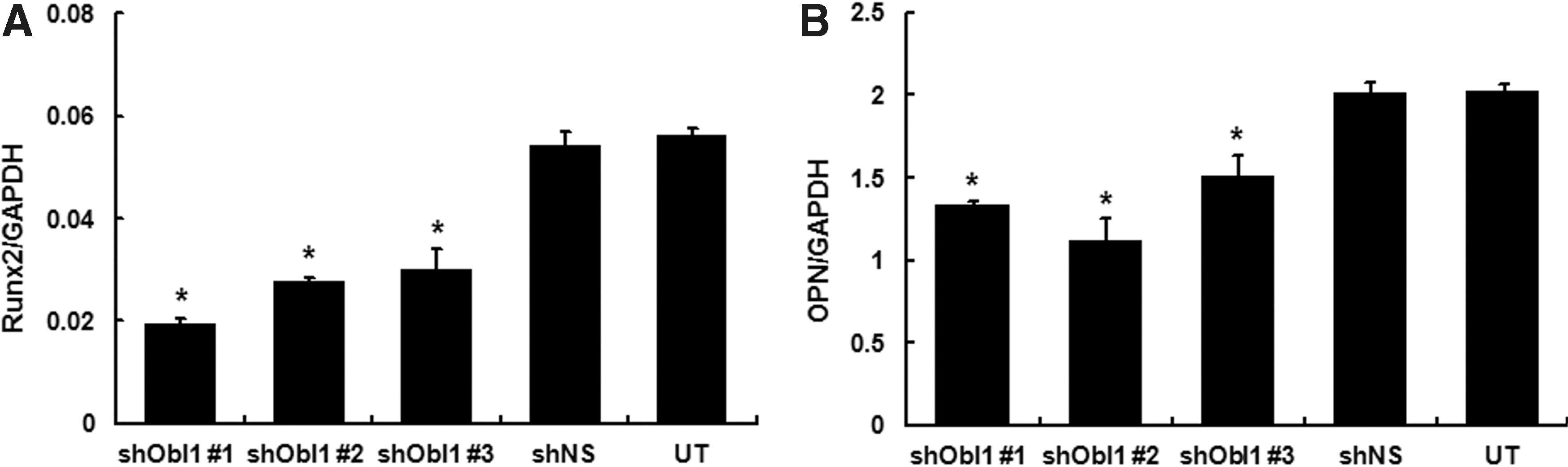
ObI-1 silencing reduces the expression of osteogenic differentiation markers. Real-time PCR analysis was performed to evaluate expression profile of early osteogenic differentiation markers in W20-17 cells and reveals a significant decrease in
We then evaluated the effects of ObI-1 silencing on osteoprogenitors proliferation, which represents the first step of osteogenic differentiation. Interestingly, ObI-1 silencing selectively impaired the proliferation of preosteoblasts, with a decreased BrdU incorporation 24 h after osteogenic induction compared with control cells, whereas the proliferation of undifferentiated MSCs was not affected (Fig. 6A, B). Silencing efficacy was confirmed by real-time PCR analysis 48 h after siRNA transfection (Fig. 6C). These findings suggest that ObI-1 specifically stimulates the proliferation of cells committed toward the osteogenic lineage, rather than being a general regulator of cell proliferation.

ObI-1 silencing selectively impairs osteoprogenitors proliferation. mMSCs were transfected with either siNS or siRNAs targeting ObI-1 (siObI1#1 or siObI-1#2).
ObI-1 expression stimulates osteoblast differentiation through a BMP-mediated mechanism
We then evaluated whether overexpression of ObI-1 could enhance osteoblast differentiation in mMSCs. At this aim, we induced the osteogenic differentiation in mMSCs stably transfected with the ObI-1 Flag encoding plasmid and observed an increased mineralized matrix deposition compared with control cells (Fig. 7A–F and Supplementary Fig. S7A). Similar results were obtained when cells were cultured in medium supplemented only with ascorbic acid 2-phosphate and glycerol 2-phosphate without the osteogenic inducer dexamethasone, further indicating that ObI-1 expression enhances mMSCs commitment and/or differentiation toward the osteogenic lineage (Fig. 7B, E, and Supplementary Fig. S7A). In agreement with this finding, ObI-1 Flag transfected mMSCs showed increased levels of Runx2 and OPN compared with controls (Supplementary Fig. S8). Therefore, it is conceivable that ObI-1 favors osteoblastogenesis by directly or indirectly stimulating Runx2 expression.
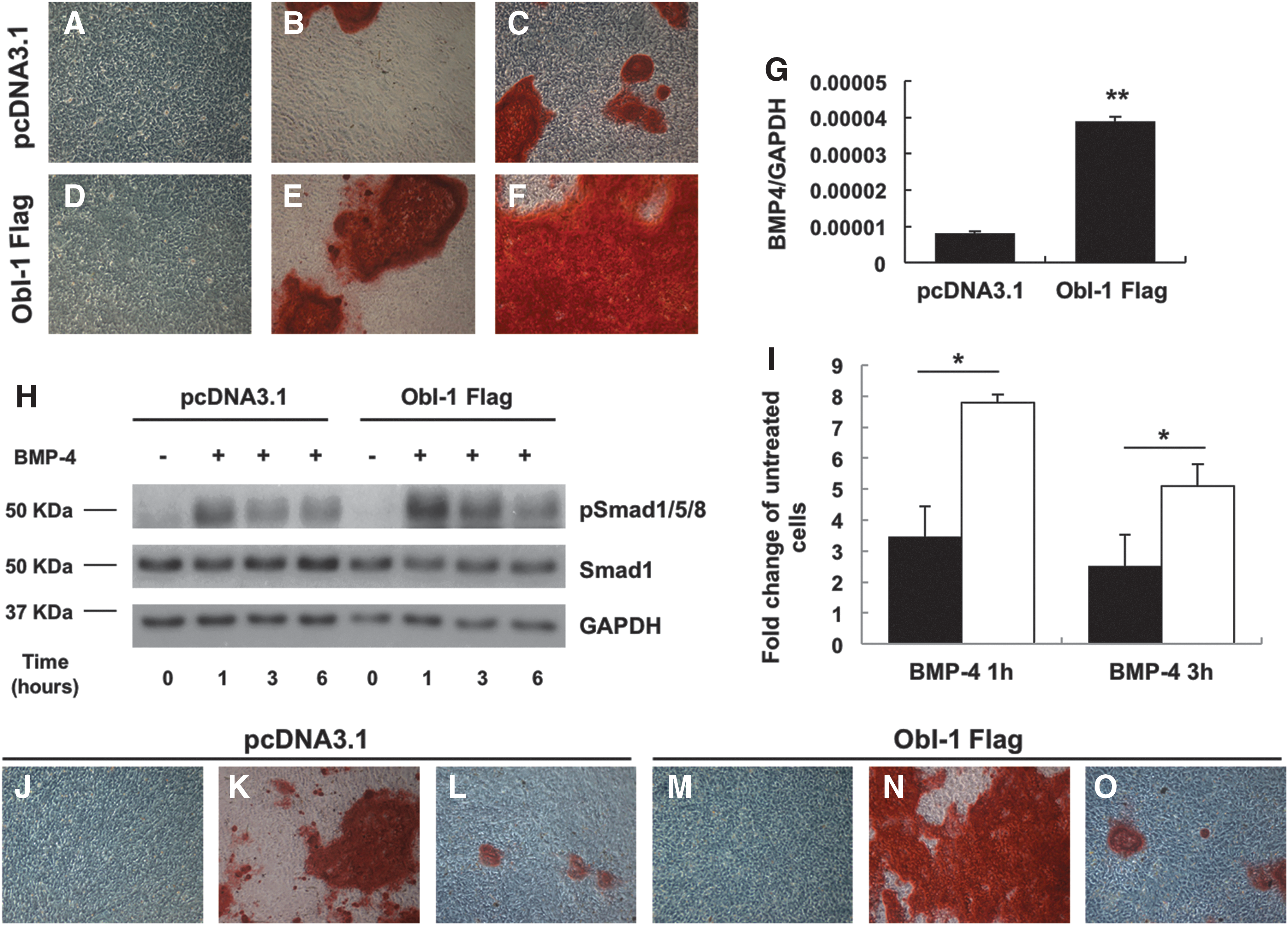
ObI-1 modulates osteoblastogenesis through a BMP-mediated pathway. ObI-1 overexpression increases osteoblast-mediated calcium deposition after 14 days of differentiation. mMSCs stably transfected with
BMPs are among the most well-studied factors known to induce osteoblasts and chondrocytes differentiation also through the increase of Runx2 levels [17,37,38]. To investigate whether ObI-1 affects the BMP pathway, we evaluated BMP-4 expression by RT-PCR and found that its transcript levels were significantly upregulated in mMSCs overexpressing ObI-1 compared with control cells (Fig. 7G). Conversely, ObI-1 silencing induced a decrease in BMP-4 levels (Supplementary Fig. S9).
We then evaluated whether ObI-1 expression influences BMPs signaling and Smad1/5/8 phosphorylation and, therefore, activation. mMSCs, stably transfected with either the ObI-1 Flag expression plasmid or a pcDNA3.1 empty vector, were treated with 40 ng/mL BMP-4 and Smad1, and phospho(p)Smad1/5/8 levels were assessed at different time points. Western blot analysis showed that ObI-1 overexpression significantly increases the activation of the Smad pathway (Fig. 7H, I). We can hypothesize that ObI-1 also acts on other proteins involved in BMPs signaling.
In addition, we observed that BMP signaling inhibition counteracts ObI-1-mediated stimulation of osteogenic differentiation. Indeed, treatment with the BMPR-I antagonist LDN193189 during osteogenic differentiation of mMSCs transfected with ObI-1 Flag completely abolished ObI-1-mediated enhancement of differentiation (Fig. 7J–O and Supplementary Fig. S7B).
Collectively, these findings suggest that ObI-1 regulates osteogenic differentiation, at least in part, by stimulating BMP-4 expression and function.
Discussion
Despite intensive investigation, our knowledge of the molecular mechanisms driving MSCs differentiation toward the osteogenic lineage is still incomplete. A better comprehension of this biological process is of crucial importance since bone-related pathologies constitute an increasing health care burden in industrialized countries and require safer and more effective therapeutic strategies [39]. MSCs represent a promising cell source for regenerative medicine applications and an excellent model to study pathways relevant in osteoblast differentiation. These cells, indeed, present several advantages over other adult stem cells: they can be extensively expanded in vitro; they are immune-privileged and able to produce trophic factors [40] and several anti-inflammatory and immunomodulatory cytokines [8,41,42]. Therefore, the identification of the molecular bases of osteogenic differentiation is mandatory to regulate and optimize the process and to fully exploit MSC therapeutic potential.
The present report focuses on the characterization of ObI-1, a novel putative transcriptional regulator of osteogenic differentiation. ObI-1 belongs to the family of KRAB/zinc-finger transcription factors, the largest family of transcriptional regulators in mammals. These transcription factors are present exclusively in tetrapod vertebrates and are involved in the regulation of several biological processes, such as cell proliferation and differentiation (including osteogenic), apoptosis, and neoplastic transformation [28,43]. The role of ObI-1 as transcription factor is also supported by its nuclear localization. Our finding is also consistent with a previous article that describes the same gene as a transcriptional repressor in mouse gonads [20].
As most regulatory proteins, ObI-1 levels and function seem to be tightly regulated by Ub-mediated proteasomal degradation. Proteasomal degradation, in addition to its role in protein turnover and degradation of misfolded products, plays an important regulatory role in fine-tuning the activity of proteins involved in a myriad of processes, from cell cycle regulation to DNA repair [44]. Since gene expression needs to be finely modulated in time and space and in response to external signals, it is not surprising that transcription factors and chromatin-modifying proteins constitute major targets for the Ub-proteasome system [34,45].
Moreover, most transcriptional factors, in particular those regulating cell growth and proliferation, are highly unstable proteins targeted by the Ub-proteasome system [46]. The proteasomal degradation of ObI-1, as well as its very short half-life, could indicate the requirement of tightly regulated levels of this protein within cells. Although we could not detect the ObI-1 protein by western blot analysis in stably transfected mMSCs, we were clearly able to appreciate the positive effects of its overexpression on osteogenic differentiation in ObI-1 transfected cells compare with the control. This could indicate that this protein is rapidly degraded after stabilization in response to a stimulus or undergoes cycles of stabilization and destabilization, hampering our ability to detect it. Further studies are required to elucidate the mechanisms regulating ObI-1 stability, the E3 ubiquitin ligases and deubiquitinating enzymes involved, as well as the specific stimuli inducing its stabilization.
Our data suggest that ObI-1 is required in the early steps of osteoblast differentiation: in fact, its silencing impairs osteoprogenitors proliferation resulting in a reduction of both ALP expressing mature osteoblasts and mineral matrix deposition. Conversely, its overexpression enhances the differentiation process.
Osteoblast differentiation requires the early expression of the transcription factor Runx2, considered the master gene of bone formation [9,10]; this gene is expressed early during mouse embryonic development in the cells prefiguring the future skeleton and is able to regulate the expression of most bone-specific proteins [14]. Runx2 protein levels are also tightly regulated at transcriptional and translational levels as well as through posttranslational modifications, including phosphorylation, ubiquitination, and acetylation [47]. We observed that ObI-1 knockdown strongly reduces Runx2 early expression, while its overexpression increases it; these data suggest that ObI-1 role in osteoblastogenesis is partly mediated by Runx2 and that ObI-1 may directly or indirectly regulate Runx2 levels.
To identify a possible mechanism of action of ObI-1, we decided to evaluate whether BMPs-mediated signaling was involved. BMPs play a crucial role in bone development; these growth factors, locally synthesized by the skeleton, play an autocrine role in osteoblast differentiation acting on a large number of pathways and cellular functions [5,37]. Among other genes, BMPs are able to induce Runx2 expression that is essential to activate osteoblast differentiation, and Runx2-Smads cooperation is believed to mediate the activity of BMPs in modulating gene transcription in cells of the osteoblastic lineage [17,38,48].
Our data suggest that ObI-1 role in osteogenic differentiation is, at least in part, mediated by the BMPs pathway. The following lines of evidence support this hypothesis: (1) endogenous ObI-1 silencing reduces BMP-4 transcript levels, whereas its over-expression increases them, respectively; (2) ObI-1 overexpression enhances BMP-4 stimulation of osteoblast differentiation increasing Smad1/5/8 phosphorylation; and (3) treatment with a BMPR-I antagonist completely abrogates ObI-1-mediated stimulation of osteogenic differentiation. BMPs act through pathways that involve several players and are subject to multiple types of regulation [37,49]; therefore, additional studies will be required to better characterized ObI-1 role and mechanism of action in modulating BMP signaling.
Despite their name, BMPs expression and function are not limited to bone. In fact, these proteins constitute a group of morphogens with pivotal roles during embryonic development and organogenesis as well as in adult tissue homeostasis [50]. These multifunctional growth factors regulate proliferation, differentiation, and function of a wide variety of cell types and are involved in processes as diversified as fracture repair, vascular remodeling, spermatogenesis, and iron metabolism [51 –54]. We observed that ObI-1 is expressed in several mouse adult tissues and organs suggesting additional roles besides osteogenic regulation. ObI-1 is expressed at high levels in lung, and the significance of this finding is currently uncharacterized. However, BMP signaling has been implicated in the regulation of lung morphogenesis, lung stem cell differentiation, adult lung homeostasis, and tissue-injury repair [55 –58]. Therefore, it is tempting to speculate that ObI-1 could modulate BMPs effects in this organ.
According to bioinformatic analysis, ZNF717 is the best candidate as ObI-1 ortholog in humans. ZNF717 is present in humans as well as in other nonhuman primates and encodes a KRAB zinc-finger transcription factor. Our preliminary evaluation is consistent with the hypothesis that ZNF717 may be the ObI-1 human ortholog; ZNF717 is expressed in hMSCs and its levels increase during osteogenic differentiation with an expression profile comparable to what observed for ObI-1 in mMSCs. Furthermore, Håkelien et al. performed the first genome-wide analysis of Runx2 target genes in human osteoblasts. Interestingly, chromatin immunoprecipitation-seq analysis revealed that ZNF717 transcription seems to be regulated by Runx2 [59]. However, further studies are required to confirm the role of ZNF717 in human osteogenic differentiation as well as to elucidate its mechanisms of action and a possible modulation of BMP signaling. Recent studies also indicate that ZNF717 is recurrently altered in gastric tumors and hepatocellular carcinomas, and may be regulated by methylation [60 –62].
In conclusion, we have identified a novel transcriptional regulator of osteogenic differentiation able to stimulate MSC commitment toward the osteogenic lineage and osteoblast maturation partly through BMP-mediated activation of Runx2. Further studies will be required to fully elucidate ObI-1 mechanism of action in osteoblast development and other biological processes.
Footnotes
Acknowledgments
We thank Daniela Sarnataro for technical help and assistance with confocal microscopy. This work was supported by the MIUR (grant no. PRIN prot. N. 2010B5B2NL_004).
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.