
Abstract
Eye is a complex organ with a highly specialized tissue structure. The establishment of human pluripotent stem cells (hPSCs) has allowed the simulation of eye development in vitro. Most differentiation works of hPSC-derived ocular cells focus on a single, tissue-specific lineage, however, that faces difficulty in reflecting the complexity of eye development. Recently, the generation of a self-formed ectodermal autonomous multi-zone of ocular cells availably mimics the process of whole-eye development. In this study, we developed a rapid defined method to induce the differentiation of multi-zone ocular cells (MZOCs) from human induced pluripotent stem cells, which specifically experienced the key progenitor stages of anterior neuroectoderm and eye field stem cells by a 2.5-dimensional culture. These differentiated cell types spanned neural retina, retinal pigment epithelium, surface ectoderm, and neural crest and lens cells. In addition, the surface ectoderm zone of MZOCs could be mechanically isolated and induced into corneal epithelial cells, and the isolated neural crest zone could be directed into corneal endothelial cells. This in vitro differentiation process vividly mimics the development of vertebrate eye, and it provides a promising model for the study of ocular morphogenesis, as well as an ideal resource of seed cells for corneal regenerative medicine.
Introduction
The development of vertebrate eye is a complex and multistep process that is associated with complicated and ordered interactions between different tissues during embryonic development. Immediately after the onset of gastrulation, eye field is specified in the anterior neuroectoderm. The walls of the diencephalon evaginate to form the optic vesicles (OV). The OV comes into close contact with the surface ectoderm where the lens placode is formed. The OV gives rise to optic cup and further into the retina, retinal pigment epithelium (RPE), and optic stalk. The surface ectoderm gives rise to the lens and corneal epithelium. The periocular mesenchyme cells (neural crest) migrate between the lens and corneal epithelium to form the corneal endothelium and stroma [1 –4].
The establishments of human embryonic stem (hES) cell and human induced pluripotent stem (hiPS) cell culture have realized the in vitro simulation of eye development. Several groups have introduced the in vitro induction methods of the ocular-characteristic cells, such as retinal cells, RPE, and lens or corneal cells, from hES and iPS cells [5 –9]. Moreover, hES and iPS cells were also reported to generate the three-dimensional (3D) retinal, lens, or corneal tissues. However, these studies generally focus on one single or specific tissue, which fails to reflect the complexity process of whole-eye development. More recently, a self-formed ectodermal autonomous multi-zone (SEAM) of ocular cells from hiPS cells was developed. With the pluripotency of generating ocular surface ectoderm, lens, retina, and RPE cells, the differentiation of SEAM availably mimics the eye development process and provides a promising resource for the study of ocular morphogenesis [10,11].
As the outermost layer of the eye, cornea is one of the last structures specialized during eye development, and it provides significant refractive and barrier functions [4]. Various corneal diseases are major factors that induce blindness with the suffering population at >10 million worldwide [12]. Although keratoplasty is effective for the treatment of corneal dysfunction, global shortage of donor corneas is not able to meet the keen demand [13]. Corneal tissue engineering has emerged as a practical strategy to develop corneal tissue substitutes. However, this therapeutic strategy is restricted by limited sources of seed cells, including deficient autologous limbal stem cells (LSCs), immune rejection of allogenic LSCs, and the limited proliferative ability of human corneal endothelial cells (CEnCs) [14,15]. Therefore, it is of great significance and necessary to exploit renewable corneal cell sources.
Here, we developed a rapid and efficient method for the induction of multi-zone ocular cells (MZOCs) from hiPS cells under a defined culture condition. To emulate the natural development process of the eye, hiPS cells were first differentiated into anterior neuroectoderm by a 2.5-dimensional (2.5D) culture technique, then transformed into eye field stem cells (EFSCs), and further differentiated into MZOCs spanning neural retina, RPE, surface ectoderm, and neural crest and lens cells. The ocular surface ectoderm and neural crest zones of MZOCs could be mechanically isolated and directed into corneal epihtelial cells (CEpCs) and CEnCs in vitro, respectively.
Materials and Methods
hiPS cell culture
hiPS cell line DYR0100 was generously provided by Stem Cell Bank, Chinese Academy of Sciences, and cultured on plates coated with growth factor reduced Matrigel (BD354277; BD Biosciences, San Jose, CA). We used serum-free medium mTeSR1 (STEMCELL Technology, Vancouver, Canada) as the stem cell culture medium, and we manually passaged hiPS cells once every 4 to 5 days with Collagnase IV (Gibco, Grand Island, NY). All the cells were incubated at 37°C in a humidified atmosphere containing 5% carbon dioxide. The medium was changed every day.
MZOCs differentiation from hiPS cells
The differentiation culture for hiPS cells was performed as indicated in Fig. 1A. First, hiPS cells were seeded on 1% Matrigel-coated dishes and cultured in eye field differentiation medium [EDM; DMEM/F12 (Gibco) and neuralbasal medium (Gibco) (1:1) supplemented with 2 mM L-GlutaMAX (Gibco), 0.1 mM MEM nonessential amino acids (NEAA; Gibco), 0.1 mM β-mercaptoethanol (Gibco), and 1% N2 (Life Technologies, Grand Island, NY)] mixed with 2% Matrigel for 2 days; then, 2% Matrigel was removed. After another 5 days' culture in EDM, the culture medium was changed to ocular cell differentiation medium [ODM; DMEM/F12 supplemented with 10% knockout serum replacement (KSR; Gibco), 2 mM L-GlutaMAX, 0.1 mM NEAA, and 0.1 mM β-mercaptoethanol] for 3 to 4 weeks.
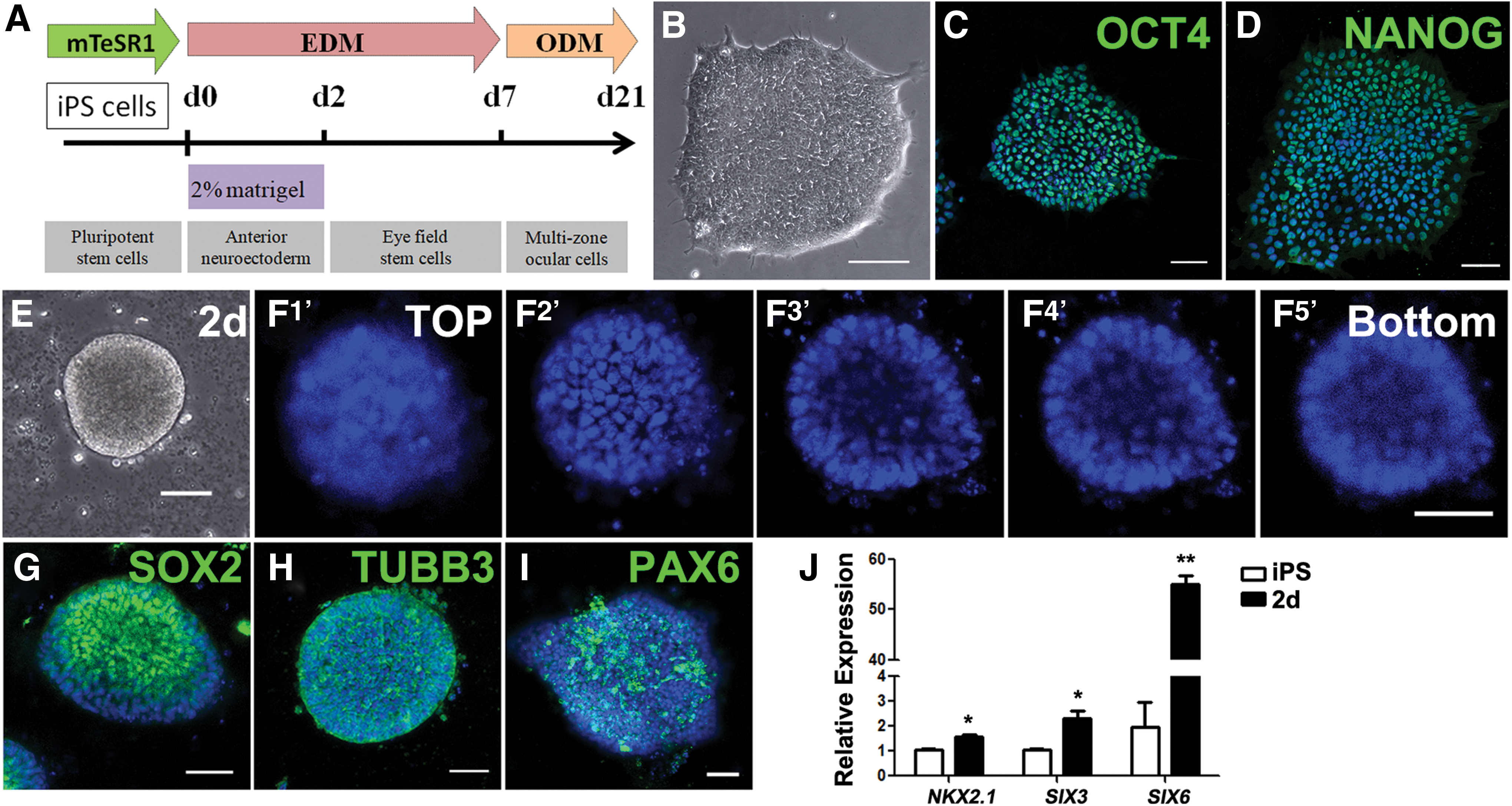
Differentiation of anterior neuroectoderm from hiPS cells.
Isolation and differentiation of hiPS-derived corneal epithelial and endothelial cells
For induction and enrichment of CEpCs and CEnCs from MZOCs, the cells in third and fourth zones were mechanically isolated by a glass needle (Sigma, St. Louis, MO) under microscopy along the visible boundaries, respectively. Isolated cells were dissociated with 0.25% trypsin-EDTA (Sigma) for 5 min at 37°C and gently triturated into a single-cell suspension. Isolated zone 3 cells were seeded on 1% Matrigel-coated dishes and cultured in corneal epithelial differentiation medium [CEpDM; DMEM/F12, 1% N2, 10 μM Y-27632 (Sigma), 20 ng/mL keratinocyte growth factor (KGF; PeproTech, Rocky Hill, NJ), 2 mM L-GlutaMAX, and 0.1 mM NEAA]. Isolated zone 4 cells were seeded on 1% Matrigel-coated dishes and cultured in corneal endothelial differentiation medium [CEnDM; defined keratinocyte serum free medium (Gibco)/DMEM/F12 (1:1), 0.5% DMSO (Sigma), 1% insulin-transferrin-selenium (ITS; Invitrogen, Carlsbad, CA), 1 nM Cholera toxin (Sigma), 0.2% Primocin (Invivogen, California, USA), 5% KSR, 2 ng/μL human epidermal growth factor (EGF; R&D, San Diego, CA), and 10 μM Y-27632].
Fluorescence staining
The differentiated hiPS cells were fixed in 4% paraformaldehyde (PFA; Boster, Wuhan, China). The stratified hiPS-derived CEpCs were fixed, embedded in optim alcutting temperature compound (Sakura, Netherland), and sectioned at 10-μm serial sections. The samples were blocked with 5% normal serum for 30 min at room temperature; they were treated with the primary antibodies overnight at 4°C (Table 1) and, subsequently, with Alexa Fluor 488- and 594-conjugated secondary antibody (1:200; Invitrogen) for 1 h at 37°C. Nuclei were stained with 4,6-diamidino-2-phenylindole (0.1 μg/mL; Beyotime Biotechnology, Shanghai, China) before fluorescence microscopy (Nicon, Japan, and Zeiss, LSM800, Germany).
A List of Primary Antibodies Used for Immunostaining
RNA isolation and real-time PCR
Total RNA was extracted from the differentiated hiPS cells by using Nucleospin RNA Kits (BD Biosciences). Complementary DNA (cDNAs) were synthesized by using the Primescript™ First-Strand cDNA Synthesis kit (TaKaRa, Dalian, China) according to the manufacturer's protocol. Real-time PCR was carried out by using SYBR® Green reagents and the Applied Biosystems 7500 Real-time PCR System (Applied Biosystems, Foster City, CA). The cycling conditions were 10 s at 95°C followed by 40 two-step cycles (15 s at 95°C and 1 min at 60°C). The quantification data were analyzed with the Sequence Detection System software (Applied Biosystems) by using GAPDH as an internal control (Table 2).
Primer Sequences for Quantitative Reverse Transcription-Polymerase Chain Reaction
Colony-formation assay
Isolated CEpCs were seeded onto Mitomycin (MMC; Haizheng, Zhejiang, China)-treated 3T3 feeder cells at a density of 2,000 cells per well and cultured in corneal epithelial mature medium [DMEM (Sigma), Ham F-12 (Sigma), 10% fetal bovine serum (Invitrogen), 10 ng/mL EGF, 0.4 ng/mL hydrocortisone succinate (Wako, Shanghai, China), ITS, 1% NEAA, 0.1 nM cholera toxin (Sigma), and 2 nM 3,305-Triiodo-Lthyronine Sodium salt (Sigma)] for 2 weeks. The colonies were fixed with 4% PFA and then stained with Giemsa (Beyotime).
Statistical analysis
Data in this study are representative of at least three independent experiments and are presented as means ± standard deviation. Statistical analysis was performed by using SPSS 17.0 software (SPSS, Chicago, IL). The differences between the control and experimental groups were tested with Student's t-test. A P value of <0.05 was considered statistically significant.
Results
Differentiation of anterior neuroectoderm from hiPS cells
To emulate the development of vertebrate eye in vitro, we established a stepwise and chemically defined approach to induce hiPS differentiation (Fig. 1A). hiPS cell line DYR0100, used for differentiation, was positive for the typical pluripotent markers OCT4 and NANOG by immunostaining (Fig. 1B–D). After 2 days culture in 2% Matrigel-mixed EDM, differentiating hiPS cells turned into 2.5D hollow vesicle-like structures (Fig. 1E, F). Immunostaining analysis showed that this 2.5D structure was positive for the neuroectoderm markers SOX2 and TUBB3, and the neural progenitor marker PAX6, whose expression is more abundant in the anterior regions of the embryonic neuroectoderm [16] (Fig. 1G–I). Besides, real-time PCR analyses showed that there was significant upregulation of the forebrain markers NKX2.1, SIX3, and SIX6 in 2.5D differentiating cells compared with undifferentiated cells (Fig. 1J). It is known that forebrain includes the structures of eyes and visual tracts [17]. These results suggested that hiPS cells differentiated into hollow anterior neuroectoderm-like cells, the progenitor stage of eye development, by 2.5D culture.
Differentiation of EFSCs from hiPS-derived neuroectoderm cells
The eye field is specified during forebrain morphogenesis [3]. To investigate whether EFSCs can be induced from hiPS-derived anterior neuroectoderm cells, the differentiating hiPS cells were cultured in only EDM for another 5 days; 2.5D anterior neuroectoderm-like cells differentiated into EFSCs (Fig. 2A). The induction of EFSCs was confirmed by immunostaining of key early eye field transcription factors. The majority of cells were positive for PAX6, RAX, OTX2, and LHX2, expressed by early eye field progenitors (Fig. 2B–E). Besides, we compared the expressions of eye field marker genes between the differentiating cell under 2% Matrigel-mixed EDM (2.5D culture) and only EDM [two-dimensional (2D) culture]. Real-time PCR analysis showed that the expression of the pluripotent marker OCT4 declined more rapidly and the process of eye field differentiation was promoted by 2.5D culture. The 2.5D culture significantly increased the expression of eye field markers RAX, SIX3, and OTX2 after 7 days of differentiation (Fig. 2F). These results suggested that EFSCs could be induced from hiPS-derived anterior neuroectoderm cells, and the 2.5D culture system could promote the differentiation process of EFSCs from hiPS cells.

Differentiation of eye field stem cells from hiPS cells.
Differentiation of MZOCs from hiPS-derived EFSCs
To investigate whether hiPS-derived EFSCs can give rise to different sublineages of ocular cells in culture, the medium of EFSCs was changed to ODM, in which the differentiating cells spontaneously and progressively formed five identifiable zones in additional 3 weeks (Fig. 3A–G). Zones 1 and 2 formed first, and these were followed by zones 3 and 4. The visible zone 5 appeared at last. Different zone cells had distinctive morphology and a visible boundary between zones (Fig. 3D–G). After 2 weeks' differentiation, cells in zone 1 expressed the neural retina markers PAX6 and CHX10, whereas cells in zone 2 were positive for the typical RPE markers PAX6 and MITF (Fig. 3H–J). Three weeks later, zone 3 cells expressed the surface ectodermal markers PAX6 and P63, and they also expressed CK14, a marker of epithelial progenitor cells, in the peripheral region (Fig. 3K, L). Zone 4 cells primarily expressed the neural crest markers P75 and SOX10, but some P75+ cells were found to appear in the adjacent zone 2, and some SOX10+ cells in the adjacent zone 3 (Fig. 3M–O). This suggested that zone 4 cells exhibited the capacity of migration of the neural crest cells. Cells in zone 5 specifically expressed the lens marker α-CRYSTALLIN (Fig. 3P). After 5 weeks' differentiation, MITF+ and PAX6+ cells in zone 2 became pigmented, and they acquired a cobble-like polygonal morphology, which was highly characteristic of native RPE cells in vivo (Fig. 3Q). Collectively, as shown in Fig. 3R, these findings imply that the differentiation process in this study generates presumptive ocular cells, such as neural retina, RPE, surface ectoderm, neural crest, and lens, and mimics the interaction of the different cell lineages of eye development in situ.

Differentiation and characterization of multi-zone ocular cells.
Induction and characterization of corneal epithelial cells from MZOCs
Next, we determined whether the presumptive surface ectoderm zone could differentiate to CEpCs in vitro. After 3 weeks' differentiation, zone 3 cells were mechanically isolated from MZOCs and cultured in CEpDM. After an additional 2 to 3 weeks of culture, the differentiating cells appeared to be compact and exhibited a cobblestone-like morphology (Fig. 4A). To further examine whether zone 3 cells differentiated to the corneal epithelial lineage, we performed immunostaining for typical corneal epithelial markers. These epithelial-like cells were labeled positively for PAX6 and P63, and partially for CK14 and cornea-specific CK12 (Fig. 4B–D). Cultured on the MMC-treated 3T3 feeder cells for 10–14 days, the epithelial-like cells formed the compact colony (Fig. 4E, F). Besides, after an additional 3 to 5 weeks' culture, the cells formed the stratified epithelia consisting of four to six layers, which were labeled positively for the mature CEpCs markers CK12 and CK3 (Fig. 4G, H). In addition, real-time PCR analysis was also carried out. Compared with hiPS cells, a significant increase in the expression of corneal epithelial markers, PAX6, CK12, and CK3, was observed in the third zone-derived CEpCs. However, these expressions were still lower than the expressions of mature human CEpCs (Fig. 4I).
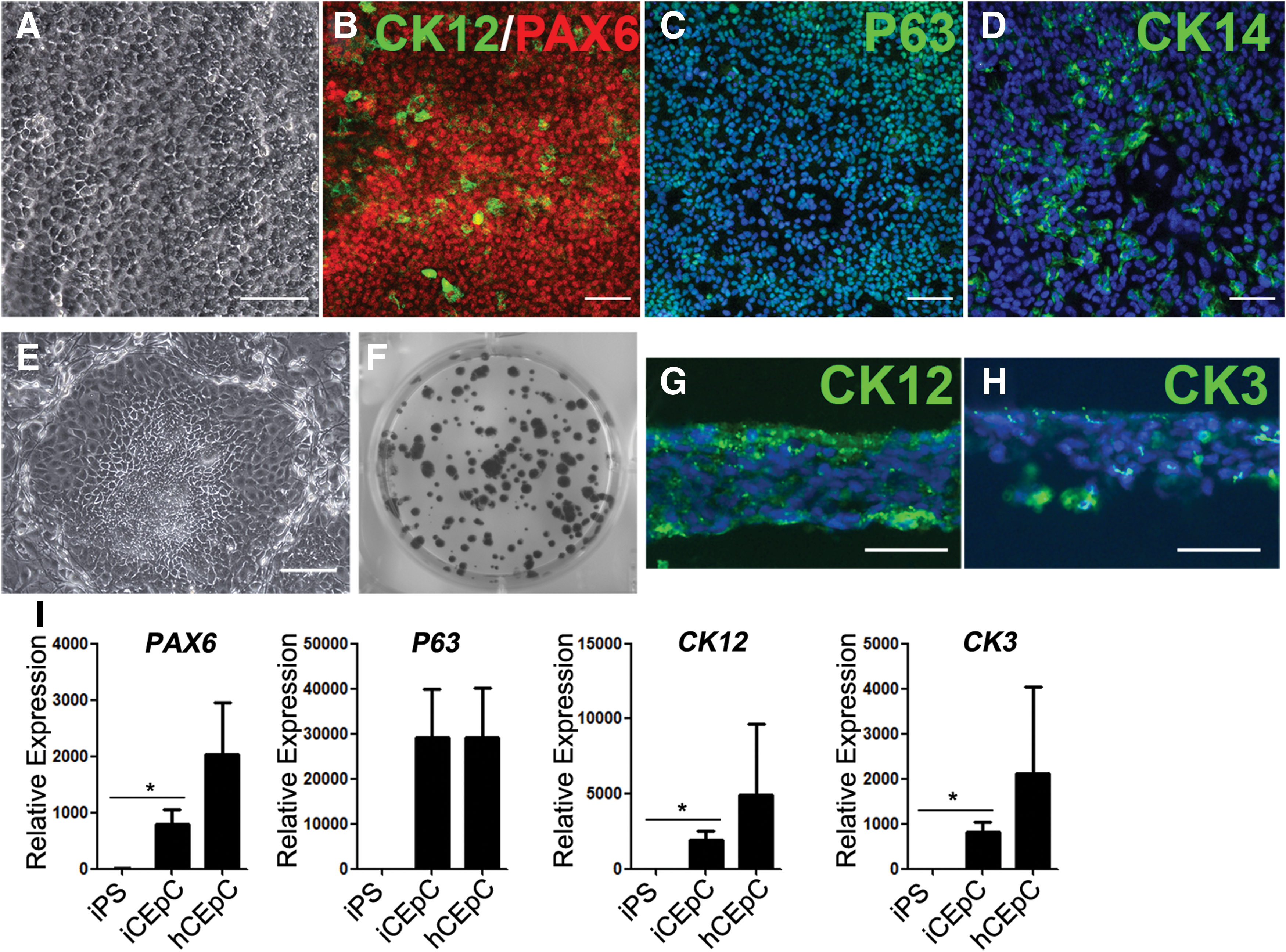
Morphology and characterization of CEpCs derived from the isolated surface ectoderm cells.
Induction and characterization of CEnCs from MZOCs
To test whether CEnCs could be induced from the presumptive neural crest zone in vitro, we mechanically isolated the fourth zone cells from MZOCs after 3 weeks' differentiation. The isolated cells were cultured in CEnDM. Within 1 week of induction, the cells displayed a regular polygonal morphology (Fig. 5A). The cell density was ∼3,360 ± 372 cells/mm2, which was significantly higher than the corneal endothelial decompensation threshold of ∼500 cells/mm2 [18]. Immunostaining showed that the differentiating cells were labeled positively for the typical CEnC markers ZO-1 and Na+/K+-ATPase (Fig. 5B, C). Flow cytometric analysis revealed that 96.9% of the differentiating cells were Na+/K+-ATPase + cells (Fig. 5D). In addition, we also confirmed the increased messenger RNA expression of ZO-1, Na+/K+-ATPase, AQP-1, N-cadherin, SLC4A4, and SLC16A4 by real-time PCR in the neural crest-derived CEnCs versus hiPS cells, but lower than expression of mature human CEnCs, except ZO-1, which suggested that the hiPS-derived CEnCs here were immature (Fig. 5E).
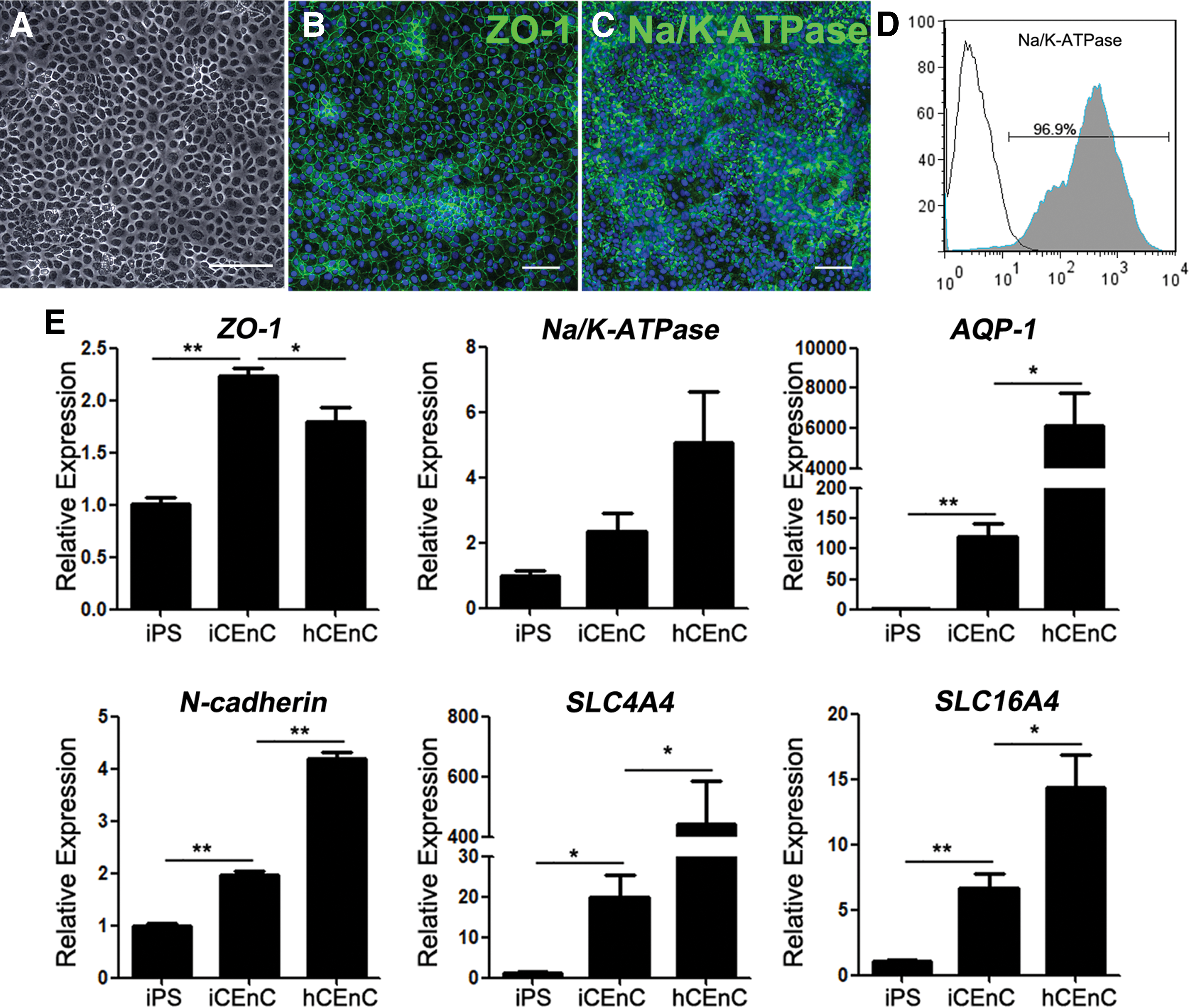
Morphology and characterization of CEnCs derived from the isolated neural crest cells.
Discussion
In this study, we developed a simple differentiation protocol that induces the hiPS cells to differentiate into MZOCs (neural retina, RPE, surface ectoderm, neural crest, and lens) after the differentiated stages of the anterior neuroectoderm and EFSCs. In addition, the surface ectoderm zone of MZOCs induced by this differentiation protocol could be isolated and turned into CEpCs, and the neural crest zone could be isolated and induced into CEnCs.
Previous studies have successfully demonstrated the in vitro induction of ocular cells from hES and hiPS cells [5 –9]. Recently, the generation of SEAM from hiPS successfully mimicked the whole-eye development process, and it served as a promising model of ocular morphogenesis [10,11]. Different from this culture system of SEAM, we first induced anterior neuroectoderm, a key stage of eye development, from hiPS cells by 2.5D culture. Along the anteroposterior axis, anterior neuroectoderm develops into the forebrain, which includes telencephalon, hypothalamus, prethalamus, as well as eyes and visual tracts [17]. Several studies have induced neural cells of neuroectoderm from hES cells by inhibition of Activin/Nodal, bone morphogenetic protein (BMP), or by activation fibroblast growth factor (FGF) signaling [19 –21]. In this study, hiPS cells spontaneously differentiated into vesicle-like anterior neuroectoderm after 2 days' induction with 2% Matrigel-mixed EDM. This hiPS-derived anterior neuroectoderm is defined by the expression of the typical anterior neuroectoderm and forebrain markers (SOX2, TUBB3, PAX6, NKX2.1, SIX3, and SIX6), and it further differentiates into EFSCs.
Most of the previous data regarding hES differentiation are derived from experiments performed in 2D conditions [6,11,22,23]. The differentiating cells grow as monolayers, and receive an equal amount of nutrients and growth factors, resulting in polarized cell adhesion and 2D contact with neighboring cells. These physical characteristics of 2D culture do not fully reflect the essential physiology of natural process. Compared with 2D culture, 3D methods involve formation of spheroids, including culture in suspension, encapsulated, or grown on the top of a 3D matrix, which can mimic a developmental process through its heterogeneity, and better translate the complex physiological features in vitro [5,24,25]. Here, we used a system of a 2.5D culture. It possesses the characteristics of both 2D and 3D cultures, including grown on rigid materials, spheroid formation, and uniform spheroid size. And our data indicated that the formation of 2.5D anterior neuroectoderm (2.5D culture) significantly increased the expression of eye field markers (RAX, SIX3, and OTX2), compared with the neural induction without 2% Matrigel-mixed EDM (2D culture).
Instead of four zones appearing during SEAM induction, five zones, including neural retina, RPE, surface ectoderm, neural crest, and lens, were generated by our induction procedure. All five zones exhibited separating distribution and visible boundaries, and each zone had specific differentiation fate for ocular cells. Besides, four zone cells of SEAM were characterized during 2–6 weeks, whereas all five zones of MZOCs in this study were characterized within 4 weeks. Therefore, the approach established here emulates the development of vertebrate eye in vitro, providing a promising resource for the study of ocular morphogenesis and regenerative medicine.
The development of the vertebrate cornea needs to achieve the inductive interactions between surface ectodermal and mesenchymal (mostly neural crest) tissues. Meanwhile, presumptive corneal epithelium was differentiated from surface ectoderm [4]. Most previous studies indicated that the generation of CEpCs from hES relies on the usage of feeder cells, undefined conditioned media, or amniotic membrane [8,22,26,27]. Recently, the hES-derived CEpCs by small molecules have become available through blocking transforming growth factor-β or Wnt-signaling pathways, as well as via activating FGF or BMP signalings [28,29]. In this study, interactions among the different ocular cell lineages develop to zone 3 surface ectoderm cells, which were isolated and seeded onto CEpDM. KGF and Y27632 are reported to induce and maintain the expression of CEpC-specific markers [10,11,30]. With application of 20 ng/mL KGF and 10 μM Y27632, CEpCs were efficiently induced from isolated surface ectoderm cells and expressed typical marker of P63, PAX6, K12, and K3, which exhibited the capacities of colony formation and stratified arrangement.
Previous studies have revealed that corneal endothelium is solely derived from neural crest cells [4,31]. Several reports have described the generation of neural crest cells from hES, involving coculture on PA6 or M5 feeder layers, embryoid body formation, inhibition of Smad and Activin A signaling, and activation of Wnt signaling [23,32 –34]. Here, our result showed that neural crest cells were spontaneously generated in the process of MZOC differentiation. Then, the isolated neural crest cells could be seeded onto CEnDM. In addition, the application of 10 μM Y27632 was reported to successfully induce neural crest-derived CEnCs, which expressed typical corneal endothelium markers (ZO-1, Na+/K+-ATPase, AQP-1, N-cadherin, SLC4A4, and SLC16A4) within 1 week [9]. It was known that primary human CEnCs had limited regenerative capacity [35]. Here, the hiPS-derived CEnCs exhibited limited proliferative capacity in continuous passage culture (data not shown). Therefore, we would further optimize the differentiated CEnCs culture system for long-term expansion.
Based on the principles in generating viable cornea tissue equivalents, corneal engineering starts from generating epithelium, stroma, and endothelium layers, and it culminates in full-thickness cornea tissue equivalents. Many seed cells and scaffolds were reported to construct the full-thickness cornea substitute, including human, bovine, and fetal pig corneal cells, acellular human corneal stroma, collagen-chondroitin sulfate foam, collagen gel, and type I collagen [36 –39]. Here, both CEpCs and CEnCs were generated in our established differentiation system. Previously, we reported the use of native porcine conjunctiva to prepare xenogeneic acellular conjunctiva matrix as a scaffold of tissue-engineered corneal epithelium [40]. However, in the future, we plan to seed hiPS-derived CEnCs and CEpCs on the acellular porcine cornea to construct the full-thickness cornea equivalent for animal studies for a later functional test. We believe that our work will hopefully provide an optimal method to obtain adequate CEpCs and CEnCs in vitro, and it could be applied to clinical studies in future.
Footnotes
Acknowledgments
This work was partially supported by the Shandong Provincial Nature Science Fund (grant nos. ZR2015HQ005, ZR2017PH036, ZR2017PH009, ZR2017PH065, and ZR2018LH008) and the National Natural Science Foundation of China (grant no. 81700811). W.S. and Q.Z. are partially supported by the Taishan Scholar Program (grant nos. 20150215 and 20161059) and the Innovation Project of Shandong Academy of Medical Sciences.
Author Disclosure Statement
No competing financial interests exist.