
Abstract
Tumors, traumata, burn injuries or surgeries can lead to critical-sized bony defects which need to be reconstructed. Mesenchymal stem cells (MSCs) have the ability to differentiate into multiple cell lineages and thus present a promising alternative for use in tissue engineering and reconstruction. However, there is an ongoing debate whether all MSCs are equivalent in their differentiation and proliferation ability. The goal of this study was to assess osteogenic and adipogenic characteristic changes of adipose-derived stem cells (ASCs) and bone marrow-derived stem cells (BMSCs) upon Myostatin inhibition with Follistatin in vitro and in vivo. We harvested ASCs from mice inguinal fat pads and BMSCs from tibiae of mice. By means of histology, real-time cell analysis, immunohistochemistry, and PCR osteogenic and adipogenic proliferation and differentiation in the presence or absence of Follistatin were analyzed. In vivo, osteogenic capacity was investigated in a tibial defect model of wild-type (WT) mice treated with mASCs and mBMSCs of Myo−/− and WT origin. In vitro, we were able to show that inhibition of Myostatin leads to markedly reduced proliferative capacity in mBMSCs and mASCs in adipogenic differentiation and reduced proliferation in osteogenic differentiation in mASCs, whereas proliferation in mBMSCs in osteogenic differentiation was increased. Adipogenic differentiation was inhibited in mASCs and mBMSCs upon Follistatin treatment, whereas osteogenic differentiation was increased in both cell lineages. In vivo, we could demonstrate increased osteoid formation in WT mice treated with mASCs and mBMSCs of Myo−/− origin and enhanced osteogenic differentiation and proliferation of mASCs of Myo−/− origin. We could demonstrate that the osteogenic potential of mASCs could be raised to a level comparable to mBMSCs upon inhibition of Myostatin. Moreover, Follistatin treatment led to inhibition of adipogenesis in both lineages.
Introduction
Tumors, trauma, burns, or surgery can lead to critical-sized defects of various body tissues, for example, bone, cartilage, muscle, or fat that need to be reconstructed. For instance, in case of bone defects, current treatment options include autologous grafts, allografts, or synthetic prosthesis. However, downsides are limited availability, donor-site morbidity, and high costs [1 –3] that leave the need to explore alternative options. Tissue engineering provides the possibility to replace damaged or lost tissue using various scaffolds and culture-expanded mesenchymal stem cells [2,4,5].
Mesenchymal stem cells (MSCs) are immature tissue precursor cells that have the ability to differentiate into multiple lineages independent of their tissue of origin, such as adipocytes, chondrocytes, and osteoblasts. They can be harvested out of bone marrow by puncture and aspiration of bone marrow from the iliac crest. However, they can also be isolated out of adipose tissue by means of minimally invasive procedures such as liposuction. MSCs can then be expanded abundantly in culture while retaining their growth and differentiation potential. Given these characteristics, mesenchymal stem cells are highly suitable for use in clinical regenerative medicine and tissue engineering.
There is an ongoing debate whether all MSCs are equivalent in their differentiation and proliferation ability or whether they have differing qualities. While some studies have shown adipose (ASC) and bone marrow-derived stem cells (BMSC) to be comparable in osteogenesis and adipogenesis [6 –9], other authors report evidence of MSCs retaining certain characteristics of their tissue of origin and superior osteogenic potential in BMSCs [10,11]. Furthermore, it has been shown that BMSCs and ASCs also differ in their immunophenotypic profiles: Cells from both lineages present a common expression profile, example, surface antigens CD29, CD49c, CD147, CD166, and HLA-abc. However, differences have been reported in the expression of CD34, PODXL, CD36, CD49f, CD106, and CD146, facilitating BMSC to differentiate into hematopoietic and vascular cells [11]. Some groups report comparable levels of adipogenesis and osteogenesis in both cell lineages, other groups report a superiority of BMSCs in their differentiative capacity [6,10,12 –14].
Myostatin (MSTN) and Activin are ligands of the TGF-β superfamily that are negative key regulators in skeletal muscle growth. They also show well-established antiosteogenic characteristics [15], leading to increased bone resorption and inhibition of osteogenic differentiation [15 –18]. Blockage of MSTN leads to an increase of muscle mass and enhances the secretion of osteogenic growth factors, for example, BMP-2 and IGF-1. In the absence or blockade of MSTN, an increase of osteogenic differentiation and bone mineralization as well as a reduced adipogenesis has been observed [15,17,19,20].
The protein Follistatin is a potent inhibitor of MSTN, rendering it inert by direct binding [15,21] and thus enhances osteogenesis and muscle growth. Follistatin also antagonizes Activin A and GDF 11 and is therefore a nonselective inhibitor [15,18,22].
The goal of this study was to assess the osteogenic and adipogenic characteristic changes of ASC and BMSC upon MSTN inhibition. We hypothesize that blocking the MSTN pathway through application of Follistatin will advance the osteogenic potential of ASC to the level of BMSC while reducing adipogenic differentiation concomitantly.
Materials and Methods
Isolation of mouse ASCs and mouse BMSCs
All animal experiments were approved by the IACUC LANUV NRW. C57BL/6J mice were obtained from Jackson Laboratory (No. 000664). C57BL/6J female littermates at the age of 12 to 14 weeks were used for all experiments. C57BL/6J-Mstnlean/J mice obtained from Jackson Laboratory (No. 009345) of the same age were used for acquisition of Myo−/− mouse ASC (mASC) and mouse BMSC (mBMSC). Mice were anesthetized using inhalation anesthesia with isoflurane and buprenorphine. mASCs were harvested from inguinal fat pads as previously described [9,23]. Inguinal fat pads were excised and washed in betadine/PBS. Thereafter, fat pads were minced under sterile conditions and digested with 0.1% Collagenase A (Roche Diagnostics, Mannheim) for 30 min at 37°C in a shaking water bath, centrifuged down, washed, and plated. Culture conditions were performed with DMEM GlutaMAX, 10% FBS, and 100 IU/mL penicillin/100 IU/mL streptomycin for in vitro experiments [24].
For isolation of mBMSCs, tibiae of mice were harvested after euthanasia (cervical dislocation). Bone marrow was washed out utilizing a 16 Gauge syringe and PBS. Accordingly, mBMSCs were processed analogously to mASCs.
Osteogenic differentiation
At passage 1 and 80% confluence, osteogenic differentiation was initiated. Osteogenic differentiation medium (additional β-Glycerophosphate 1%, ascorbic acid 0.1% modified from Neuhuber et al. [25]) was changed every third day accordingly. Cells were incubated at 37°C with 5% CO2.
Adipogenic differentiation
At passage 1 and 80% confluence, adipogenic differentiation was initiated. Adipogenic differentiation medium (mASC: additional Insulin 320 nM, Dexamethasone 1,000 nM; mBMSC: additional Insulin 5,000 nM, Dexamethasone 100 nM [26]) was changed every third day with glucose concentration accordingly. Cells were incubated at 37°C with 5% CO2.
Tibial defect model
Female littermates at age 12 to 14 weeks were used for all surgical experiments. Surgical procedures were performed under inhalation anesthesia with isoflurane and buprenorphine. An established murine tibial defect model was performed as previously described [9]. Briefly, after shaving and disinfecting the left leg, an incision was made on the proximal anterior skin surface over the tibia. After splitting the anterior tibial muscle, the tibia was properly exposed. A unicortical defect was created on the anterior tibial surface using a 1-mm drill bit. The five animal groups included (1) wild-type (WT) animals treated with DMEM (control), (2) WT animals treated with 1.5 × 105 mASCsWT, (3) WT animals treated with 1.5 × 105 mASCsMyo−/Myo−, (4) WT animals treated with 1.5 × 105 mBMSCsWT, and (5) WT animals treated with 1.5 × 105 mBMSCsMyo−/Myo−. Lactic acid films were used as protective cover over the inserted medium, modified as previously described [27]. Briefly, cells were collected in 1 μL DMEM and inserted into the defect through a microsyringe.
To allow tracking of mASC and mBMSC in vivo, cells were labeled with a turboGFP lentiviral construct (GE Healthcare Life Sciences, Piscataway, NJ,
Wound closure was performed with 6-0 Prolene interrupted sutures. The anterior tibial muscle was reset into its anatomical position. Euthanasia was performed after 3 and 7 days according to National and International Laws and Guidelines. Briefly, cervical dislocation was performed after thorough anesthesia to harvest tissue.
Follistatin treatment
In all experiments with Follistatin application, Follistatin protein (concentration: 2 μg/mL) (PeproTech Germany, Hamburg, Germany) was diluted into the medium.
Histological stainings
Alizarin Red staining (28 days after osteogenic differentiation) was performed as previously described [15]. Oil Red O staining was performed according to published standardized methods after 2 weeks of adipogenic differentiation [28].
Quantification of bone formation
Every sixth section was used to characterize bone formation with aniline blue staining as previously described [16,27]. Images were taken with a brightfield microscope (Zeiss Axiovert 100) and the following settings with AxioVision 4.8: objective × 2.5, exposure time 614 ms, dimensions 3,900 × 3,090 pixels (Px), scanned color. Histomorphometric measurements of Aniline Blue-stained sections were performed in Adobe Photoshop (Adobe, San Jose, CA) with modifications [16,29]. Briefly, a 2,000- × 2,000-Px-dimensioned selection box was placed to cover the entire defect area. By using the Adobe Magic Wand Tool (settings: tolerance 60%; noncontiguous), new osteoid formation was selected semiautomatically. Tonal separation in two steps and deselecting existing cortical bone resulted in highlighted pixels reliably corresponding to bone formation area.
Real-time cell analysis
For real-time cell viability analysis, mBMSC and mASC were seeded in two 8-well plates with an integrated microelectronic sensor array (iCELLigence Real-Time cell analyzer; CEA Biosciences, San Diego). Cultivated cells were incubated with either Follistatin (2 μg/mL) or PBS and osteogenic and adipogenic differentiation medium or proliferation medium. Cell proliferation and survival were monitored real-time by measuring cell-to-electrode responses of the seeded cells. The cell index (CI) was calculated for each E-plate well using RTCA software 1.2 (Roche Diagnostics, Meylan, France), as previously described [20]. CI was calculated for each E-plate well using RTCA Software. Graphs are real-time generated outputs from the iCELLigence system.
Quantitative determination was achieved by choosing a region of interest of 2,000 × 2,000 Px and automatically selecting immunohistochemically positive stained pixels using Adobe Magic Wand Tool (settings: tolerance 60%; noncontiguous). The number of nuclei in the selected area of interest was counted manually and the number of positively stained pixels was given in 1 × 103 pixels and put in relation to the number of nuclei in the selected area.
Immunohistochemistry
For immunofluorescence staining of RUNX-2, a master gene of osteogenesis and a marker for osteoblast differentiation (mouse, monoclonal, sc-101145, 1:50, Runx2 (27-K); Santa Cruz Biotechnology), Osteocalcin, a marker for late osteogenic differentiation (mouse, monoclonal, sc-365797, 1:50, Osteocalcin (G-5); Santa Cruz Biotechnology), PCNA, a marker for proliferation (mouse, monoclonal, sc-25280, 1:50, PCNA (F-2); Santa Cruz Biotechnology), MSTN (Myostatin) (mouse, monoclonal, sc-393335, 1:50, GDF8/11; Santa Cruz Biotechnology), and PPARγ, a marker for adipogenic differentiation (rabbit, 2435S, 1:800, PPARγ; Cell Signaling) were carried out. Incubation with primary monoclonal IgG κ (Kappa light chain) antibodies was performed overnight in blocking solution at 4°C. After washing with PBS, a Maus IgG kappa-binding protein conjugated to CruzFluor™ 488 (CFL 488; Santa Cruz Biotechnology) or CruzFluor 680 (CFL 680; Santa Cruz Biotechnology) was used for detection. All cells have been counterstained with DAPI. Sections were subsequently mounted with Fluoromount Aqueous Mounting Medium (Sigma-Aldrich). Images for immunofluorescence were taken with a fluorescence microscope (Olympus IX83). Measurement of stained sections were performed as described before [15]. Briefly, analogous to quantification of bone formation, a region of interest was selected in four slides per animal (2,000 × 2,000 Px). Immunohistochemically positive stained pixels were automatically selected by using the Adobe Magic Wand Tool (settings: tolerance 60%; noncontiguous) and divided by DAPI-positive nuclei.
RNA preparation and cDNA synthesis
The homogenization of harvested cells was achieved with 1 mL TRIzol reagent (Life Technologies, Darmstadt, Germany) on ice. Tissue was processed as described before [30]. Subsequently, RNA purification was performed with the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Synthesis of cDNA was performed by means of the High-Capacity cDNA Reverse Transcription Kit with RNase inhibitor (Life Technologies) using 200 ng total RNA per reaction.
Quantitative real-time PCR
Quantitative determination of relative gene expression was performed on Applied Biosystems 7900HT Fast Real-Time PCR System (96-well plates) using TaqMan® gene expression assays (Acvr2b, Mm00475713_m1; Alp, Mm00475834_m1; Bmp-2, Hs00154192_m1; Follistatin, Mm00514982_m1; Igf-1, Mm00439560_m1; Mstn, Hs00976237_m1; NfκB1, Hs00765730_m1; Pcna, Mm00448100_g1; Pparγ, Mm00440940_m1; Runx2, Mm00501584_m1) and TaqMan Universal Master Mix (Applied Biosystems, Darmstadt, Germany). For each reaction, 2 ng cDNA was used. Data were analyzed according to the manufacturer's ΔΔCt method (Applied Biosystems). 18 S was used as a reference gene.
Statistical analysis
Results of the study are presented as mean ± standard error of the mean (SEM) of at least three independent experiments. P values were calculated by student's t-test comparing two groups and ANOVA if comparing more than two groups. Statistical significances were set at a P value <0.05.
Results
Follistatin enhances proliferation of mBMSCs in osteogenic differentiation, whereas reducing proliferation of mBMSCs and mASCs in adipogenic differentiation
To investigate the effect of Follistatin on the proliferative capacity of mASCs and mBMSCs during osteogenic and adipogenic differentiation, we performed a scratch assay with a defined gap of 500 μm. After 24 h of cell settlement and differentiation, the two-well silicone insert with a defined cell-free gap was removed. Pictures were taken after another 24 h in the adipogenic or osteogenic differentiation medium.
Upon adipogenic differentiation, mBMSC showed a decrease of the gap size of 29% in the control group, whereas Follistatin treatment led to an increase of 2% (P < 0.001). In mASCs, we were able to show a decrease of the gap size of 92%, whereas a decrease of 39% was observed in the Follistatin treatment group (P < 0.001). Taken together, adipogenic differentiation led to an increase of proliferation in both mASCs and mBMSCs, whereas treatment with Follistatin reduced the proliferative capacity significantly.
During osteogenic differentiation, mBMSC showed a 21% gap size increase, whereas treatment with Follistatin lead to a decrease of 12% (P < 0.01). During osteogenic differentiation of mASCs a gap size decrease of 99% was observed in the control group, whereas Follistatin treatment showed a decrease of 69% (P < 0.01). Thus, Follistatin led to increased proliferation of osteogenic differentiating mBMSC, whereas proliferation of mASCs was reduced (Fig. 1).

Scratch Assay (500 μm) of mASCs and mBMSCs during adipogenic and osteogenic differentiation. During adipogenic differentiation, both mASCs and mBMSCs exhibited a decreased proliferative capacity in Follistatin-treated cells (left column). Osteogenic differentiation led to an increase of proliferative capacity in mBMSCs with Follistatin treatment but not in smASCs (right column). Results are shown as means ± SEM of change compared with the initial gap size. Scale bar: 100 μm. P value: ** <0.01; *** <0.001; (two sample t-test). mASCs, mouse adipose-derived stem cells; mBMSCs, mouse bone marrow-derived stem cells; mASCs, mouse adipose-derived stem cells; SEM, standard error of the mean.
Follistatin enhances proliferation of mBMSCs, whereas reducing proliferation of mASCs in osteogenic differentiation
Next, we wanted to take a closer look on proliferation of both mASCs and mBMSCs during osteogenic differentiation with or without Follistatin treatment. iCELLigence Real-Time Cell Analyzer allows observation of cell proliferation in real time. Cells were seeded with proliferation and differentiation medium (with or without Follistatin).
We observed a 1.4-fold increase of proliferating mBMSCs (P < 0.01) and a 1.2-fold increase of differentiating mBMSCs upon treatment with Follistatin in osteogenic proliferation/differentiation medium.
mASCs revealed a reduction of proliferation upon Follistatin application in both differentiation [8% (not significant)] and proliferation media [22% (P < 0.05)] (Fig. 2).
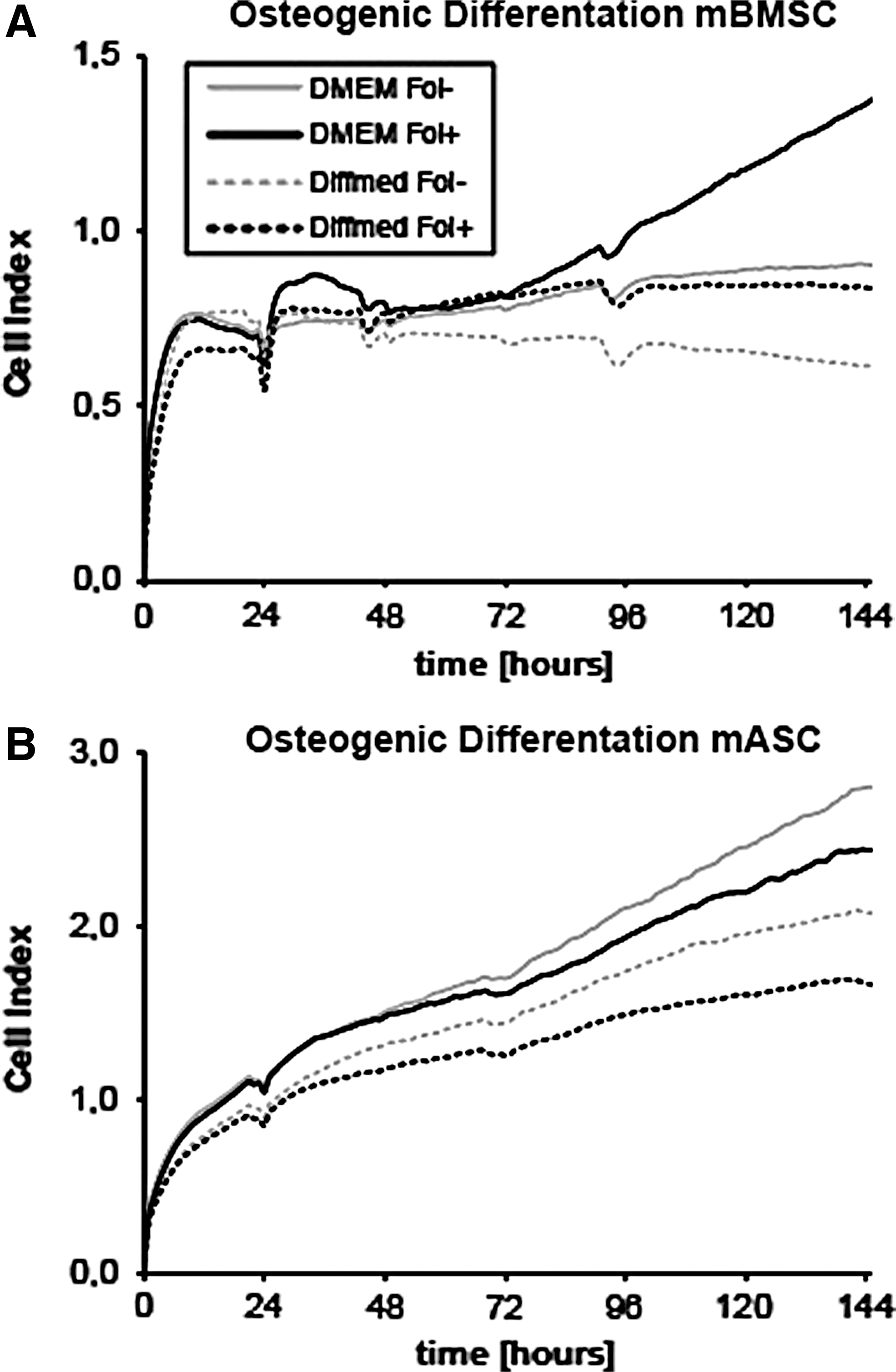
Real-time analysis of proliferation during osteogenic differentiation of
Follistatin enhances the osteogenic potential of mASCs and mBMSCs
To identify underlying mechanisms of Follistatin interplay in osteogenic differentiation, we measured expression levels of key genes for proliferation, osteogenic differentiation, osteoblastogenesis, osteoclastogenesis, and Myostatin (24 h after initiating differentiation). We showed no alteration of Mstn expression in neither mASCs nor mBMSCs with Follistatin treatment compared with control. Pcna expression as indicator for proliferation was significantly increased in Follistatin-treated mBMSCs (2.4-fold, P < 0.001) but not mASCs compared with control medium. Runx-2 was significantly increased in both mASCs (1.48-fold, P < 0.001) and mBMSCs (2.3-fold, P < 0.001) upon Follistatin treatment compared with control. Fstn (Follistatin) expression was downregulated in mBMSCs with Follistatin treatment (reduction of 61%, P < 0.01) but not mASCs compared with control medium. Nf-κB as a key gene in regulating osteoclastogenesis was less expressed in mASCs (reduction of 54%, P < 0.05) and mBMSCs (reduction of 78%, P < 0.001) upon Follistatin administration. Alp expression was increased in both mASCs (2.9-fold, P < 0.05) and mBMSC (2.73-fold, P < 0.01) with Follistatin added to the differentiation medium. Igf-1 expression was significantly increased in mASC (2.8-fold, P < 0.001) and mBMSC (1.8-fold, P < 0.01) treated with Follistatin compared with the control group. Bmp-2 expression was significantly increased in mASC (1.6-fold, P < 0.01) and mBMSC (1.9-fold, P < 0.001) treated with Follistatin in comparison to the control group. Expression of Activin receptor 2b (Acvr2b) was not significantly deviated upon treatment with Follistatin (Fig. 3A).
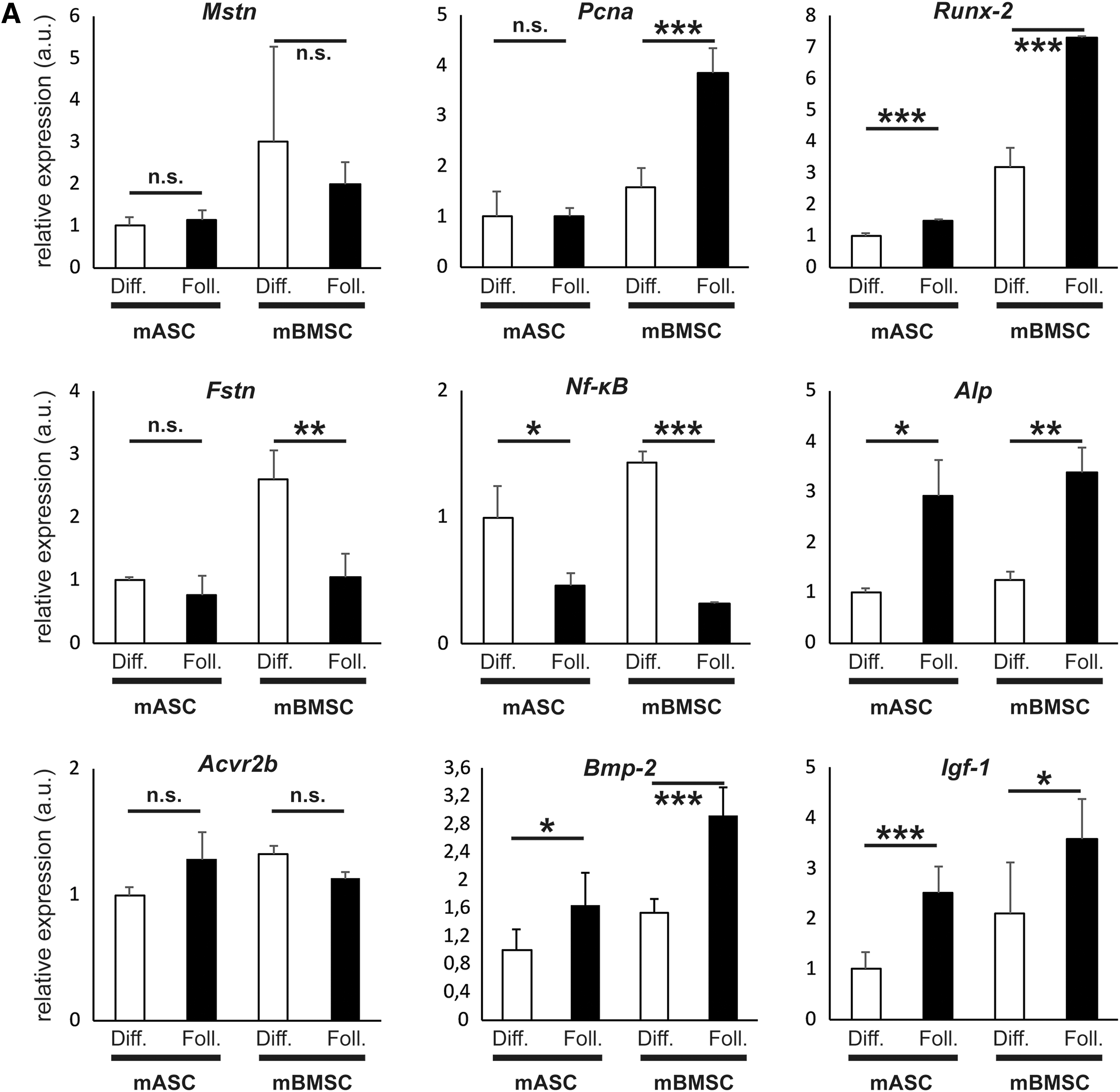
(Continued).
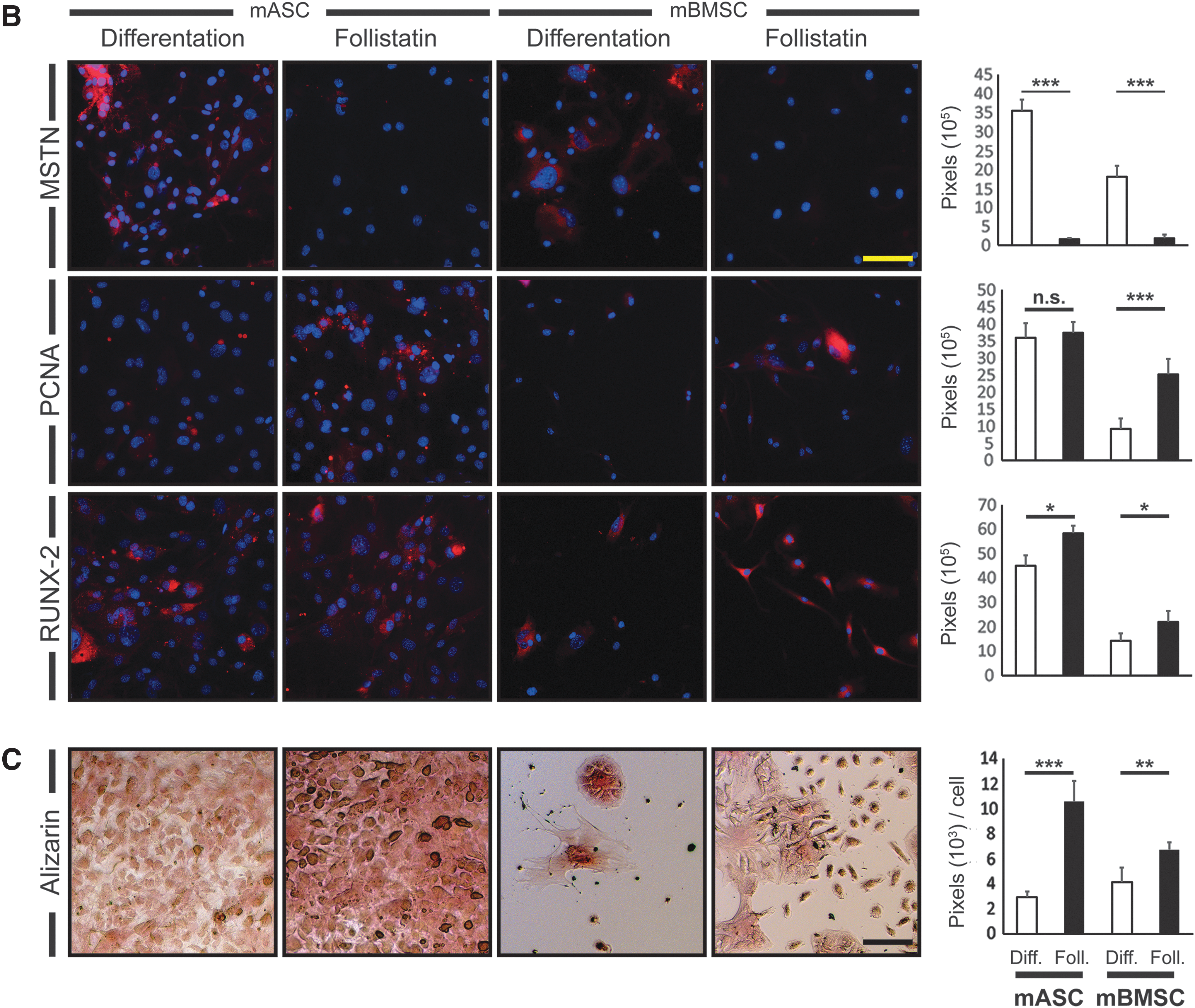
Proliferation and osteogenic differentiation was further evaluated through immunofluorescence. We observed a massive reduction of MSTN signal in Follistatin-treated osteogenic differentiating mASCs and mBMSCs. PCNA was not affected by Follistatin administration in mASCs but significantly increased in mBMSCs (P < 0.001). RUNX-2 was significantly increased in mASCs (P < 0.05) and mBMSCs (P < 0.05) treated with Follistatin (Fig. 3B).
mASCs (3.6-fold, P < 0.001) and mBMSCs (1.6-fold, P < 0.01) showed enhanced extracellular calcification upon Follistatin administration during osteogenic differentiation for 28 days (Fig. 3C).
Follistatin inhibits adipogenic differentiation of mASCs toward osteogenesis
We further investigated the impact of Follistatin on adipogenic differentiation. Therefore, we performed real time quantitative PCR (qRT-PCR) to measure expression of Mstn, Pcna, Runx-2, and Pparγ during adipogenic differentiation with and without administration of Follistatin. We showed no difference in Mstn expression during adipogenic differentiation between cells treated with Follistatin and cells in the control group. Pcna expression was reduced in mASCs treated with Follistatin (reduction of 38%, P < 0.05) but not mBMSCs. Runx-2 expression was enhanced during adipogenic differentiation in both mASCs (2.8-fold, P < 0.01) and mBMSCs (1.8-fold, P < 0.05) treated with Follistatin. Expression of Pparγ as master gene for adipocytic differentiation was significantly reduced in mASCs (reduction of 91%, P < 0.001) and mBMSCs (reduction of 72%, P < 0.001). Igf-1 expression was significantly increased in mASC (1.9-fold; P < 0.01) and mBMSC (3.1-fold; P < 0.001) treated with Follistatin compared with the control group. Bmp-2 expression was significantly increased in mASC (3.1-fold; P < 0.05) but not mBMSC treated with Follistatin compared with the control group. Expression of Activin receptor 2b (Acvr2b) was significantly increased in mASC (1.4-fold; P < 0.01) upon treatment with Follistatin compared with the control group (Fig. 4A).

qRT-PCR
Through immunohistological reactions for detection of MSTN, RUNX-2, and PPARγ, we were able to show a significantly reduced signal for MSTN (427-fold reduction, P < 0.001) and PPARγ (7,055-fold reduction, P < 0.001) upon application of Follistatin compared with control in both mASCs and mBMSCs. The signal for RUNX-2 was significantly increased in the Follistatin group (221-fold, P < 0.001) (Fig. 4B).
For measuring lipid accumulation during adipogenic differentiation, Oil Red O staining was performed after 14 days of differentiation. After Follistatin administration, mASCs showed a dramatic reduction of the amount of lipids by 99% (P < 0.001) compared with the untreated group. In mBMSCs, lipid accumulation after adipogenic differentiation was less pronounced than in mASCs and it was completely inhibited after administration of Follistatin (Fig. 4C).
Myo−/− leads to increased osteoid formation in mASC and mBMSC and osteogenic differentiation and proliferation in mASC in vivo
To investigate the effects of Myostatin blockade observed in vitro, we transplanted mBMSC and mASC of WT and Myo−/− origin into murine WT unicortical tibial defects. We were able to demonstrate increased osteoid formation through Aniline Blue staining in both mASCMyo −/− (2.6-fold; P < 0.01) and mBMSCMyo −/− (1.5-fold; P < 0.05) compared with cells of WT origin in tibial defects 7 days postoperatively (Fig. 5A). Immunohistochemistry against Osteocalcin showed increased mineralization in mASCMyo −/− (1.5-fold; P < 0.01) compared with cells of WT origin in tibial defects 7 days postoperatively, whereas there was no significant increase in mBMSCMyo −/− or WT origin (Fig. 5B). Through immunohistochemistry for MSTN, PCNA, and RUNX-2, we were able to reproduce a reduction of MSTN in tibial defects transplanted with mASCMyo −/− (53-fold reduction; P < 0.01) and mBMSCMyo −/− (15-fold reduction; P < 0.01 compared with cells of WT origin 3 days postoperatively. PCNA signal was increased in defects transplanted with mASCMyo −/− (2.1-fold; P < 0.05) compared with cells of WT origin 3 days postoperatively. RUNX-2 signal was increased in defects transplanted with mASCMyo −/− (3.3-fold; P < 0.05) compared with cells of WT origin 3 days postoperatively (Fig. 5C). GFP labeling of the injected cells demonstrates the involvement of the transplanted cells in the formation of new bone in the defect area (Fig. 5C).

Aniline Blue Staining
Discussion
The aim of this study was to investigate osteogenic and adipogenic effects of Myostatin blockade on mouse ASCs and mouse BMSCs in vitro and in vivo. We were able to show that inhibition of Myostatin leads to generally reduced proliferative capacity in mBMSCs and mASCs in adipogenic differentiation and reduced proliferation in osteogenic differentiation in mASCs, whereas proliferation of mBMSCs was increased upon osteogenic differentiation. Osteogenic differentiation was increased in both cell lineages upon Myostatin blockade.
In adipogenic differentiation, we could show that Follistatin treatment led to inhibition of adipogenic differentiation and a general enhancement of osteogenesis in both cell lineages.
We were able to further emphasize these results in an in vivo model. In WT mice with a tibial defect treated with mASCMyo −/− and mBMSCMyo −/− and of WT origin, we could demonstrate increased osteoid formation in mASCMyo −/− and mBMSCMyo −/−, as well as increased osteogenic differentiation, mineralization, and proliferation in mASCMyo −/− compared with control. Furthermore, we observed that mBMSCs are much more fragile to transplant which might explain the lack of changes in mineralization and expression levels of Runx-2 in defects treated with mBMSCMyo −/−.
In earlier studies, it has been shown that Follistatin blocks the Activin Type II receptor and thus inhibits Activin A, Myostatin, and GDF11, and is also a partial antagonist of BMP. However, it also directly binds Myostatin rendering it into an inactive form. Moreover, in a previous study, we could demonstrate that treatment with either Follistatin or Myostatin neutralizing antibody showed significantly enhanced osteogenic regeneration in osteogenic differentiation in vitro, whereas selective neutralization of Activin A showed less osteogenic effects [15]. Moreover, we were able to show a significant decrease of Myostatin and its downstream targets p38 and SMAD2/3 upon application of Follistatin in vivo and thus demonstrating a sufficient blockade of the Myostatin signaling pathway. Therefore, Follistatin is not very selective but it has been proven to be a very potent inhibitor of Myostatin [21,31 –33]. However, Activin A and GDF11 also play an important role in bone regeneration and remodeling and further studies are needed to differentiate the exact role of Follistatin.
Previously, it could be shown that Myostatin knockout mice exhibited an enlarged fracture callus and an increased total bone area and callus strength postosteotomy as opposed to wild-type mice [16,17,19]. Upon application of Follistatin, enhanced expression of ALP, RUNX-2, and PCNA, as well as increased calcification and a higher trabecular area and trabecular bone mineral content has likewise been shown [15,16,19]. Furthermore, there is no significant difference in Acvr2b receptor expression levels upon Follistatin treatment compared with control, hence there is no compensatory upregulation upon blockade of the Acvr2b receptor through Follistatin. We only see an upregulation in Acvr2b-expression in mASC during adipogenesis under Follistatin treatment. This could be due to a high blockade concentration of Follistatin to the receptor and wasting of the receptor [31,32]. Thus, in accordance with our findings, inhibition of Myostatin pathways does not only lead to increased muscle mass but also enhances osteogenic differentiation and bone repair.
In adipogenic differentiation, Myostatin leads to an upregulation of adipogenic factors, such as C/EBPα and adiponectin, and an increase of lipid accumulation as seen in Oil Red O-stained adipocytes. Respectively, blockade of Myostatin induces a downregulation of adipogenesis [34]. These findings correspond to the results of our study. As we were able to show an increased expression of Runx-2, Igf-1, BMP-2, and ALP in mASCs in adipogenic differentiation after application of Follistatin, we conclude that myostatin inhibition in adipogenic differentiation not only suppresses adipogenesis but alters adipogenic differentiation toward osteogenesis.
There is an ongoing debate whether all MSCs are equivalent in their proliferative and differentiative capacities. In contrast to our findings, some studies suggested similar or superior proliferative and differentiative potential in osteogenesis and adipogenesis in human [6] and animal [7,8] ASCs and BMSCs. However, most studies agree on a superiority of osteogenic potential in BMSCs compared with ASCs based on AP activity, matrix calcification, and gene expression in human cells [9,10,12 –14,35,36]. These findings have been confirmed in in vitro studies with animal cells [37 –39]. Pachón-Peña et al. [11] demonstrated a superiority of ASCs in adipogenesis, however, reported no differences in osteogenesis in human ASCs and BMSCs.
Although in most studies, a superiority of osteogenic potential has been shown for BMSCs, there are other factors to consider in choosing the optimal stem cell source for clinical application: While BMSCs are harvested through aspiration from the iliac crest, which can be quite painful and frequently requires spinal or local anesthesia, ASCs can easily be harvested through lipoaspiration or surgically resected adipose tissue. Lipoaspiration usually yields a sufficient amount of cells for therapeutic application. In contrast, BMSCs require extensive in vitro passaging before enough material for clinical application is gained. Moreover, long-term in vitro passaging leads to a reduced proliferation rate, erosion of telomeres, and increased β-galactosidase activity, and is also time and labor consuming and represents a higher risk of possible contamination [40 –43].
Furthermore, it has been demonstrated that osteogenic capacity of BMSCs decreases with age: In BMSCs harvested from older donors, an increase of β-galactosidase-positive cells, a marker commonly associated with senescence, has been observed [40,44,45]. Even though the number of harvested cells and matrix mineralization has been comparable in cells from young, old, and osteoporotic donors [44,46], there has been a longer population doubling time, more apoptotic cells, a decreased life span, decreased matrix mineralization, and a decrease of AP activity and expression of Osterix in older cells compared with cells from younger donors [40,44,45].
Furthermore, estrogen depletion, as seen in age-associated diseases like osteoporosis, leads to an increased bone resorption through Ros- and TNF-β-mediated upregulation of T cells and thus increased expression of TNF and IFNγ stimulating osteoclastogenesis [47 –50]. In aging, commitment of BMSCs favors adipogenesis and ultimately results in fatty marrow [48,49]. In addition, in mice exhibiting shortened telomeres, associated with aging and osteoporosis, a reduced amount of harvested BMSCs, inhibited growth, and decreased osteogenic differentiation has been shown [49,51 –53].
In contrast, ASCs seem to retain their osteogenic capabilities with increasing age [54]. ASCs from all age groups maintain their rapid proliferation rate and the quantity of cells harvested in adipose tissue [55]. More importantly, no differences in gene expression and matrix mineralization could be shown between ASCs from adult and elderly patients [54 –56].
In our study, we could show that treatment with the Myostatin inhibitor Follistatin leads to an increased osteogenic potential of mASCs and mBMSCs and an inhibition of adipogenesis in both cell lineages. One could stipulate that treatment with Follistatin could thus attenuate the effects of estrogen depletion in aging.
In addition to osteoporosis and general changes associated with aging, elderly patients exhibit a reduced muscle mass. Recently, it has been shown that in addition to a biomechanical relationship [57,58], paracrine signaling also plays an important role in the muscle–bone interaction [57,59 –62]. It has been shown that Myostatin expression in muscle is decreased after aerobic exercise [63], thus treatment with Myostatin inhibitors could imitate the effects of aerobic exercise and hereby stimulate osteogenesis.
Thus, hypothesizing that the osteogenic differentiation potential of ASCs can be raised to the level of that in BMSCs using additional treatment such as Myostatin inhibitors, growth factors, exercise, or a combination of such, we conclude that ASCs are a valid source of stem cells for clinical application in bone defects.
Conclusion
Our study demonstrated that blockage of MSTN by Follistatin promotes osteogenesis in both mBMSCs and mASCs and generally inhibits adipogenesis, whereas increasing the differentiative potential of mASCs toward osteogenesis. However, mBMSCs still prove to be superior in their osteogenic potential. We conclude that although cells from both lineages share the capability to differentiate and proliferate into osteogenic precursor cells, they do retain characteristics of their tissue of origin and do not respond equally to treatment with Follistatin. Yet, MSTN blockage by Follistatin could prove to be a promising therapeutic approach in the treatment of bone defects caused by for example, tumors, burns, trauma, or surgery.
Footnotes
Acknowledgment
This work was supported by a grant of FoRUM (Research grant by the Ruhr University Bochum, School of Medicine).
Author Disclosure Statement
No competing financial interests exist.