
Abstract
Dental pulp has been revealed as an accessible and a rich source of mesenchymal stem cells (MSCs) and its biological potential is currently under intense investigation. MSCs from dental pulp stem cells (DPSCs) have been indicated as a heterogeneous population oriented not only in repairing dentine but also in maintaining vascular and nervous homeostasis of the teeth. We sought to verify the phenotype of cells isolated from dental pulp of young donors and to investigate in vitro their role as pericyte-like cells. Specifically, we evaluated how culture conditions can modulate expression of pericyte markers in DPSCs and their capacity to stabilize endothelial tubes in vitro. DPSCs cultured in standard conditions expressed MSC markers and demonstrated to contain a population expressing the pericyte marker NG2. These DPSCs were associated with low sprouting capacity in extra-cellular (EC) Matrix and limited ability in retaining tubes formed by endothelial cells in a coculture angiogenesis model. When cultured in endothelial growth medium (EGM)-2, DPSCs significantly upregulated NG2, and partially alpha-smooth muscle actin. The resulting population conserved the stem marker CD73, but was negative for calponin and endothelial markers. EGM-2-conditioned DPSCs showed a higher sprouting ability in EC Matrix and efficient association with human umbilical vein endothelial cells allowing the partial retention of endothelial tubes for several days. Among growth factors contained in EGM-2 we identified basic fibroblast growth factor (bFGF) as mainly responsible for NG2 upregulation and long-term stabilization of endothelial tubes. According to the in vitro analysis, DPSCs represent an effective source of pericytes and the appropriate culture conditions could result in a population with a promising ability to stabilize vessels and promote vascular maturation.
Introduction
Mesenchymal stem cells (MSCs) are multipotent stem cells that conserve in the adult the ability to differentiate into cells of several mesodermal tissues, including cartilage, bone, skeletal, and cardiac muscles. MSC's differentiative potential is under intense investigation to exploit its role in facilitating the repair of injured tissues; indeed, the inoculation of MSCs has been already proposed as therapeutic method in various diseases [1 –3].
Despite many efforts, the difficulty of the in vitro precise characterization and the variability in tissue uptake significantly limit the clinical use of MSCs. The recent finding of dental pulp as a potential source of pluripotent stem cells [dental pulp stem cells (DPSCs)], has increased the number of potential tools for regenerative medicine [4,5]. DPSCs originate from the neural crest, and they are physiologically involved in the homeostasis of dentine [6], but it has been also demonstrated that they contribute to bone remodeling [4]. In vitro, DPSCs have several advantages that make them good candidates as a source MSCs: noninvasive and easy isolation procedure, and a high proliferative rate. However, several studies have suggested that fresh isolated DPSCs are a quite heterogeneous stem cell population that includes several cell subpopulations with specific characteristics that can express different surface antigens; this characteristic probably results from embryonic origin of DPSCs. Indeed, neural crest cells originate in the ectoderm and after a phase of epithelial–mesenchymal transition and migration, they settle down in different parts of the body, including dental pulp. The ectodermal origin of DPSCs is confirmed by their potential to differentiate into neural cells [7]. In particular, DPSC subpopulations have been found to express CD271, a receptor for neurotrophins, a family of protein growth factors that stimulate the survival of neuronal cells [8]. Several studies indicate that intrinsic positional information can determine final cell phenotype [9] and that neural crest populations can produce also mesenchymal derivatives suggesting the importance of environmental signals in regulating neural crest development and differentiation [10].
Even though the DPSCs have been phenotypically and morphologically investigated in several studies, there are no specific and widely accepted biomarkers available for their characterization [5]. It is well known that these cells express typical markers of mesenchymal and bone marrow stem cells, including STRO-1 and CD146, as well as the embryonic stem cell marker, OCT4. Among typical surface markers expressed by MSCs, DPSCs express also CD44, CD73, CD90, and CD105 [11,12].
DPSC's differentiation into endothelial lineage has been proved in numerous in vitro and in vivo models [13,14]. In addition, it has been postulated an angiogenic role of DPSCs through a paracrine mechanism [15]. More intriguingly, a perivascular origin has been reported for DPSCs that seem to express typical perivascular markers, including alpha-smooth muscle actin (α-SMA), NG2, platelet-derived growth factor receptors (PDGFR-β), and CD146 [16]. In particular, Shi and Gronthos demonstrated that the majority of DPSCs expressed the pericyte marker, 3G5, confirming their similarities with perivascular mural cells [17]. In parallel, pericytes seem to have multipotent stem cell-like characteristics and some authors have suggested that pericytes themselves are precursors of MSCs in vivo [18].
It is well known that pericytes have a fundamental role in contributing to angiogenesis during several phases of development, in tissue regeneration, and in pathological conditions [19]. Pericytes, when activated, can migrate through the basal lamina and differentiate into smooth muscle cells or other progenitor cells; also, activated pericytes can migrate toward endothelial cells and interacting with them allow vessel stabilization [20]. Similarly, specific MSCs obtained from adipose tissue human adipose stem cells (hASCs) and adult human bone marrow stem cells (hBMSCs), when cocultured in vitro with endothelial cells, support their organization into perivascular-like structures differentiating themselves into mural cells [21].
Although pericytes have represented the focus of several investigation, to date there is no concordance about their univocal molecular characterization. Usually pericyte phenotype prediction was based on negative expression of hematopoietic and endothelial surface markers (CD45 and CD31/CD34) and positive expression of CD146, NG2, and α-SMA [22]. Also, to distinguish three subsets of human pericytes, the combined analysis of NG2 and α-SMA was proposed, associated with capillaries (NG2+αSMA−), venules (NG2−αSMA+), and arterioles (NG2+αSMA+) [18].
In our study, we aimed to explore the phenotypic and functional characteristics of human MSCs isolated from dental pulp (DPSCs). In particular, we investigated the ability of different culture media conditioning DPSCs toward a pericyte-like phenotype with particular attention about pericytes–endothelial cell interaction that is a paradigm of vessel stabilization.
Materials and Methods
Cell isolation and culture
Human DPSCs were obtained from dental pulp harvested from third molar of randomly selected young patients (13–19 years old) recruited at Department of Science Dentistry and Maxillofacial, “Sapienza” University of Rome. All samples were obtained after written informed consent of patients or, in case of minors, from the next of kin or caretakers. The study was approved by the Ethics Committee of the “Sapienza University,” Rome, Italy (RIF:CE:4336). DPSCs were isolated as previously described [23,24]. In brief, the pulp was gently removed from the tooth under sterile conditions, placed in a Petri plate, and divided into small pieces with surgical scalpel. Hank's solution (Life Technologies, Monza, Italy) was added to the specimen for 2 h at 37°C in the presence of 5% CO2, and then incubated with 0.1% type IV collagenase (Sigma-Aldrich, Milan, Italy) for 15 min at 37°C in 5% CO2. After washing, the pellet was suspended in completed DMEM-L 10% fetal bovine serum (FBS) and seeded in 25-cm2 flasks (Sarstedt, Verona, Italy). After 3 days, several adherent cell clones were observed. When the adherent cells reached the confluence, approximately within 7 or 12 days, the cells were passed for expansion and immunophenotyped by flow cytometric analysis. DPSCs were utilized only if positive for CD90, CD44 (Millipore, Milan, Italy), CD73 (Cell Signaling Technology, Danvers), and CD105 (BD Biosciences, Milan, Italy), and negative for CD14, CD19 (Millipore), and CD34 (Miltenyi Biotec). DPSCs were used from passage 6 to 12. DPSCs (from sixth passage) were cultured in 10% FBS supplemented with Dulbecco's modified Eagle's medium (DMEM) until the 10th passage. Some plates were exposed to different culture conditions for 2 weeks using complete endothelial medium (EGM-2) (EGM-2-conditioned dental pulp stem cells (DPSC) [E-DPSC]) or cultured in 10% FBS-supplemented DMEM (DMEM-conditioned DPSC [D-DPSC]). EGM-2 was prepared using endothelial basal medium (EBM)-2 basal medium with the supplementation of hydrocortisone, human bFGF, vascular endothelial growth factor (VEGF), insulin-like growth factor (IGF)-1, ascorbic acid, human epidermal growth factor (EGF), gentamicin/amphotericin (GA)-1000, and heparin following medium instructions. In addition, DPSCs was differently incubated with the endothelial basal medium EBM-2 in which were routinely added IGF-1, ascorbic acid, GA-1000, and heparin enriched or not with EGF, fibroblast growth factor (FGF), and VEGF that we singularly added to the medium. Also, we treated D-DPSCs with transforming growth factor-β (TGF-β) (10 ng/mL) for 48 h. Human umbilical vein endothelial cells (HUVECs) (Lonza, Walkersville) were cultured in EGM-2 growth medium (Lonza). The HUVECs were used from passage 3 to 12.
Immunomagnetic separation of NG2+ subpopulation of DPSCs
DPSCs treated with EGM-2, EBM-2+ FGF, and DMEM were immunomagnetic separated by the MACS Cell Separation Kit (Miltenyi Biotec). Briefly, after passing DPSCs through 30 μm nylon mesh to remove cell clumps, 107 total cells were counted and incubated with MicroBeads conjugated to monoclonal human NG2 antibody for 30 min at 4°C under slow and continuous rotation. Subsequently, MACS Column was placed in the MidiMACS separator, cell suspension was passed through the column, and unlabeled cells were collected. The column was removed and NG2+-labeled cells were collected. After separation, DPSCs were immunophenotyped by flow cytometric analysis as described below.
Quantitative assessment of islet-like structures on EC Matrix
Early DPSC (6 and 7 passages), D-DPSC and E-DPSC suspensions were placed on extra cellular (EC) Matrix (Millipore) following polymerization of wells in specific media with low serum concentration. After 18 h, cells formed islet-like structures. These structures were cultured until 24 h, 48 h, and 6 days and then fixed in 4% paraformaldehyde. Sprouts were analyzed using a plugin for sprout analysis developed and available from the angiogenesis update site within ImageJ as described [25].
We measured the mean number of sprouts on pictures taken on day 1, 2, and 6 of human dental pulp stem cells (hDPSCs), D-human dental pulp stem cells (D-hDPSCs), and E-human dental pulp stem cells (E-hDPSCs) in EC Matrix.
Fluorescence-activated cell sorting analysis
DPSCs, treated or untreated, were fixed with 4% paraformaldehyde and characterized by flow cytometry analysis. After washing, cells were incubated with mouse allophycocyanin (APC)-conjugated anti-CD34 moAb (Miltenyi Biotec), mouse anti-CD90 moAb (Millipore), mouse anti-NG2 moAb (Santa Cruz Biotechnology, Germany), rabbit anti-calponin moAb (Abcam, Cambridge) and rabbit anti-CD73 moAb (Cell Signaling Technology), for 1 h at 4°C, followed by phycoerythrin (PE)-conjugated anti-mouse IgG H&L or Cy5-conjugated anti-rabbit IgG H&L (Abcam) for additional 45 min. Alternatively, DPSCs, treated or untreated, were used to perform a double staining with rabbit anti-CD73 moAb (Cell Signaling Technology) and mouse anti-NG2 moAb (Santa Cruz Biotechnology) for 1 h at 4°C, followed by PE-conjugated anti-mouse IgG H&L and Cy5-conjugated anti-rabbit IgG H&L (Abcam) for additional 45 min.
Moreover, human DPSCs, immunomagnetic separated with NG2 microbeads as described above, were fixed with 4% paraformaldehyde and after washing, were incubated with mouse anti-NG2 (Santa Cruz Biotechnology), mouse anti-CD146 (Millipore), and mouse anti-PDGFRβ (Santa Cruz Biotechnology) followed by PE-conjugated anti-mouse IgG H&L (Abcam) for additional 45 min.
Also, multipotent mesenchymal stromal-specific surface antigens were quantified in DPSCs by flow cytometry analysis. In brief, DPSCs after 28 days of the pulp separation were fixed with 4% paraformaldehyde. After washing, cells were incubated with mouse anti-CD105 moAb (BD Biosciences) anti-CD44, anti-CD90 (Millipore), or rabbit anti-CD73 moAb (Cell Signaling Technology) for 1 h followed by PE-conjugated anti-mouse IgG H&L (Abcam) or Cy5-conjugated anti-rabbit IgG H&L (Abcam) for additional 45 min. Mouse anti-CD14, anti-CD19 moAb (Millipore), and APC-conjugated anti-CD34 moAb (Miltenyi Biotec), were used as negative control.
All samples were analyzed with a FACScan cytometer (BD Accuri C6 Flow cytometer) equipped with a blue laser (488 nm) and a red laser (640 nm). At least 20,000 events were acquired.
Western blotting
Whole cell lysates, obtained from all experimental groups of HUVEC and DPSC populations, were processed for western blot analysis. Cells were suspended in a buffer containing 1% Triton, 0.1% sodium dodecyl sulfate (SDS), 2 mM CaCl2, and 100 mg/mL PMSF. Protein content was determined using the Protein Assay Kit 2 (Bio-Rad Laboratory, Hercules, CA). Thirty micrograms of protein extract from each sample were separated in 8% SDS-polyacrylamide gel and then transferred to a nitrocellulose membrane (Whatmann, Dassel, Germany). The following antibodies (Abs) were detected: mouse anti-human NG2 (Santa Cruz Biotechnology), rabbit anti-alpha-SMA (Cell Signaling Technology), mouse anti-N-cadherin (Santa Cruz Technology), and mouse anti-von Willebrand factor (vWF) (Santa Cruz Biotechnology). horseradish (HRP)-conjugated anti-mouse secondary antibody was from Cell Signaling Technology. The membranes were visualized using a chemiluminescent detection system (Euroclone, Milan, Italy). Anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody was used as control of protein loading.
In vitro angiogenesis and binding assay
For tube formation assay, HUVECs (1 × 104 cells/well) were seeded alone or in coculture with E-DPSCs and D-DPSC (3 × 103 cells/well) on polymerized EC Matrix (Millipore) 15-well microslides (10 μL/well) of ibidi (Munich, Germany) following the manufacturer's instructions in EGM-2 media (Lonza). The cells were incubated at 37°C, 5% CO2 in a humidified atmosphere, vascular network photographed at indicated time points using a camera-equipped microscope (Nikon), and quantified using ImageJ angiogenesis analysis software. In particular, we determined and compared pictures taken on various times, the total tube length, the Mesh index (total master segments length/number of master segments), and the percentage of retention of total tube length of HUVEC alone and in coculture with D-DPSC and E-DPSC. For some experiments, to distinguish the different behavior of HUVEC, as single cells or in coculture, we used prelabeled E-DPSC. Cells were washed with phosphate-buffered saline (PBS), centrifuged, and resuspended in PBS with Calcein (2 μg/mL) for 10 min. After incubation, cells were centrifuged, washed with PBS, and seeded in coculture with HUVEC. Then, they were investigated for their ability to form and/or stabilize vascular networks with a Life Technology Floyd microscope.
Immunofluorescence
Coculture of HUVEC and E-DPSC were seeded on EC Matrix. After 18 h, they were fixed with paraformaldehyde 4% solution for 10 min, washed with PBS and permeabilized with a solution of 0.1% Triton-X (Sigma-Aldrich) for 10 min. After washing, cells were incubated overnight with primary antibody directed against NG2 (Santa Cruz Biotechnology). After extensive washings with PBS, cells were treated with fluorescein-labeled IgG secondary antibody (1:100 in PBS containing 3% bovine serum albumin) for 30 min at room temperature. After several rinses, cells were mounted with VECTASHIELD mounting medium containing DAPI and photographed with a fluorescence microscope (AXIOPHOT; Zeiss).
Transwell migration assay
NG2+ DPSCs subpopulation separated by MACS was maintained in EGM-2, EBM-2+ FGF, and DMEM for 2 weeks. The three NG2+DPSC experimental groups were treated or not for 24 h with the inhibitor of fibroblast growth factor receptor (FGFR), AZD4547, and migration ability was determined by using transwell chamber assay (24-well plate, 8-μm pore size). DPSCs (NG2+ DPSCs as experimental groups and NG2− DPSCs as control) were resuspended in serum-free medium and adjusted to a density of 1 × 105 cells/mL. One hundred fifty microliters of cell suspensions were added to the upper chamber of the transwell insert placed in a companion plate. We diluted FGFR inhibitor at 1 and/or 2 μM in serum-free medium and we added 150 μL of this solution to the cells in the top chamber of the insert to give a final volume of 300 μL. EC serum-free medium was added to untreated inserts. To the bottom chamber of each insert, we added 800 μL of DMEM containing 5% FBS as chemoattractant. Cells were allowed to migrate through coated filters for 16–18 h. The cells attached on the lower membrane surfaces were fixed with methanol and stained with 0.1% Crystal Violet w/v in 0.1 M borate, pH 9.0, and 2% ethanol v/v for 20 min at room temperature. Cells were counted at 400 × magnification with standard optical microscopy and the average number of cells per field in five random fields was recorded. Triplicate filters were used and the experiments were repeated three times.
Statistical analysis
Mean values and standard deviation for vessel and sprout counts, mesh index, and tube length were determined for each analysis. All experiments were done at least in triplicates and data analyzed by Student's t-test.
Results
Different culture conditions modify DPSC phenotype
More than 90% of cells isolated from single dental pulp, when analyzed at sixth passage, resulted CD44+, CD105+, CD73+, CD90+, and CD14−, CD19−, CD34−, in accordance with MSC-like phenotype. In our experiments we used only DPSCs that, initially, after flow cytometry analysis, displayed MSC phenotype (early DPSCs) (Supplementary Fig. S1). Following the sixth passage, DPSCs (from sixth passage) were cultured in 10% FBS supplemented DMEM until 10th passage and then distributed in two different culture conditions: 10% FBS-supplemented DMEM (D-DPSCs), or EGM-2 medium (E-DPSCs). The cells were cultured in these conditions for 2 weeks, then, sprouting ability of D-DPSCs and E-DPSCs from the same passage (12th) was analyzed (Fig. 1A). E-DPSCs demonstrated higher sprouting ability with respect to both low-passage DPSCs (early DPSCs) and D-DPSCs with a significant higher number of sprouts, as early as 24 h (Fig. 1B). After few days, E-DPSCs began to form tube-like structures also, with several islets connected with cell bridges, and these networks remained visible for up to 7 days of culture. Tube networks formed by E-DPSCs in EC Matrix were not present in early DPSCs or D-DPSCs. To verify the phenotypic characteristics distinguishing E-DPSCs from D-DPSCs, we performed flow cytometry and western blot analyses for selected markers. As reported in Fig. 2A both, D-DPSCs and E-DPSCs were CD34−. However, E-DPSCs resulted significantly different from D-DPSCs for the reduction in CD90 (17% vs. 58%) and the upregulation of NG2 (71% vs. 45%). In addition, calponin, a marker of smooth muscle cells, was absent in both cell populations. Although in E-DPSC population the expression of CD73 was slightly decreased with respect to D-DPSCs (80% vs. 94%), the number of NG2+CD73+ cells increased (75% vs. 52%) (Fig. 2B). Western blot analysis confirmed a higher expression of NG2 in E-DPSCs with respect with D-DPSCs and early DPSCs (Fig. 2C). In addition, E-DPSCs with respect to D-DPSCs, expressed more α-SMA and high levels of N-cadherin, whereas vWF antigen was nearly absent in both cell types. E-DPSC phenotype resembled that of DPSCs treated for 48 h with TGF-β, although the latter expressed a higher level of α-SMA. These results suggested that EGM-2 was able to stimulate the enrichment of the initial DPSC population with pericyte-like cells.
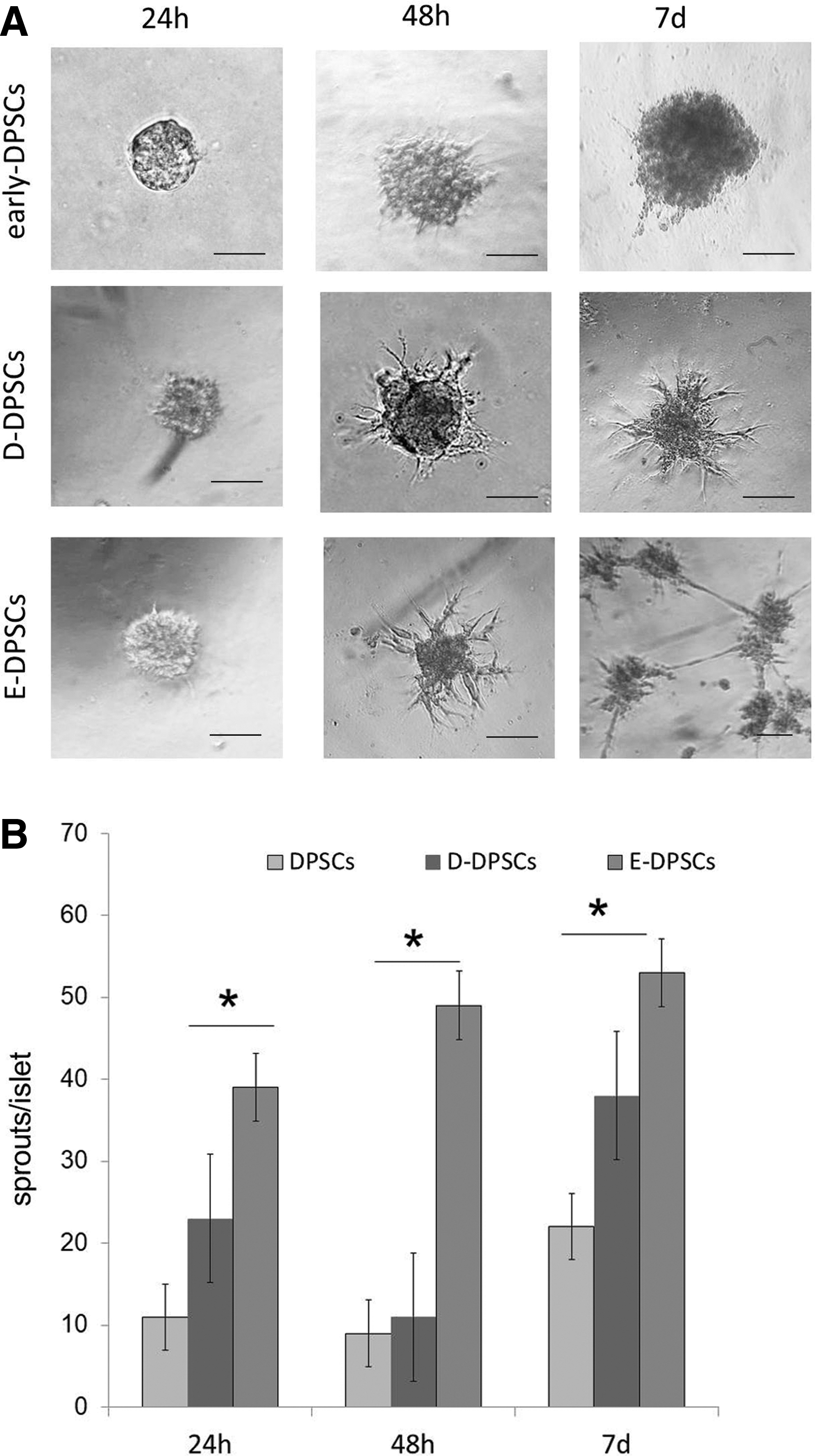
Sprouting ability of DPSCs.

Characterization of DPSCs cultured in different media.
E-DPSCs support endothelial tube formation
To test the hypothesis that E-DPSCs can support endothelial cells to form and stabilize vascular structures, we evaluated the effect of DPSCs during formation of tubes by HUVECs. As shown in Fig. 3A and B, HUVECs alone when cultured in EC Matrix formed tube network composed by lined cells that disappeared after 24 h. The coculture with E-DPSCs that were added 30 min after HUVECs, determined the stabilization of some HUVEC tubes and these stabilized tubes remained intact for up to 7 days (Fig. 3C, D). The new tubes appeared thicker and formed by juxtaposed cells (Fig. 3G). On the contrary, when D-DPSCs were added to HUVECs, very few tubes remained visible after 24 h and 7 days, and in addition, these tubes were prevalently formed by single lined cells (Fig. 3E–H). Quantitative analysis of tube formation was performed considering the mesh index (total master segment length/number of master segments) and the percentage of retention of total tube length of HUVEC alone and in coculture with D-DPSC and E-DPSC (Fig. 3I).
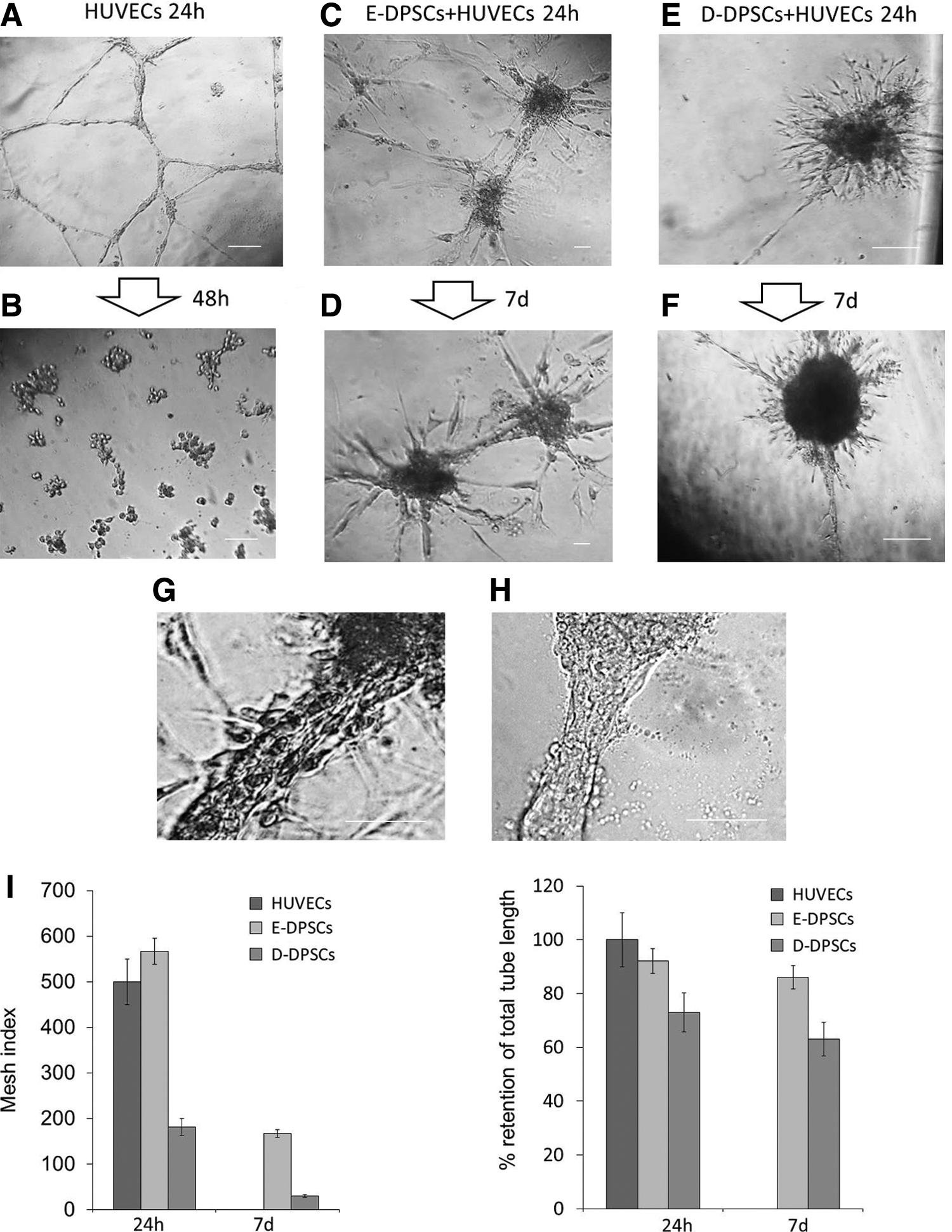
Vascular tube stabilization potential of DPSCs. HUVECs were cultured in EC Matrix alone
To confirm the collaboration between DPSCs and HUVECs in forming and stabilizing some tubes, we stained E-DPSCs with green fluorescent dye, calcein, and performed the coculture test with HUVECs in EC Matrix (Fig. 4). Analysis of images obtained from merging the phase contrast and fluorescence digital photos at different times revealed that, fluorescent E-DPSCs preferentially adhered on HUVECs, and, in particular, they attached to network nodes and then migrated along HUVEC tubes (Fig. 4, E-DPSCs+HUVECS 48 h vs. 7 days). Furthermore, in Fig. 5A, it is shown that after 24 h E-DPSCs bind preferentially to HUVECs and tended to align along tubes. Nonstained HUVECs and fluorescent DPSCs are visible as tight interacting cells localized both in network nodes and in tubes.
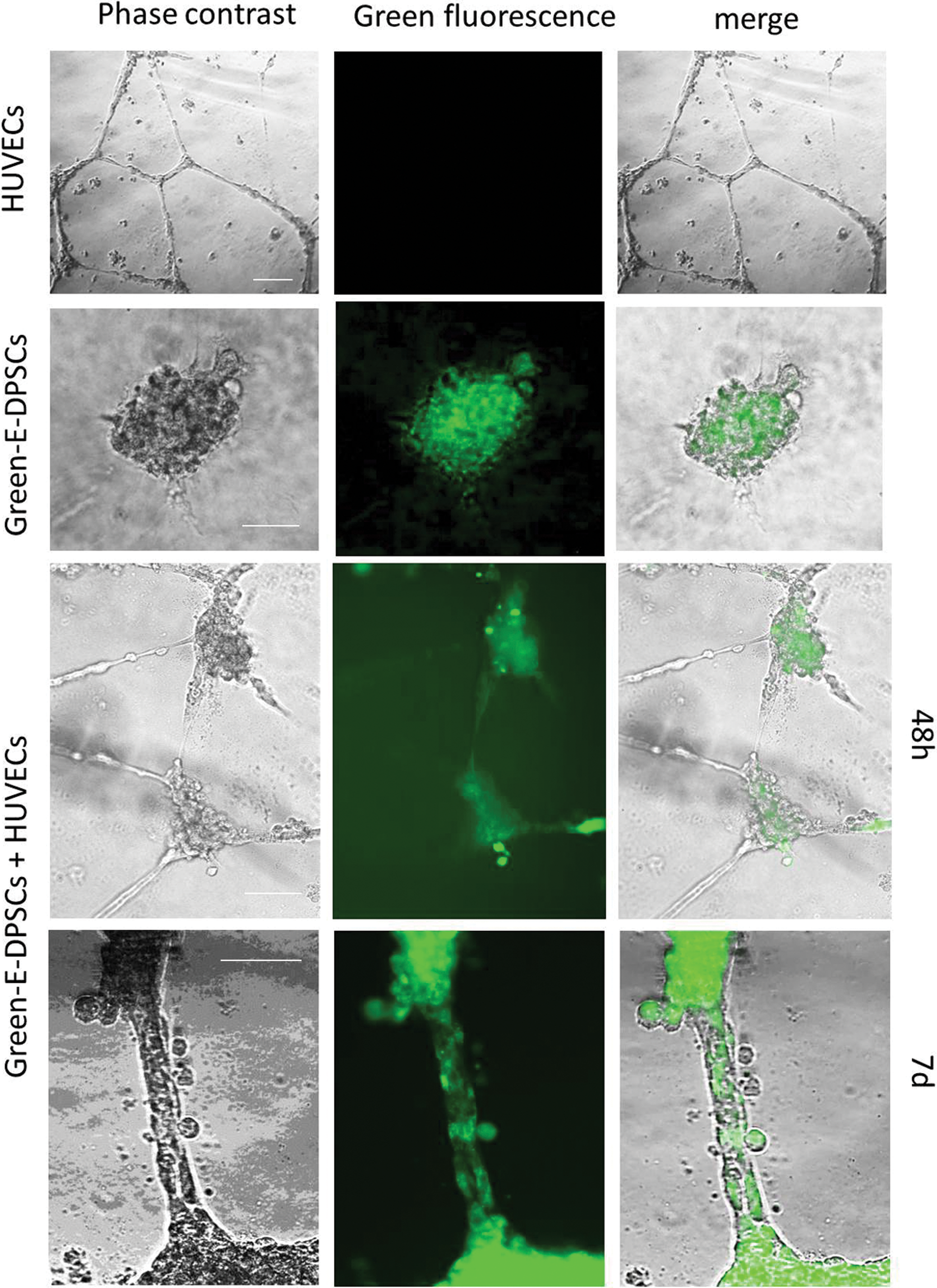
Vascular tube stabilization potential of E-DPSCs. HUVECs (images in the first line) and E-DPSCs (second line) were cultured in EC Matrix alone or cocultured (third and fourth lines). E-DPSCs were previously incubated with the fluorescent dye calcein. The same optical field was photographed both in phase-contrast and fluorescence light and resulting images were digitally merged (last column). For coculture, images taken at different time points (48 h and 7 days) are shown. Scale bar 100 μm. Color images are available online.
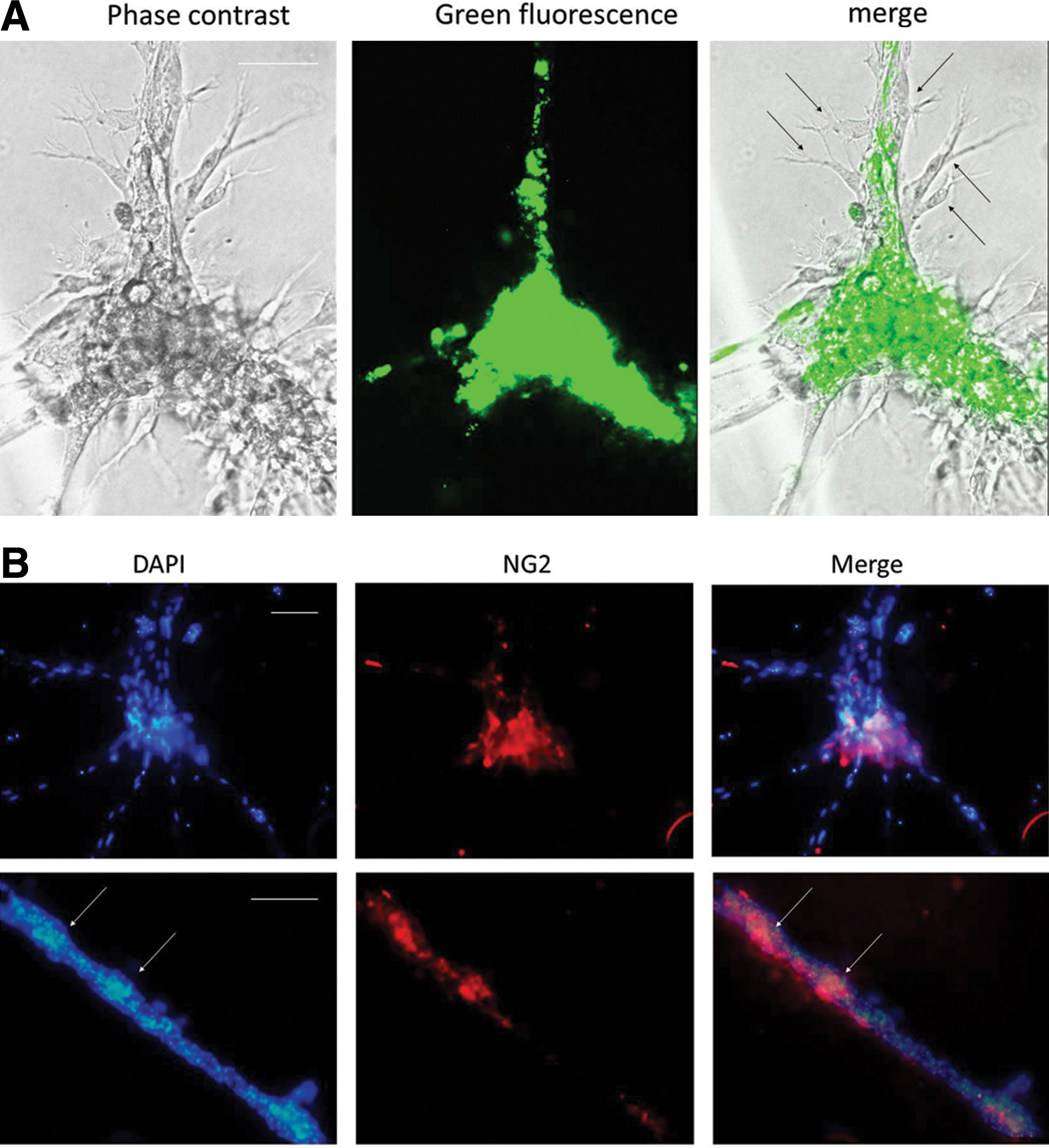
To verify the pericytic phenotype of E-DPSCs along the tubes, we immunostained cocultures for NG2, marker not usually expressed by HUVECs as shown also in Fig. 6A (Fig. 5B). NG2-positive cells were found on the tubes, mainly in more dense cellular zones and also in the nodes, mainly near the starting point of tubes.
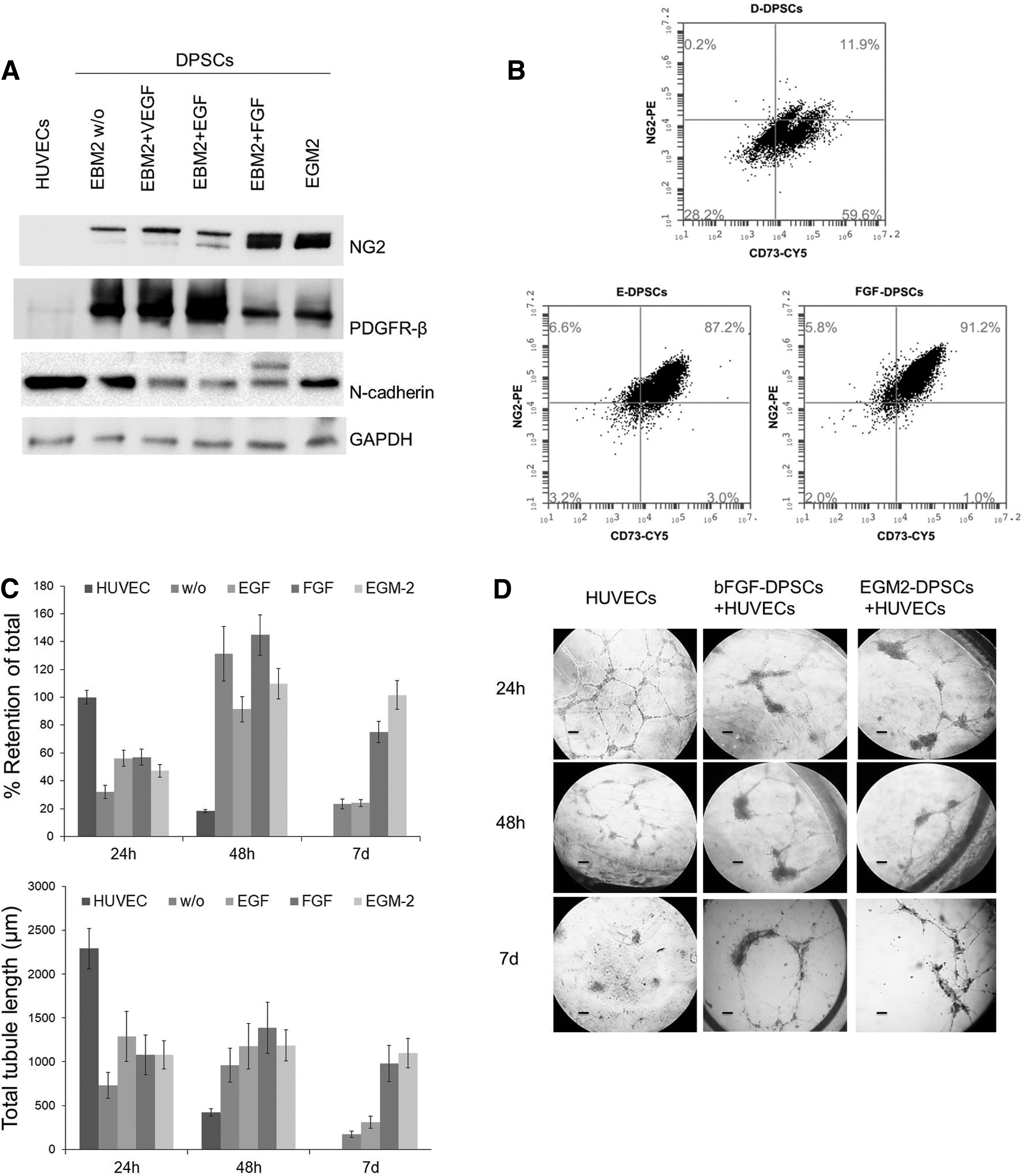
Characterization of DPSCs cultured with different growth factors.
bFGF is a key player in tube stabilization by DPSCs
To verify the impact of the different growth factors contained within EGM-2, we cultured DPSCs with EBM-2 (without supplements), or EBM-2 supplemented with single factors, FGF, VEGF, and EGF. Figure 6A shows results from representative western blots performed with total cell lysates from DPSCs in different culture conditions considering the expression of NG2, PDGFR-β, and N-cadherin. Single growth factors demonstrated a differential effect on selected markers and bFGF stimulated the highest expression of NG2 in the same way as EGM-2. PDGFR-β was expressed mainly in the absence of growth factors and was upregulated after the treatment with EGF. On the contrary, both bFGF and EGM-2 partially inhibited PDGFR-β expression. N-cadherin was expressed in all experimental condition and single growth factors were not associated with an increase with respect to EGM-2. The cytofluorimetric analysis for CD73 and NG2 confirmed that the treatment with bFGF induced the upregulation of NG2 in CD73+ DPSCs generating a phenotype similar to that observed in DPSCs cultured in EGM-2 medium (Fig. 6B). To verify the functional activity of DPSCs treated with bFGF, we performed a tube-supporting assay with HUVECs and calculated the total tube length and the percentage of retention of total tube length at different time points. bFGF-conditioned DPSCs were able to support the stabilization of HUVEC tubes for a longer time with respect to all other experimental condition and in a similar way as E-DPSC (Fig. 6C, histograms and representative images). The retention of endothelial tubes induced by bFGF and EGM-2 was particularly evident after 7 days of coculture.
Then, we sought to evaluate the potential role of bFGF in supporting the NG2+ cell phenotype in DPSCs. By MACS immunomagnetic separation we isolated NG2+ and NG2− DPSCs obtaining highly purified subpopulations then characterized for some stem and pericytic markers by fluorescence-activated cell sorting (Supplementary Fig. S2). Then, we evaluated the immunophenotype of these subpopulations after conditioning with EGM-2 and EBM-2 plus bFGF (Fig. 7A). bFGF and EGM-2 treatment in NG2− cells was able to induce a significant increment in the percentage of NG2+ cells (from 3.2% to 19.5% and 10.5%, respectively). At the same time, bFGF (or EGM-2) did not modify significantly the proliferative rate of NG2+ DPSCs determined by staining with Crystal Violet with respect to basal culture medium. However, FGF-conditioned cells required addiction on bFGF for their growth as demonstrated by the treatment with FGFR inhibitor AZD4547 (Fig. 7B). NG2+ cells were particularly responsive to bFGF treatment in their migratory ability. Indeed, FGF-conditioned DPSCs demonstrated an increased migratory capacity with respect to DPSCs cultured in basal culture conditions. The use of FGFR inhibitor counteracted the migratory phenotype induced by bFGF. Thus, FGF treatment was responsible for the differentiation of DPSCs toward NG2+ phenotype, and supports proliferation and migration of the resulting pericyte-like population.
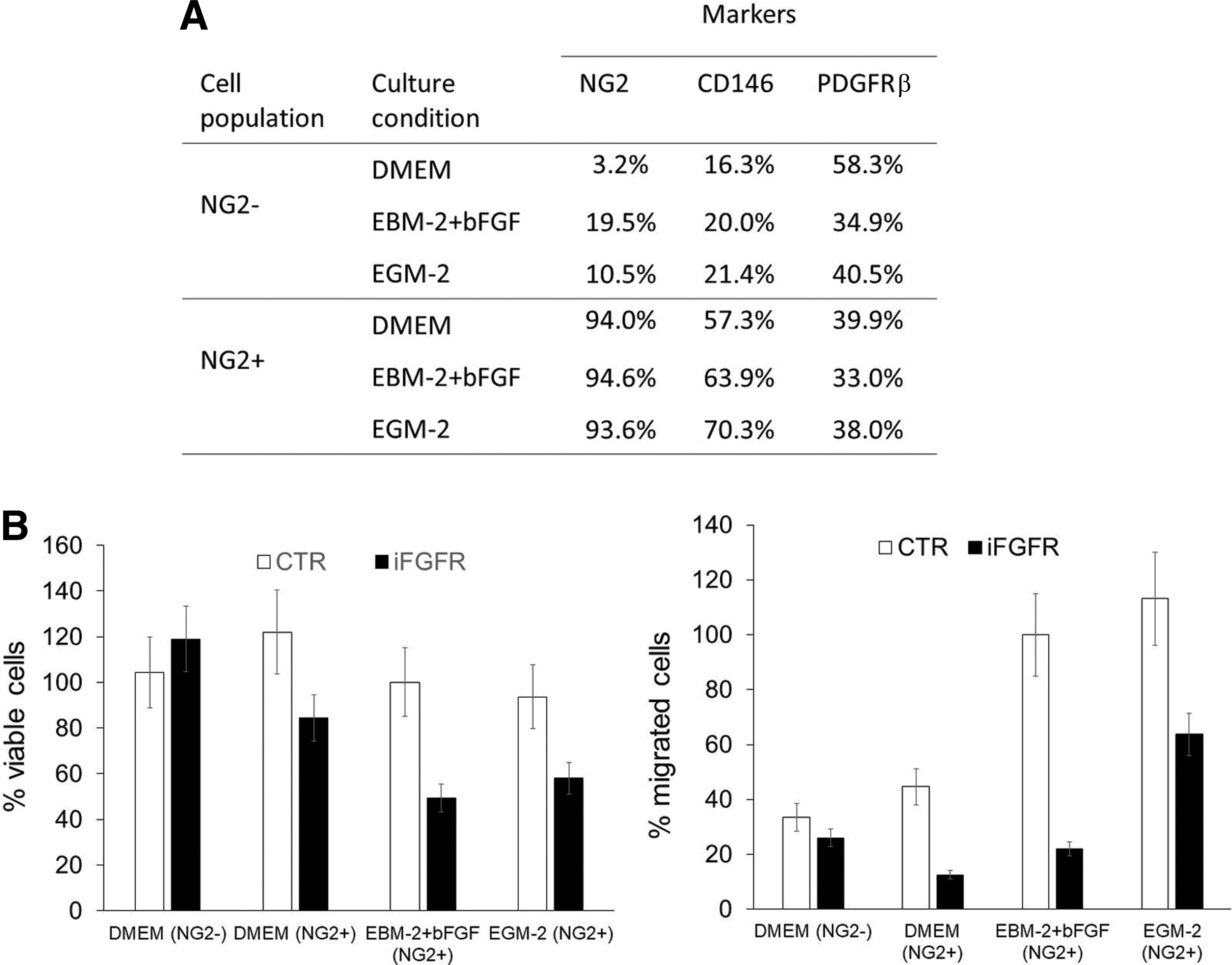
Phenotypic and functional characterization of DPSC subpopulations isolated by magnetic immunodetection of NG2 expression.
Discussion
Dental pulp plays a key role in the homeostasis and repair of tooth, first for maintaining the mineralized tissue; for many aspects, it resembles bone marrow. In both tissues the common determinant for the presence of MSCs is derived from the need for a multipotent cell population at the crossroad of many different cell types. However, according to ontogeny, DPSCs appear more enigmatic with respect to bone marrow MSCs, because they include stem cells of dual origin, neural glia and ectomesenchymal cells, suggesting a landscape in which different cell populations identified by specific combinations of cell surface protein coexist. The potential heterogeneity in tooth stem cells is exemplified by the description of different stem population according to their tissue of origin (eg, from periodontal ligament, apical papilla, dental follicle, and gingival tissue). Current data do not permit to exclude that dental stem populations of different origin are able to differentiate, in vitro and in vivo, in MSCs with similar potency, according to the physiological need or to culture conditions. It is disputable whether heterogeneity of dental stem cells can prevent differentiation obtaining reproducible results. In our procedure, we exclusively isolated dental pulp cells utilizing chemical digestion of the tissue obtained from young donors. The resulting cell population grew initially as isolated islets that rapidly proliferated generating DPSCs with elevated expression levels of the “classical” MSC markers, including CD44, CD90, CD105, and CD73. Although culture in 10% FBS-supplemented DMEM assured a high proliferation rate, isolated DPSCs in appropriate culture conditions were able to differentiate into osteogenic, adipogenic, chondrogenic, and also neural cells [26]. The presence of MSC phenotype warranted to obtain reproducible results in our experiments, also without considering further characterization.
Multipotent stem cells expressing the MSCs phenotype have been isolated from multiple human organs suggesting a ubiquitous healing role. For this reason, it is plausible that MSCs can orchestrate and/or contribute directly to the formation of the granulation tissue [27]. It is well known that blood vessels are contributed by nonvascular cells derived from multilineage progenitor cells, including hematopoietic cells [28] and myogenic cells [29]. Among these, endothelium-associated cells, known as pericytes, represent a key cell population, which surrounds endothelial cells in capillaries and microvessels [30]. The therapeutic contribution of MSCs to vasculature repair includes the direct differentiation into endothelial cells, smooth muscle cells, and pericytes. For example, in a swine model of chronic ischemic cardiomyopathy, the transplanted MSCs were found to differentiate also in SMCs and endothelium [31]. In a significant way, it was found that undifferentiated MSCs combined with HUVECs, seeded on a graft, were able to grow in vivo and to function as pericytes wrapping around the endothelial tubes [32]. MSCs can contribute in a similar functional manner to pericytes in coculture models stabilizing endothelium and its physical characteristics [33].
Our data indicated that DPSCs, when cultured in standard conditions (DMEM +10% fetal calf serum [FCS]) and analyzed as early as 3 weeks after isolation, contain a significant but high variable of NG2+ population (from 30% to 60% of positive cells). Intriguingly, in the last 10 years several data have accumulated about an apparent identity between MSCs and pericytes both in vitro and in vivo. Pericytes can originate from MSCs [32] and themselves are a source of MSCs, sharing the ability to differentiate in different cell lineages [34]. However, importantly, data in vivo seem to indicate that pericytes do not behave as MSCs in their regenerative function [35]. Collectively, available data suggest that pericytes are a highly plastic cell type showing functional and expression profiles common to MSCs and fibroblasts.
DPSCs cultured in DMEM behaved differently with respect to E-DPSC, with a lower sprouting ability and a lower capacity in stabilizing HUVEC tubes. These functional aspects were supported by a different phenotype associated with immature or specific subsets of pericytes. To date, pericytes are still defined according to their ability to elongate and wrap along endothelial cells. In fact, accumulating results in the last years are showing that pericytes are highly heterogeneous population in terms of ontogeny, molecular phenotype and, probably functions. Indeed, previous studies have demonstrated the existence of different pericyte subpopulations [36,37]. Differential expression level of common, but not specific, pericyte markers, α-SMA, PDGFR-β, and NG2, could identify multiple cell populations with different roles in angiogenesis [38].
The culture in EGM-2 determined a significant increase in the expression of NG2 but also a decrease in stem markers such as CD90. Significantly, all NG2+ cells were also CD73+. DPSCs cultured in EGM-2 expressed also PDGFR-β, N-cadherin, but low levels of α-SMA and did not express calponin. Among these markers only NG2 resulted coherently upregulated in parallel with functional gaining of DPSCs in EGM-2, thus supporting for this marker a key role in pericyte phenotype. In addition, we demonstrated that EGM-2 and bFGF were responsible in inducing the expression of NG2 in NG2− DPSCs. In an embryonal model, Kumar et al. observed in vitro the differentiation of two pericyte subpopulations different in PDGFR-β expression. Interestingly, in this study, in accordance with our results, CD73, but not CD90, expressed in both immature pericytes and mature pericyte subpopulations. The paracrine communication through PDGF-B/PDGFR-β allows the recruitment of pericytes by endothelial cells [39]. Our data indicate that high expression of PDGFR-β, as induced in DPSCs by EGF or VEGF, was not sufficient to determine a functional pericyte status in DPSCs. Thus, PDGFR-β expression looks to be a basal condition in pericyte lineage, but pericyte maturation seems to be associated mainly with the expression of NG2. NG2 is highly expressed in vivo in pericytes along capillaries and during angiogenesis when it appears implicated in capillary sprouting [40,41]. A tumor model confirmed that only a subpopulation of PDGFR-β+ pericyte progenitors was able to differentiate into more mature NG2+ or α-SMA+ phenotype [42].
While the direct association of pericytes with endothelial cells has been long described, their functional roles in angiogenesis have been largely underinvestigated. We know that during capillary sprouting, vascular pericytes are actively recruited by endothelial cells and their presence is critical for physiological angiogenesis. First, in vivo, pericytes may serve as a source of vasculature-associated stem cells, able to orchestrate the regeneration in different tissues. Indeed, pericytes play a leading role both in endothelial cell survival and migration and in avoiding vessel regression. This function is suggestively exemplified by results obtained in dystrophic mouse muscles, where transplanted pericytes demonstrated a regenerative potential higher than myoblasts and endothelial cells [22]. The role of transplanted pericytes could be primarily oriented in repairing host vascular networks facilitating the subsequent healing phases. Pericytes from human cord blood induced complete restoration of kidney function better that bone marrow derived MSCs after chemotherapy-induced acute renal failure [43]. In addition, pericytes were able to ameliorate ischemia after transplantation into mouse models by modulating several phases of recovery [44].
These data can indicate a double role of pericyte in injured tissue: promoting tube formation in its early phases, during the sprouting of endothelium; stabilizing formed vessels in unstable microenvironment. Our functional model, in which DPSCs were added on tube network, could offer information about the stabilization capacity of DPSCs. We observed an early ability of DPSCs to adhere to HUVECs, localizing preferentially on network nodes. Then DPSCs migrated along some tubes, forming complex structures that were stable for several days. The capacity of DPSCs to bind to endothelial cells is modulated by expression of specific membrane protein, including N-cadherin. Both HUVECs and DPSCs expressed N-cadherin allowing the homotypic and heterotypic binding between the cell types. DPSCs cultured in EGM-2 showed high expression of N-cadherin suggesting a particular capacity of these cells in selecting partner cells, such as endothelial cells. This aspect could be relevant in an in vivo model where DPSCs are transplanted in the host tissue, determining their predilection to adhere to vessels.
Conclusion
Our data confirm that dental pulp is an effective source of MSCs, represented by a cell population with a consistent MSC protein profile expression and high proliferative capacity in standard culture conditions. Specific culture conditions enriched DPSCs with a population expressing pericyte markers that in vitro demonstrated the capacity to stabilize endothelial tubes. Further molecular characterization of DPSCs and their in vivo transplantation will permit to verify regenerative potential of DPSCs in defective angiogenesis models.
Footnotes
Acknowledgment
The authors gratefully acknowledge the MIUR and ALCLI “Giorgio e Silvia,” a nonprofit association, for the financial support.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.