
Abstract
Spinal muscular atrophy (SMA) is caused by the mutation or deletion of the survival motor neuron 1 (SMN1) gene. Only ∼10% of the products of SMN2, a paralogue of SMN1, are functional full-length SMN (SMN-FL) proteins, whereas SMN2 primarily produces alternatively spliced transcripts lacking exon 7. Reduced SMN protein levels in SMA patients lead to progressive degeneration of spinal motor neurons (MNs). In this study, we report an advanced platform based on an SMN2 splicing-targeting approach for SMA drug screening and validation using an SMN2 splicing reporter cell line and an in vitro human SMA model through induced pluripotent stem cell (iPSC) technology. Through drug screening using a robust cell-based luciferase assay to quantitatively measure SMN2 splicing, the small-molecule candidate compound rigosertib was identified as an SMN2 splicing modulator that led to enhanced SMN protein expression. The therapeutic potential of the candidate compound was validated in MN progenitors differentiated from SMA patient-derived iPSCs (SMA iPSC-pMNs) as an in vitro human SMA model, which recapitulated the biochemical and molecular phenotypes of SMA, including lower levels of SMN-FL transcripts and protein, enhanced cell death, and reduced neurite length. The candidate compound exerted strong splicing correction activity for SMN2 and potently alleviated the disease-related phenotypes of SMA iPSC-pMNs by modulating various cellular and molecular abnormalities. Our combined screening platform representing a pMN model of human SMA provides an efficient and reliable drug screening system and is a promising resource for drug evaluation and the exploration of drug modes of action.
Introduction
Spinal muscular atrophy (SMA) is an inherited autosomal recessive neuromuscular disease characterized by progressive degeneration of spinal motor neurons (MNs). Clinically MN loss in SMA patients leads to subsequent muscle weakness and atrophy [1]. SMA is mainly caused by low levels of the survival motor neuron (SMN) protein, which is ubiquitously expressed and regulates RNA processing [2]. Two SMN genes, SMN1 and SMN2, encode human SMN protein [3]. The SMN1 gene primarily produces the full-length SMN (SMN-FL) protein and is deleted or mutated in most patients with SMA [4]. The SMN2 gene is present in most SMA patients, and an inverse correlation between SMN2 copy number and clinical severity of SMA has been shown [5,6]. Although SMN2 encodes SMN protein, the majority of SMN2 gene products are truncated unstable proteins lacking exon 7 (SMNΔ7) with only low levels of functional SMN-FL protein produced due to inefficient splicing of exon 7 [7].
Although the antisense oligonucleotide nusinersen has recently been approved for use by the FDA [8,9], there is still much progress to be made and other new and promising approaches to explore. Until recently, potential treatment strategies have been developed based on the molecular pathophysiology of SMA, such as pharmacological compounds targeting SMN2 to increase SMN-FL protein or to facilitate correct splicing of SMN2 [10], the introduction of SMN1 using vector-mediated gene delivery [11], and non-SMN targeting approaches to protect MNs [12]. Many studies have shown therapeutic efficacy in patient-derived fibroblast-based assays and model animals, such as yeasts, flies, zebrafish, and mice [13]. However, subsequent clinical trials reported, to date, have failed to demonstrate convincing results based on these approaches [14]. Therefore, a proper in vitro human SMA model could be a promising route for the development of effective therapies.
For the last decade, drug testing and development systems using patient-derived induced pluripotent stem cell (iPSC) models have been reported for various neurological diseases such as Parkinson's disease, Alzheimer's disease, amyotrophic lateral sclerosis, Huntington's disease, SMA, and other neurodevelopmental diseases [15,16]. Analysis of SMA patient-specific iPSCs (SMA iPSCs) has revealed varying degrees of MN loss, consistent with a neurodegenerative status particularly in MNs [17 –19]. MNs derived from SMA iPSCs show increased apoptosis [20] and delayed neurite outgrowth [21]. At the later stages of differentiation, SMA iPSC-derived MNs exhibit a selective reduction in number and size compared with wild-type (WT) iPSC-derived MNs [19]. Several studies have shown that SMA disease-relevant models based on iPSC technology can be applied to develop new platforms for drug assay and developmental studies. Treatment with valproic acid (VPA) and tobramycin, SMN-inducing compounds, induced partial restoration of SMN protein in SMA iPSCs [19]. Specific inhibitors of apoptotic pathways [20], the thyrotropin-releasing hormone analog [22], and antioxidant compounds [21] have been shown to increase SMN protein levels in spinal MNs differentiated from SMA iPSCs and, thus, may have potential efficacy in treating SMA.
In this study, we describe a human in vitro SMA model based on the generation of patient-specific iPSCs. MN progenitors (pMNs) with an ability to undergo terminal differentiation into mature MNs were generated from SMA iPSCs, characterized as imitating the typical phenotypic features of SMA, and served as a reliable drug-discovery platform. Our drug screening approach for SMA has a clear advantage because we used the combined screening strategy of a luciferase reporter assay for the quantitative assessment of SMN2 splicing followed by an iPSC-based assay using pMNs derived from SMA iPSCs (SMA iPSC-pMNs). A library of small-molecule compounds was screened using a SMN2-luciferase reporter cell line to identify potential drug candidates that may restore the inclusion of the SMN2 exon 7. In addition, we validated and characterized in SMA iPSC-pMNs and showed that one candidate compound rescued the pathological disease phenotypes. Our SMA disease model system provides a platform for efficient drug screening and validation of drug candidates.
Materials and Methods
Cell culture
Primary fibroblasts from SMA type 1 patients (GM09677, GM00232) were obtained from Coriell Cell Repositories (Camden, NJ). Human WT fibroblasts (CRL-2097 and GM08333) were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and Coriell Cell Repositories, respectively. They were maintained in minimal essential medium (Gibco, Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (FBS; Gibco), 2 mM
Establishment and maintenance of human iPSCs
Healthy control (CRL2097, IMR90) and SMA patient (GM09677, GM00232) fibroblasts were reprogrammed by electroporation with Episomal iPSC Reprogramming Vectors (Cat. No. A14703; Invitrogen) encoding OCT4, SOX2, KLF4, L-MYC, LIN28, and shRNA-p53 as described previously [24,25]. Five days after electroporation, the cells were reseeded at 1 × 105/well onto Matrigel (BD Biosciences, San Diego, CA)-coated six-well plates with iPSC medium comprising DMEM/F12 medium (Gibco) supplemented with 20% knockout serum replacement (Gibco), 1 mM nonessential amino acids (Gibco), 0.1 mM β-mercaptoethanol (Sigma-Aldrich, St. Louis, MO), and 10 ng/mL basic fibroblast growth factor (R&D Systems, Minneapolis, MN). After 14–20 days, human embryonic stem cell (hESC)-like colonies were isolated and expanded for further experiments. iPSCs were passaged with clump dissociation using 1 mg/mL collagenase type IV (Gibco).
Chemicals
Small-molecule compounds, including rigosertib (Selleckchem, Houston, TX), suberoylanilide hydroxamic acid (SAHA) (Selleckchem), cycloheximide (CHX; Sigma-Aldrich), MG132 (Calbiochem, San Diego, CA), sodium butyrate (SB; Sigma-Aldrich), BI2536 (Sigma-Aldrich), GSK461364 (Selleckchem), HMN214 (Selleckchem), and BI6727 (Selleckchem), were used.
Analysis of alternative splicing of SMN pre-mRNA
SMN1-Luc and SMN2-Luc cells were generated by introducing SMN1 and SMN2 splicing minigene reporter plasmids, respectively, into C33A cells [26,27]. SMN splicing minigene reporters were designed by the following procedure. Briefly, to inactivate the authentic translation termination codon at the 3′ end of exon 7, a single nucleotide “C” was inserted immediately ahead of the termination codon (CUAA). A FL reporter gene was fused to 21 nucleotides downstream from the 5′ end of exon 8, and the initiation codon at the 5′ end of the reporter gene was modified by removing the “A,” thereby preventing any undesirable internal translation initiation and reducing background expression. To screen small-molecule compounds that modulate the splicing of SMN2 pre-mRNA, SMN2-Luc cells were split at a density of 1 × 104 cells/well in 96-well plates. A luciferase assay was performed 24 h after treatment with small-molecule compounds using a One-Glo Luciferase Assay System (Promega, Madison, WI).
Cell-based SMN immunoassay
SMA patient fibroblasts (8 × 103 cells/well) were seeded on black-well clear bottom 96-well plates (Greiner, Wemmel, Belgium) and treated with small-molecule compounds (10 μM) for 24 h. MG132, a proteasome inhibitor previously characterized as an SMN-elevating compound [28], was also included as a positive control. Cells were washed and fixed with 4% paraformaldehyde (PFA). The cells were permeabilized in 0. 5% Triton X-100 and 1% bovine serum albumin in phosphate-buffered saline and incubated with mouse monoclonal antibody against SMN (Cat. No. 610647; BD Transduction Laboratories, Franklin Lakes, NJ) for 2 h. Then, cells were incubated in Alexa Fluor 488-conjugated goat secondary antibody against mouse IgG (Invitrogen) for 1 h and stained with a Hoechst 33342 dye (Invitrogen). Cells were analyzed with a Cellomics ArrayScan instrument (Cellomics, Pittsburgh, PA). More than 300 cells for each sample were analyzed to obtain the average and the standard deviation.
Analysis of SMN2 promoter activity
To construct SMN2 promoter-driven FL reporter plasmids, the promoter and the 5′ end of 5′-UTR of the SMN2 gene (∼3.4 kb) were cloned into the pGL4.14 vector (Promega). To enhance the responsiveness of the reporter to the change in transcription, a PEST sequence was added to the C-terminus of the FL protein. 293T cells were transfected with 2 μg of pGL4.14-SMN2 promoter-Luc-PEST reporter plasmid using X-tremeGENE siRNA Transfection Reagent (Roche, Indianapolis, IN) and split at a density of 1 × 104 cells/well in 96-well plates. After 12 h, cells were treated with rigosertib, SAHA, and SB for 24 h and assayed for luciferase activity using a ONE-Glo Luciferase Assay System (Promega). Cell viability was measured in the same conditions by a CellTiter-Glo Luminescent Cell Viability assay (Promega).
Analysis of SMNΔ7 protein stability
To construct plasmids expressing FLuc-SMNΔ7 protein, the FL gene was cloned into the pcDNA3.1 vector, and SMNΔ7 was then inserted into the pcDNA3.1-Fluc vector [29]. 293T cells were transfected with 2 μg of plasmids and treated with 300 μg/mL hygromycin B for 2 weeks for stable selection. Stable cell lines were split at a density of 1 × 104 cells/well in 96-well plates. After 12 h, cells were treated with various concentrations of rigosertib in the presence of CHX (0.1 mg/mL). Proteasomal inhibitor MG132 (10 μM) was also included as a positive control. A luciferase assay was performed using a ONE-Glo Luciferase Assay System at 10 h after treatment. Cell viability was measured in the same conditions by a CellTiter-Glo Luminescent Cell Viability Assay.
Total RNA preparation and polymerase chain reaction analysis
Total RNA isolation was performed with an RNeasy Mini Kit (Cat. No. 74104; Qiagen, Hilden, Germany) according to the manufacturer's instructions, and a SuperScript IV First-Strand Synthesis System Kit (Cat. No. 18091200; Thermo Fisher Scientific, Waltham, MA) was used for cDNA synthesis. Semiquantitative real-time polymerase chain reaction (RT-PCR) was carried out under the following conditions: 3 min at 94°C; 32–35 cycles of 30 s at 94°C, 40 s at 55°C, 40 s at 72°C, and 5 min extension at 72°C. Each PCR product was loaded onto 2% agarose gel containing SYBR Safe DNA gel stain (Cat. No. S33102; Invitrogen), and Gel-Doc (Bio-Rad Laboratories, Hercules, CA) was used for imaging. Quantitative RT-PCR (qRT-PCR) was performed with SYBR green PCR Master mix (Applied Biosystems, Foster City, CA), and the reaction was detected in a 7500 Fast Real-time PCR system (Applied Biosystems) as described previously [30]. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal standard, and the provided software was used to calculate the CT values of the target genes. The used primer sequences are listed in Supplementary Table S1.
Alkaline phosphatase and immunocytochemistry
Alkaline phosphatase (AP) staining was performed using naphthol/Fast Red Violet solution (Sigma-Aldrich) as described previously [31,32]. For immunocytochemistry staining, the cells were fixed in 4% PFA and permeabilized with 0.1% Triton X-100. After blocking, cells were incubated with the respective primary antibodies at 4°C overnight, followed by incubation in fluorescence-conjugated secondary antibodies for 2 h. Slides were observed under an Axiovert 200 M microscope (Carl Zeiss, Gottingen, Germany) or a confocal microscope (Cat. No. FV1000 Live; Olympus, Tokyo, Japan). The used primary antibodies are described in Supplementary Table S2.
Confirmation of karyotype and short tandem repeat analysis
Chromosomal G-band karyotyping analysis was performed by GenDix, Inc. (Seoul, Republic of Korea). For short tandem repeat (STR) analysis, gDNA preparation of fibroblast and iPSCs was performed with DNeasy Blood & Tissue Kits (Cat. No. 69504; Qiagen) according to the manufacturer's instructions. STR genotyping was analyzed by HumanPass, Inc. (Seoul, Republic of Korea).
Embryoid bodies and teratoma formation assay
For analysis of in vitro differentiation capacity, iPSCs were dissociated using 1 mg/mL collagenase type IV (Gibco) and replated onto noncoated dishes in embryoid body (EB) medium supplemented with 10% of knockout serum replacement (Gibco) based on DMEM/F12 (Gibco). After 5 days, EBs were attached to Matrigel-coated Lab-Tek chamber slides (Nunc, Rochester, NY) and cultured for 10 days. iPSCs (1 × 106 cells) were subcutaneously injected into 6-week-old BALB/c-nude mice (Orient Bio, Inc., Seoul, Korea). The resulting teratomas were fixed by 10% formaldehyde, paraffin-embedded, serially sectioned, and stained using hematoxylin & eosin staining solution (Sigma-Aldrich). Teratoma assays were performed after approval by the Institutional Animal Care and Use Committee (IACUC) of KRIBB (approval No. KRIBB-AEC-17014).
Differentiation toward MNs
MNs were differentiated as described previously [33]. Neural induction media contained DMEM/F12 medium and neurobasal media at a 1:1 ratio supplemented with 1% P/S, 1% GlutaMAX (Gibco), 100 nM
Western blot analysis
The pMNs and MNs were harvested at indicated times. The cells were lysed with RIPA buffer containing 1 mM protease inhibitor and 1× phenylmethylsulfonyl fluoride (PMSF). Then, 20 μg of the cell lysate was resolved by precast gels (4%–15% gradient; Bio-Rad Laboratories). After transfer, the membranes were incubated with the appropriate primary antibody, washed, and incubated with the secondary antibody. The list of primary antibodies used is given in Supplementary Table S2. The band intensity was quantified using ImageJ software (National Institutes of Health, Bethesda, MD).
Cell viability and death analysis
To detect LIVE/DEAD cells using a LIVE/DEAD Viability/Cytotoxicity Kit (Cat. No. L3224; Invitrogen), cells were washed and incubated at room temperature with LIVE/DEAD assay reagent containing 2 μM Calcein-AM and 4 μM EthD-1. After 40 min, cells were washed and visualized with a microscope (IX51; Olympus). The fluorescence was measured at 494/517 nm and 528/617 nm with Calcein-AM and EthD-1, respectively. To quantify LIVE/DEAD cells, the cells were trypsinized, and 1–2 × 106 cells were stained with LIVE/DEAD assay reagent mixed with 50 μM Calcein-AM and 4 μM EthD-1. The cells were incubated for 20 min at room temperature protected from light and evaluated by flow cytometry (BD Accuri C6; Becton-Dickinson, Mansfield, MA).
Detection of mitochondria and mitochondrial superoxide levels
To monitor mitochondrial contents and mitochondrial superoxide generation, the cells were stained with MitoTracker (Cat. No. M7514; Invitrogen) and MitoSOX (Cat. No. M36008; Invitrogen), respectively. The cells were incubated at 37°C for 15 min using 250 nM MitoTracker and 5 μM MitoSOX staining solution, protected from light. After staining, the cells were captured at 490/516 nm and 510/580 nm, respectively.
Analysis of neurite outgrowth
MN cultures on Matrigel-coated dishes were fixed in 4% PFA. The neurites were stained with TUJ1 according to the immunocytochemistry method. Images were captured on a fluorescence microscope (IX51; Olympus). Neurite outgrowth was analyzed by NeuronJ (ImageJ add-on software). Each neurite could be observed even if it was curved using NeuronJ, and 45 neurites per condition were measured and quantified for length [34].
Animal experiments
All animal experiments were performed in accordance with Korean Food and Drug Administration guidelines. All protocols were reviewed and approved by the Institutional Animal Care and Use Committee of KRIBB (institutional permit No. KRIBB-AEC-18132). A heterozygous SMA mouse [FVB.Cg-Grm7Tg(SMN2)89Ahmb Smn1tm1Msd Tg(SMN2*delta7)4299Ahmb/J, stock No. 005025 stock No. 005025] was purchased from Jackson Laboratory (Bar Harbor, ME) and mated to obtain homozygous SMA mice (SMNΔ7 transgenic mouse). Rigosertib (10 mg/kg) was intraperitoneally administered to SMNΔ7 transgenic mice once a day from postnatal day 3–10 (n = 3 for each group). On the last day of administration, the spinal cord was isolated from each mouse 5 h after injection and lysates were prepared by sonicating in RIPA buffer (Lab-Pharm Service solution, Daejeon, Korea), containing protease inhibitor cocktail set III (Calbiochem, Darmstadt, Germany). Quantitative western blot analysis was performed as described previously [35]. Quantification of SMN expressed cell population in the spinal cord tissue was performed as described previously [36]. For histological analysis, mouse spinal cords were collected, fixed with 4% PFA solution, and transferred into 30% sucrose for cryopreservation. The tissues were frozen in optimal cutting temperature compound (Tissue-Tek OCT Compound; Sakura Finetek USA, Inc., Torrance, CA) and sectioned to produce 10-μm thick sections. All sections were permeabilized in 0.01% Triton X-100 for 15 min and incubated overnight with primary antibodies at 4°C. After washing thrice, sections were incubated with secondary antibody at room temperature for 1 h. The tissues were captured using the Evos Fl Auto 2 Cell Imaging System (Thermo Fisher Scientific). The primary antibodies used are described in Supplementary Table S2.
Neuromuscular junction formation assay
To identify functionality of hiPSC-derived MNs, the C2C12 cells were plated onto Matrigel-coated confocal dish at a density of 880 cells/mm2 in maintenance medium. After 24 h, myoblast fusion was induced by transferring the cells to the differentiation medium. The media were changed every other day. On day 4, HB9+ MNs were dissociated and seeded onto myotubes at a density of 8,800 cells/mm2. The cells were cultured with ChAT+ MN, in a differentiation medium, containing neurotrophic factors (10 ng/mL insulin-like growth factor 1; PeproTech, Rocky Hill, NJ), 10 ng/mL brain-derived neurotrophic factor (PeproTech), and 10 ng/mL ciliary neurotrophic factor (R&D Systems) for 7 days. For evaluation of the neuromuscular junction (NMJ), overlapping region quantification of α-bungarotoxin-488 (α-BTX) and ChAT+ was performed. NMJ measurements were performed for every sixth fields of the coculture condition of iPSC-derived MNs and C2C12 cells.
Electrophysiological recordings
Whole-cell patch-clamp recordings were carried out to measure action potential and voltage-gated sodium/potassium currents. Terminally differentiated MNs plated on coverslips were placed in a recording chamber and continuously perfused (3 mL/min) with bath solution containing (in mM): 137 NaCl, 2.0 CaCl2, 10 HEPES, and 20 glucose (pH 7.3 with NaOH). The patch pipettes were made from borosilicate glass capillaries (Clark Electromedical Instruments, UK) using a pipette puller (PP-830; Narishige, Japan). Their resistances were 5–8 MΩ when filled with pipette solution containing (in mM): 140 K-gluconate, 5 NaCl, 1 MgCl2, 0.5 EGTA, and 10 HEPES (pH 7.25 with KOH). Voltage- or current-clamp protocol generation and data acquisition were controlled by computers equipped with an A/D converter, Digidata 1440 (Molecular Devices, Sunnyvale, CA), and pCLAMP 10.3 software (Molecular Devices). The signals were filtered at 5 kHz and sampled at 10 kHz using Axopatch 200B amplifier (Molecular Devices). Evoked action potentials were induced by injections of step currents from −0.1 to 0.2 nA in 0.02 nA increments for 500 ms. For the voltage-gated currents, we used voltage steps for 1 s from −70 to +90 mV in 10 mV increments. Clampfit (Molecular Devices), GraphPad Prism version 5 (GraphPad Software, Inc., San Diego, CA), and Origin (Microsoft, Redmond, WA) were used for data analysis.
Statistics
The statistical significance of the difference between two independent samples was analyzed using the nonparametric Mann–Whitney U-test. Experimental results were expressed as mean value ± standard error of the mean, and statistically significant differences were indicated on the graph by asterisks (ns: not significant, *P < 0.05, **P < 0.01, ***P < 0.001). All experiments were repeated at least thrice.
Results
Screening for SMN2 splicing modulators
As a splicing defect in SMN2 is a major factor that causes the disease phenotype in SMA patients, the recovery of SMN2 splicing is a promising therapeutic approach for the treatment of SMA [37]. Therefore, we developed a new platform for SMA drug development that combines a luciferase reporter assay for the quantitative assessment of SMN2 splicing followed by validation of drug candidates using a patient-specific iPSC-based assay (Fig. 1A). For a facile and quantitative measurement of SMN2 splicing, we developed a robust cell-based assay system (SMN2-luciferase splicing reporter cell line; SMN2-Luc stable cell line), which harbors an exogenously introduced SMN2 minigene consisting of exons 6 through 8 and intervening introns, fused with the firefly luciferase (FLuc) gene (Fig. 1B). In this assay system, any compound that enhances the inclusion of exon 7 would increase FLuc activity because exon 7-included mRNAs would produce a larger protein fused with FLuc protein. In contrast, exon 7-skipped mRNAs generate an in-frame translational termination codon at exon 8, resulting in the production of prematurely terminated proteins without the fusion of FLuc protein. After treatment with 88 small-molecule compounds, we identified two compounds (No. 53 and No. 65) that notably increased the luciferase activity by more than threefold of the dimethylsulfoxide (DMSO) control (Fig. 1C, Supplementary Table S3). The SMN1-Luc stable cell line, harboring the SMN1 minigene reporter, was additionally tested as a control to evaluate the specificity of the effect on SMN2 splicing, and no enhancement of luciferase activity was found (Supplementary Fig. S1).

Exploration of SMN2-specific splicing modulators.
The candidate compound rigosertib promotes splicing correction of SMN2 exon 7
Next, we examined whether these two compounds increased the amount of SMN protein in SMA fibroblasts from an SMA type 1 patient. We performed an immunoassay to specifically detect SMN protein using its specific antibody, which provided a quantitative measurement of SMN protein inside the cells. Of the two compounds, only compound No. 65 induced a significant increase in SMN protein (1.62-fold of the DMSO control) (Fig. 2A). Thus, subsequent studies were conducted only with compound No. 65, which was rigosertib (ON

Compound No. 65, rigosertib, increased SMN protein expression by inducing exon 7 inclusion.
We further investigated whether rigosertib could increase the expression of SMN protein by other mechanisms. To analyze the effect of rigosertib on SMN2 transcriptional activation, we constructed a luciferase reporter vector containing the SMN2 promoter by fusing with a PEST sequence to reduce the protein half-life of firefly luciferase. We found that there was no significant change in transcriptional activity with rigosertib (Fig. 2C), whereas the positive control histone deacetylase inhibitors, including SAHA and SB, induced marked effects without affecting cell viability (Supplementary Fig. S2A). A possible effect of rigosertib on SMN protein stability was also examined. Considering that degradation of SMNΔ7 is much more susceptible to the proteasome inhibition [29], we generated 293T cells stably expressing FLuc-fused SMNΔ7 protein (FLuc-SMNΔ7 stable cell lines). Luciferase activity was dramatically decreased by treatment with the protein synthesis inhibitor CHX alone, indicating the instability of SMNΔ7. Treatment with MG132 markedly increased the luciferase activity as a result of proteasome inhibition. In contrast, rigosertib had little effect on the degradation of SMNΔ7 (Fig. 2D) with no significant effect on cell viability (Supplementary Fig. S2B). These results clearly excluded the possible involvement of transcription and protein stability in the effect of rigosertib on SMN expression.
Generation of iPSCs from a type 1 SMA patient
To establish a physiologically representative in vitro human SMA model, we generated SMA iPSCs from type 1 SMA patient fibroblasts and differentiated SMA iPSCs into disease-relevant cell types for experimental validation of rigosertib. We reprogrammed SMA fibroblasts into iPSCs using nonintegrating oriP/EBNA-1-based episomal vectors (Supplementary Fig. S3A). The properties of SMA iPSCs were confirmed based on their hESC-like morphology and the expression of pluripotency markers, including AP, OCT4, NANOG, TRA-1-60, TRA-1-81, SSEA-3, and SSEA-4 (Supplementary Fig. S3B, C). In vitro and in vivo differentiation potential of the SMA iPSCs was confirmed by the presence of all three germ layers in EBs (Supplementary Fig. S3D) and teratomas (Supplementary Fig. S3E) derived from SMA iPSCs, respectively. STR analysis verified that SMA iPSCs were derived from the SMA patient's fibroblasts (Supplementary Fig. S3F), and cultured SMA iPSCs maintained a normal karyotype (Supplementary Fig. S3G). These results indicated the successful reprogramming of SMA fibroblasts into SMA iPSCs.
Development of in vitro SMA model using SMA iPSC-derived pMN
SMA is caused by low levels of SMN protein due to the loss of the SMN1 gene and inefficient splicing of the SMN2 gene, which primarily affects α-MNs of the lower spinal cord [39]. Therefore, to observe disease-relevant phenotypes, we differentiated WT iPSCs and SMA iPSCs into MNs through highly pure pMN (Fig. 3A and Supplementary Fig. S4A) using a previously described method with some modifications [33]. Control iPSCs derived from WT human fibroblasts and SMA iPSCs were differentiated into NEPs in the presence of SB431542 (inhibitor of activin-nodal signaling), DMH1 (inhibitor of bone morphogenetic protein signaling), and CHIR99021 (Wnt agonist), which were then treated with RA for caudalization and with Pur (SHH signaling agonist) for ventralization [40]. A robust population of OLIG2+ pMNs were isolated and suspended at 12 days after differentiation from iPSCs with almost no interneuron progenitors, labelled by NKX2.2. For terminal differentiation, OLIG2+ pMNs were treated with RA and Pur suspension to induce differentiation into HB9+ and ISL1+ premature MNs, which then were further differentiated into mature ChAT+ and SMI32+ MNs in the presence of compound E for 10 days (Fig. 3B and Supplementary Fig. S4B). Furthermore, the percentage of OLIG2+ and ChAT+ cells in the differentiated pMNs and MNs remained similar in WT and SMA groups with over 75.6% of OLIG2+ and 85.5% of ChAT+, respectively (Fig. 3C). These data imply that there was no significant difference in differentiation potential between WT and SMA groups. Importantly, most of the pMNs derived from WT iPSCs and SMA iPSCs were highly proliferative, as shown by Ki67 expression, and the pMNs were expanded for at least five passages, along with robust OLIG2 expression (Fig. 3D and Supplementary Fig. S4C). Furthermore, OLIG2+ pMNs can be frozen, thawed, and then terminally differentiated into mature ChAT+ MNs when needed.
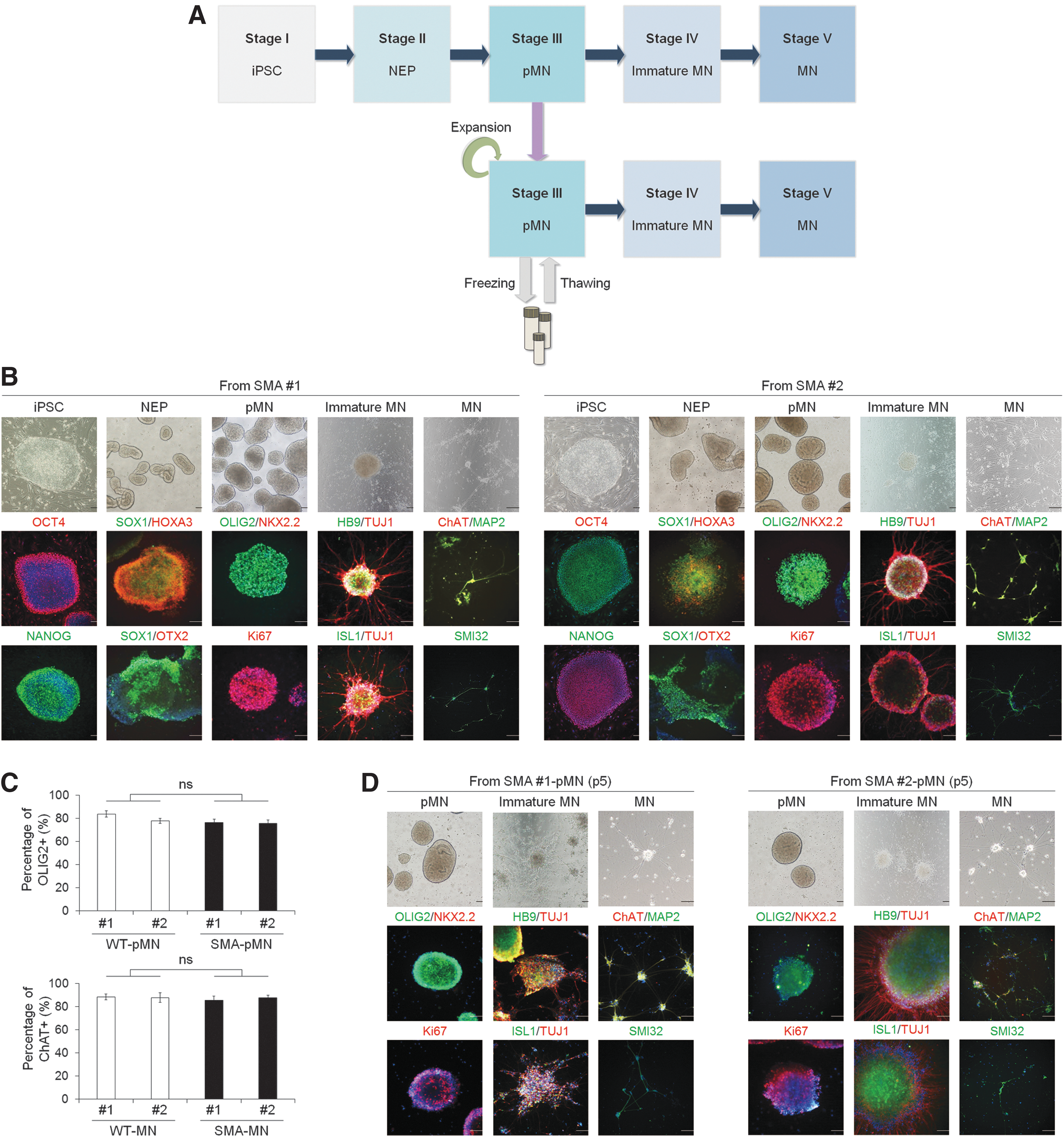
Differentiation of SMA iPSCs into pMNs and MNs.
During pMN and MN differentiation, the cells differentiated from SMA iPSCs exhibited different and even defective characteristics compared with WT cells. First, RT-PCR analysis revealed that SMA iPSC-pMNs and SMA iPSC-MNs had lower levels of SMN-FL transcripts, including SMN1 and SMN2, with SMN-FL band intensities of 21.54% in SMA#1-pMNs and 26.55% in SMA#2-pMNs compared with WT iPSC-pMNs and 33.98% in SMA#1-MNs and 25.44% in SMA#2-MNs compared with WT iPSC-MNs, respectively, and higher levels of SMNΔ7 transcripts (Fig. 4A). Second, at the protein level, the expression of SMN-FL protein in SMA iPSC-pMNs and SMA iPSC-MNs was significantly decreased, with reductions of 16.82% in SMA#1-pMNs and 35.19% in SMA#2-pMNs, compared with that in WT pMN controls and 27.73% in SMA#1-MNs and 30.27% in SMA#2-MNs, compared with that in WT MN controls, respectively, as shown in western blot (Fig. 4B) and immunocytochemistry (Fig. 4C) analyses. Nuclear gem bodies were rarely present in SMA iPSC-pMNs and SMA iPSC-MNs (Fig. 4C). These results showed a reduced level of functional SMN-FL protein in SMA iPSC-pMNs and SMA iPSC-MNs. Third, as reported in this study and previously by another group [20,41], enhanced cell death was observed in the SMA cells (1.59-fold in SMA#1-pMNs, 1.77-fold in SMA#2-pMNs and 1.71-fold in SMA#1-MNs, 1.76-fold in SMA#2-MNs, respectively) compared with WT cells (Fig. 4D). Fourth, the neurite length in SMA iPSC-MNs was much shorter (∼70.92% in SMA#1-MNs and 79.77% in SMA#2-MNs, respectively) compared with WT#1 iPSC-MNs (Fig. 4E), consistent with previous studies [17,42]. Furthermore, when terminally differentiated WT and SMA iPSC-MNs were cocultured with differentiated myotubes from mouse C2C12 cells for the formation of NMJ, we observed that aggregated α-BTX acetylcholine receptors (AChRs) on myotubes were overlapping with ChAT neurites in WT iPSC-derived MNs, suggesting the formation of NMJ. However, AChR clustering on myotubes cocultured with SMA iPSC-derived MNs was remarkably impaired (Supplementary Fig. S5). In addition, we evaluated whether the terminally differentiated MNs from WT and SMA iPSCs have functional membrane properties using electrophysiological analysis. When currents (from −0.1 to 0.2 nA with 0.02 nA steps for 500 ms) were injected into MNs, an action potential was fired (Vm >0 mV) on WT iPSC-derived MNs for up to 0.2 nA; however, the multiple action potentials were induced on SMA iPSC-derived MNs (Supplementary Fig. S6A, B). The average Na+ current (I Na) in control MNs was −54.8 ± 6.4 pA/pF for WT#1 iPSC-MNs and −53.7 ± 8.3 pA/pF for WT#2 iPSC-MNs, compared with −74.3 ± 14.7 pA/pF in SMA#1 iPSC-MNs and −85.2 ± 10.8 pA/pF in SMA#2 iPSC-MNs (Supplementary Fig. S6C–E). Thus, our data demonstrate that SMA MNs are more readily excitable than WT MNs with higher action potential firing and I Na current density in accordance with previous reports [43]. Taken together, these data imply that both SMA iPSC-pMNs and SMA iPSC-MNs in our culture system were useful for analyzing SMA pathology and could be applied to an in vitro disease model. SMA iPSC-pMNs were selected for further experiments due to their advantages, such as the more significant reduction in SMN-FL transcripts and proteins and their ability to quickly yield a sufficient quantity for performing cell culture-based assays.
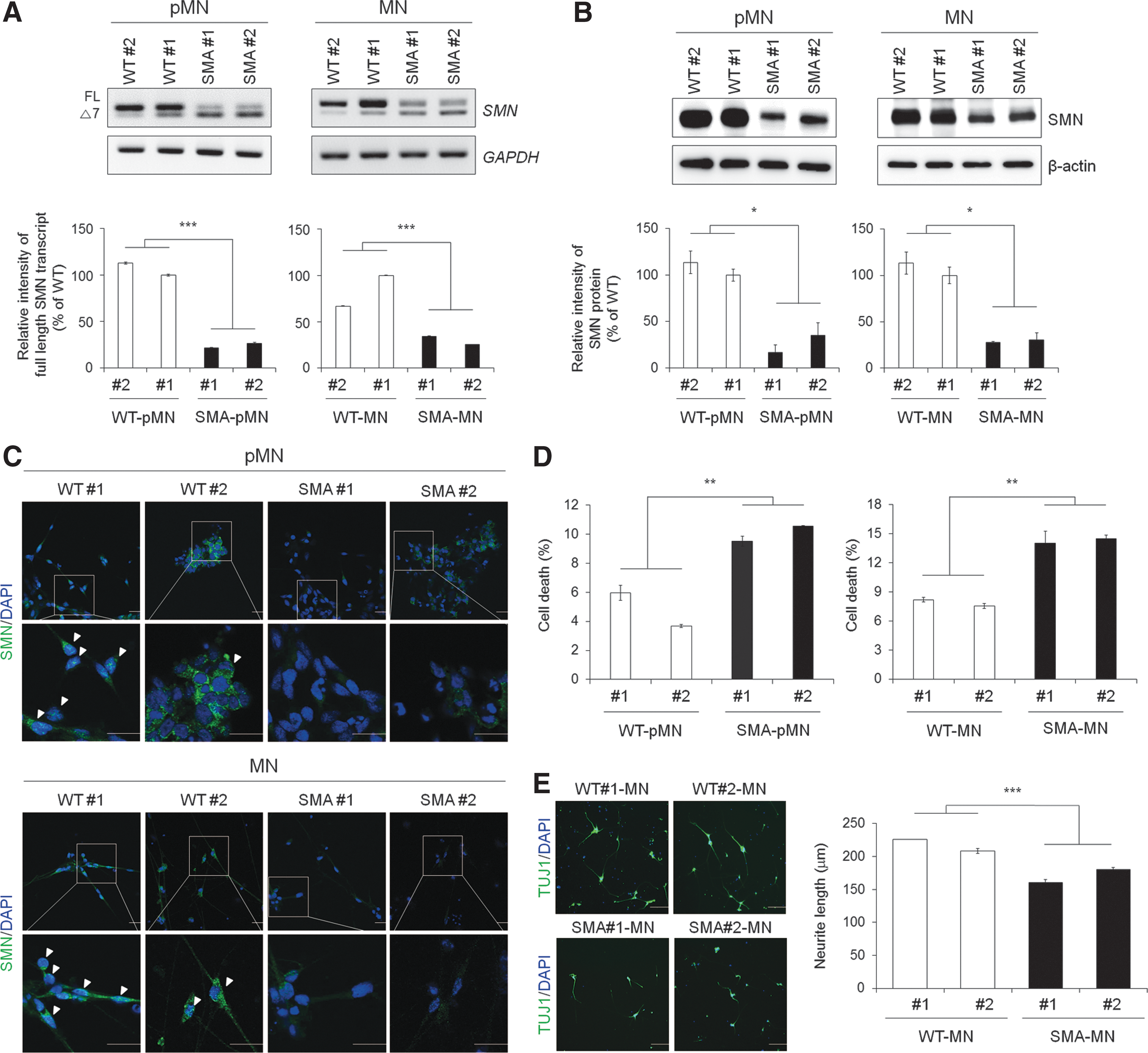
Phenotypic analysis of WT and SMA iPSC-derived pMNs and MNs.
Cellular damage in SMA iPSC-pMNs caused by the splicing defect of SMN2 can be rescued by rigosertib treatment
The selected candidate rigosertib from the primary screening using the SMN2-Luc stable cell lines was demonstrated to exert significant splicing correction activity for SMN2. Therefore, we proceeded to test whether rigosertib could alleviate the defective phenotypes by inhibiting splicing of the SMN2 exon 7 in SMA iPSC-pMNs capable of mimicking the disease phenotypes in vitro. Rigosertib increased SMN-FL mRNA levels in SMA iPSC-pMNs after 72 h of treatment (Fig. 5A). qRT-PCR analysis using the forward primer spanning exon 7 and 8 and the reverse primer for exon 8 [44] revealed that SMA#1 iPSC-pMNs and SMA#2 iPSC-pMNs had lower levels of SMN-FL transcripts with 74.21% and 50.42% reductions, respectively, compared with WT#1 iPSC-pMNs (Fig. 5B). There is no significant difference between WT controls. Rigosertib increased mRNA expression of SMN-FL, by the inclusion of exon 7 in SMA#1 iPSC-pMNs and SMA#2 iPSC-pMNs, by 2.78- and 1.46-fold, respectively. Rigosertib treatment increased the amount of SMN protein in the patient-derived pMNs, as determined by western blot analysis (Fig. 5C) and immunocytochemistry (Fig. 5D), and restored gem body formation in SMA iPSC-pMNs (Fig. 5D). To dissect the protective role of rigosertib in SMA, we examined the effect of rigosertib on the cell death of MNs derived from SMA iPSCs. Application of rigosertib significantly decreased the percentage of dead cells in SMA iPSC-pMNs from 12.86% to 7.24% in SMA#1- and SMA#2-pMNs, respectively, to 9.21% and 3.82% in rigosertib-treated SMA#1- and SMA#2-pMNs, respectively (P < 0.05) (Fig. 5E). Therefore, to understand the mechanisms underlying the cell death caused by reduced levels of SMNs, we compared mitochondrial superoxide production, which is known to be implicated in the functional defects of spinal MNs, in SMA iPSC-pMNs using MitoSOX Red, a mitochondrial superoxide indicator. Significantly more MitoSOX+ cells were observed in SMA iPSC-pMNs than in WT iPSC-pMNs (1.40- and 1.21-fold, P < 0.05) (Fig. 5F). Rigosertib treatment in SMA iPSC-pMNs decreased the generation of MitoSOX+ cells to a similar level to the WT control (P < 0.05) (Fig. 5F). Moreover, rigosertib treatment protected against SMN deficiency-induced reduction of neurite outgrowth in terminally differentiated SMA iPSC-MNs, showing a phenotype closer to differentiated WT iPSC-MNs (Fig. 5G). These data suggest that the SMN2 splicing modifier rigosertib can modulate SMN2 splicing in favor of exon 7 inclusion and thereby increase the production of SMN protein in an in vitro human SMA model and further ameliorate the cell death induced by mitochondrial oxidative stress, which is involved in the degeneration of MNs [21].
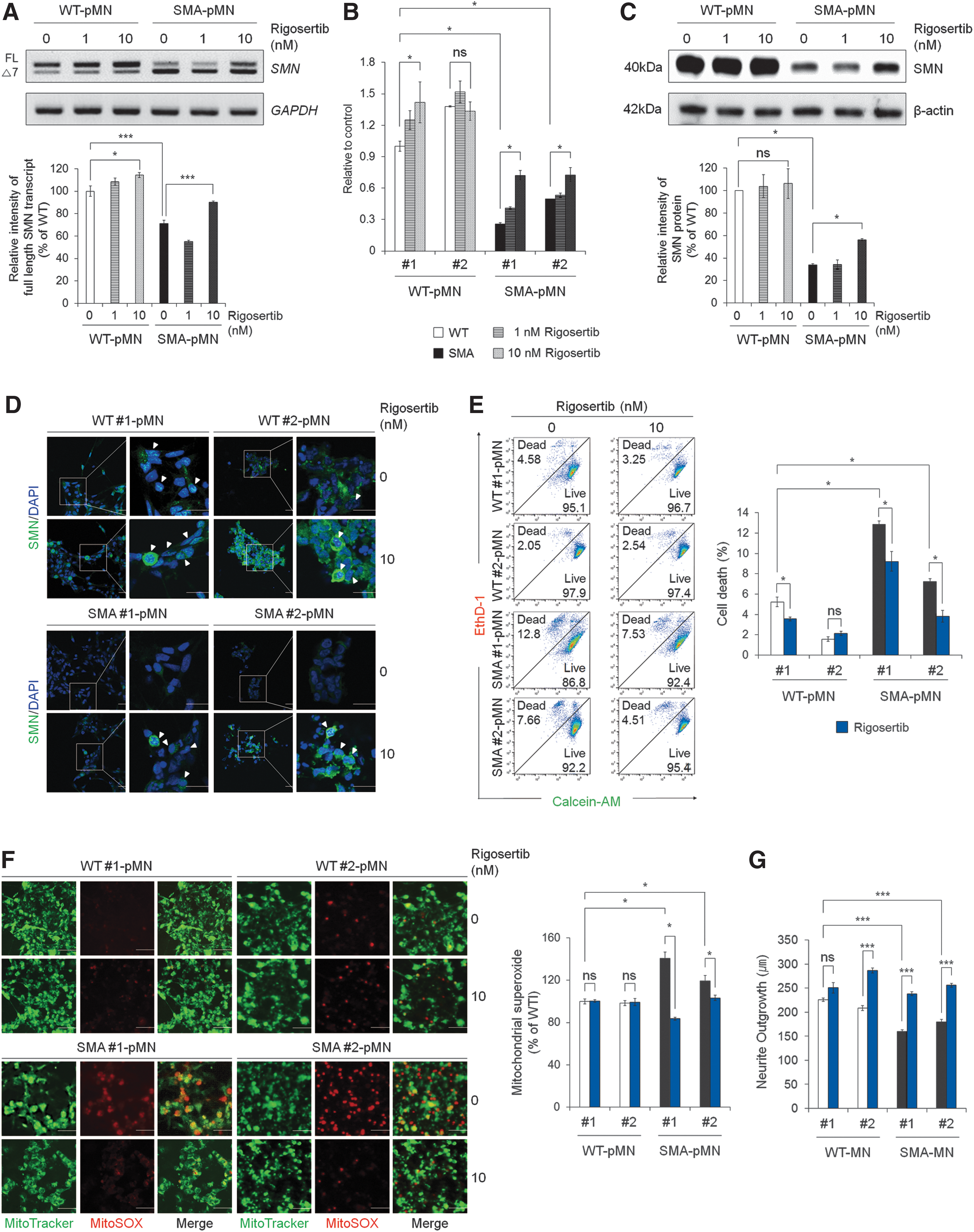
Restoration of SMN2 exon 7 splicing rescued SMA-like phenotypes in SMA iPSC-pMNs by rigosertib treatment.
Intraperitoneal injection of rigosertib increases SMN protein levels in the spinal cord of SMA mice
To examine whether rigosertib increases the amount of SMN protein in vivo, SMNΔ7 transgenic mice at postnatal day 3 were intraperitoneally administered with 10 mg/kg of rigosertib over the course of 8 days. The SMNΔ7 transgenic mouse is the most prevalently used SMA mouse model that presents a severe type of SMA and survives for only 2 weeks [45]. Five hours after the final rigosertib dosing, the spinal cord extract was prepared and subjected to quantitative western blot analysis with the SMN antibody. The amount of SMN protein in the spinal cord of rigosertib-treated SMA mice was found to be increased by 2.13-fold compared with that of SMA mice (Fig. 6A, B). In addition, we evaluated spinal cord tissue sections from the same mice, histologically. High SMN expression was observed in spinal cord tissue from heterozygous mice, whereas the SMN protein was very weakly expressed in spinal cords from the SMA mice (Fig. 6C). Consistent with western blotting analysis, SMN+ cell population in the spinal cord was significantly increased in rigosertib-treated mice compared to the SMA mice by 3.18-fold (Fig. 6C, D), indicating the effectiveness of rigosertib in vivo.
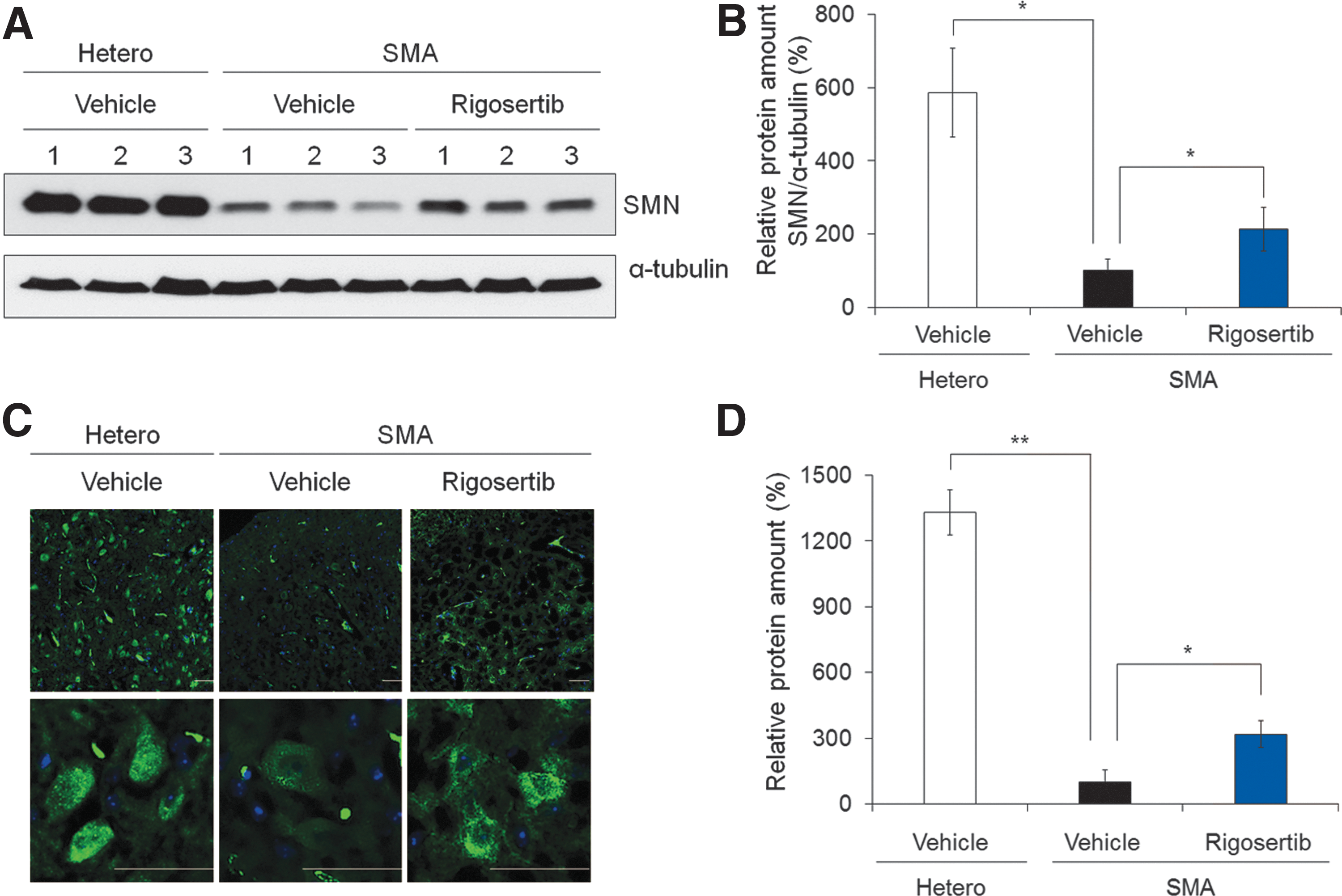
Rigosertib increases the amount of SMN protein in the spinal cord of SMA mice.
Discussion
Despite the development of promising therapeutic strategies, SMA remains a noncurative neuromuscular disease with no available FDA-approved drug treatment. Although SMA is caused by lack of the SMN1 gene, SMN2 is the primary disease-modifying gene in humans [46]. However, these two SMN genes, SMN1 and SMN2, encode SMN protein in humans, whereas most animals have only the SMN1 gene [3]. This difference makes it difficult to develop proper in vivo models capable of faithfully recapitulating human SMA disease. In addition, MNs are the primarily affected cell type in SMA, but are fundamentally difficult to obtain. Patient-specific iPSCs obtained through reprogramming technology provide a unique opportunity to model human diseases in vitro by directing iPSCs into disease-relevant cell types. Therefore, MNs differentiated from SMA iPSCs represent a useful in vitro SMA model for elucidating the molecular and pathological processes associated with SMA and for screening novel therapeutic drug candidates [47].
As SMA is fundamentally caused by reduced SMN protein levels, the recovery of SMN2 splicing is one of the most promising therapeutic approaches for treating SMA. Accordingly, the administration of small molecules [44,48,49] and antisense oligonucleotides [50,51] that promote the inclusion of exon 7 into SMN2 mRNA has been reported to correct the SMA phenotypes in mouse models. Two to six copies of the SMN2 gene have been observed in patient somatic cells [52], and SMN protein levels in patients are generally correlated with SMN2 gene copy number [53]. In this study, two SMA patient-specific iPSC lines were generated from two SMA type 1 patient's fibroblasts with homozygous deletion of exons 7 and 8 of the SMN1 and two or three SMN2 copies. This genotype did not affect the differentiation of spinal MNs, consistent with a previous report [43]. SMA iPSC-pMNs were generated and characterized as a human SMA model and were used as a testing platform for the validation of the candidate drug, a SMN2 splicing modifier. We observed relevant SMA phenotypes due to decreased amounts of SMN-FL transcript in SMA iPSC-pMNs, such as increased cell death and reduced neurite length, impaired formation of NMJ, and hyperexcitability in SMA iPSC-MNs, in line with previously observed phenotypes [17,18,20,41,42,54]. Moreover, using our model system, we identified augmented mitochondrial reactive oxygen species (ROS) production in SMA iPSC-pMNs, leading to MN death, as observed in SMA.
In this study, we report the discovery of a novel small molecule that specifically restored SMN2 splicing in a robust high-throughput cell-based assay system using SMN2-Luc stable cell lines. Out of the 88 tested small molecules, rigosertib was the most efficient compound, selectively modulating SMN2 splicing to include exon 7 and subsequently increasing SMN protein levels. Rigosertib inhibits the activity of multiple kinases, such as PLK1, by affecting multiple signaling cascades leading to cancer cell death [55,56]. Orally bioavailable rigosertib [57] has recently entered clinical trials against solid tumors and hematological malignancies such as myelodysplastic syndrome and has demonstrated promising results [58 –60]. However, our results demonstrate that its activity in SMN2 splicing modulation is not a result of PLK inhibition, the transcription of SMN2, or the stability of SMN protein. Future identification and characterization of rigosertib as an SMN2 splicing modifier will help to clarify the molecular basis of its specificity.
Increased SMN levels by rigosertib treatment ameliorated phenotypes associated with SMN1 gene deletion in a human in vitro SMA disease model. The rescue of MN death by the reduction of mitochondrial oxidative stress in SMA patient-specific pMNs treated with rigosertib suggested the possible use of this small molecule for SMA treatment. Recent studies have described dysfunction of mitochondria in mouse MNs with knocked down SMN expression using small interfering RNA, implying that SMN is required for normal mitochondrial function [61]. In addition, patients with SMA were reported to have less mitochondrial DNA [62,63] and markedly increased levels of oxidative stress [64]. The application of an antioxidant, such as N-acetylcysteine, has many beneficial effects on MNs, such as reducing mitochondrial oxidative stress in hESC-based SMA model [21]. These observations raise the possibility that mitochondrial dysfunction in SMA models may be a direct consequence of the disruption of SMN functions. Indeed, rigosertib corrected the alternative splicing defects of SMN2 exon 7 and significantly reduced mitochondrial oxidative stress. To the best of our knowledge, this is the first study reporting the effects of rigosertib treatment as an SMN2 splicing modifier in an in vitro human SMA model. Furthermore, the administration of rigosertib to a SMNΔ7 SMA mouse model led to an increase in SMN protein levels in the spinal cord. However, further experiments are needed to address the relationship between the pathogenesis of SMA by the loss of MNs and abnormal mitochondrial function in SMA and to elucidate the functions of rigosertib in recovering disease-associated phenotypes.
In summary, we established an advanced drug screening and validation platform based on an SMN2 splicing-targeting approach using the SMN2-Luc stable cell line and an in vitro human SMA model (SMA iPSC-pMNs) and showed that rigosertib can restore SMN protein levels by promoting SMN2 exon 7 inclusion. Our data provide evidence that SMA iPSC-pMNs recapitulate the disease-specific phenotypes, such as increased cell death and mitochondrial ROS production, and that such phenotypes can be reversed by rigosertib. Our findings may contribute to the development of effective therapies for SMA and other neuromuscular disease.
Footnotes
Acknowledgments
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (2015M3A9C7030128, NRF-2018M3A9H3023077, NRF-2016K1A1A8A01938649) and a grant from KRIBB Research Initiative Program. The funders had no role in the study design, data collection or analysis, decision to publish, or preparation of the article.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.